Genome-Wide Development of MicroRNA-Based SSR Markers in Medicago truncatula with Their Transferability Analysis and Utilization in Related Legume Species



Abstract
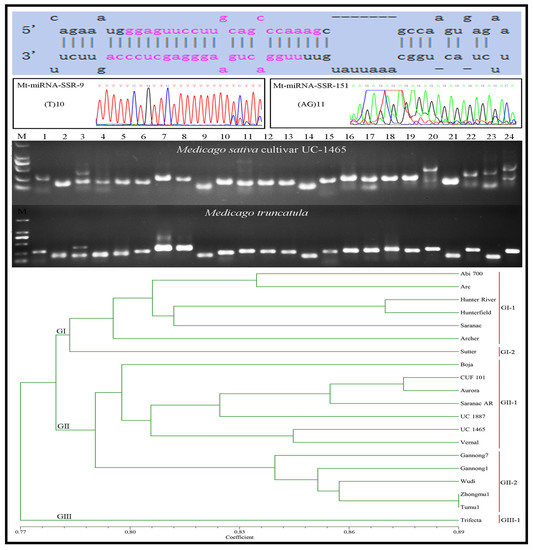
:
1. Introduction
2. Results and Discussion
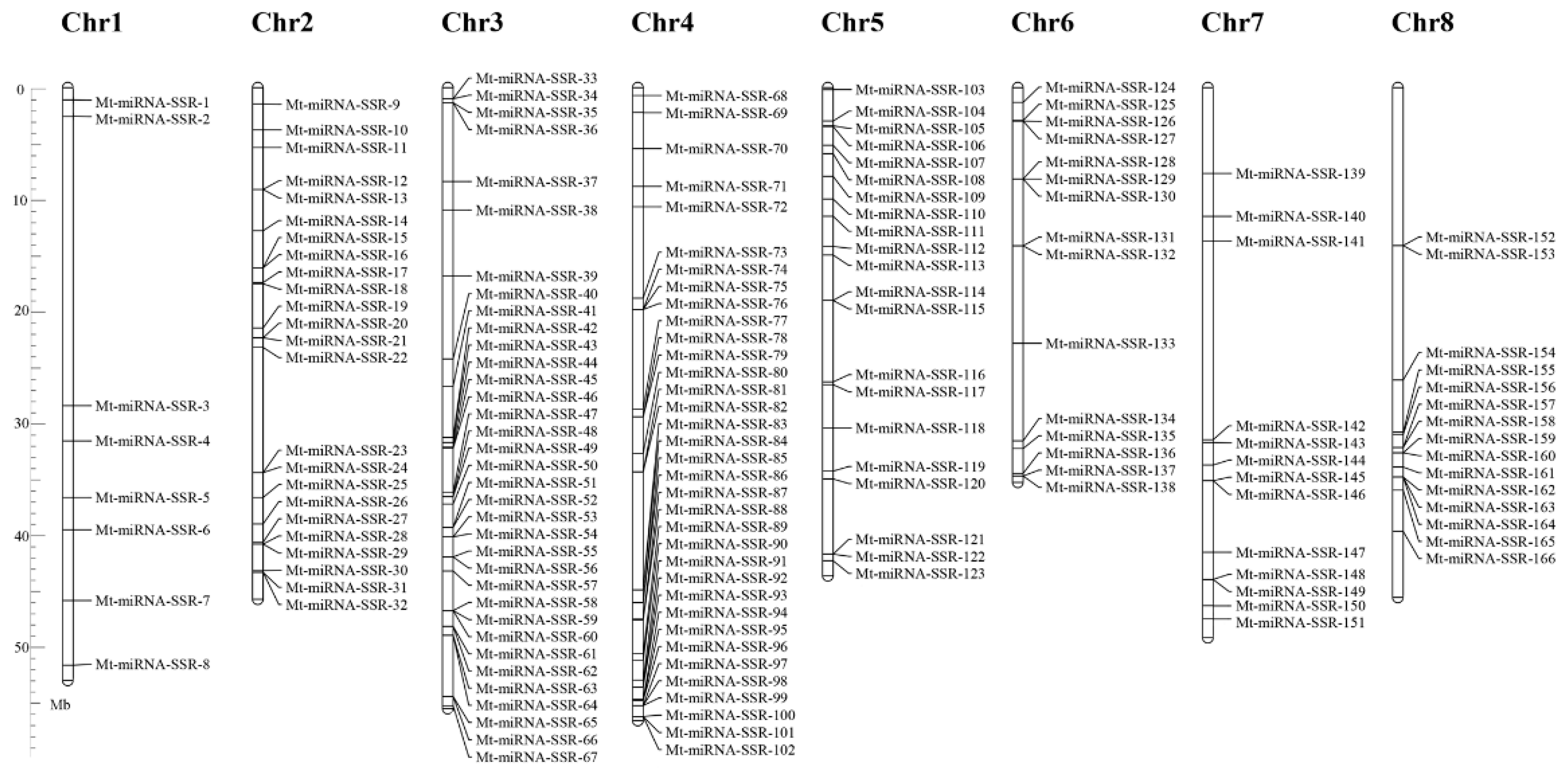
2.1. The Frequency, Distribution and Characterization of miRNA-SSRs in the M. truncatula Genome
2.2. Development of Mt-miRNA-SSR Markers
2.3. Functional Classification of SSR-Containing miRNA Genes
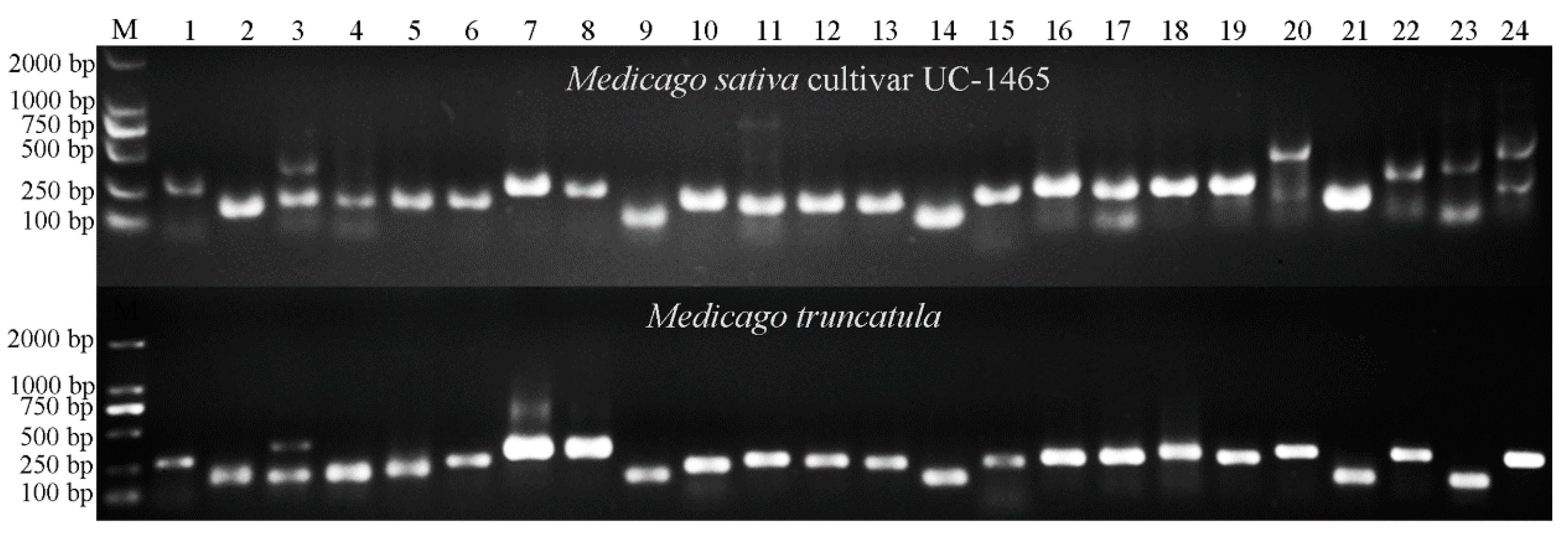
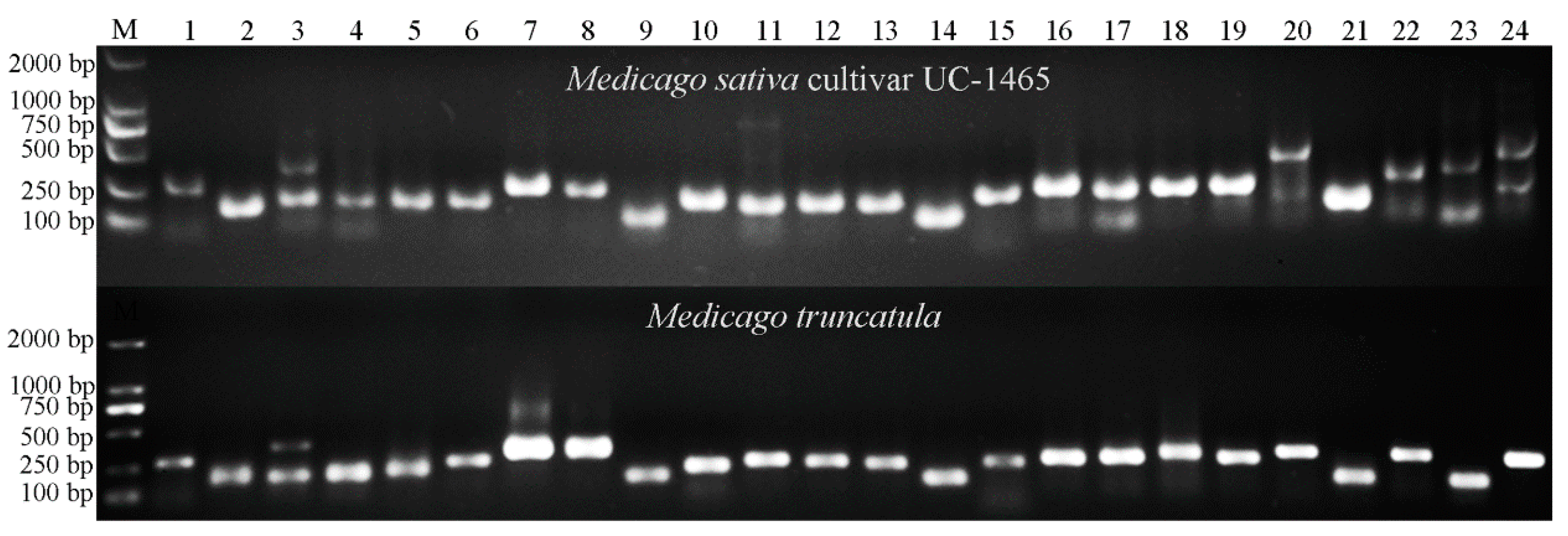
2.4. Transferability of Mt-mi-RNA-SSR Markers in Leguminous Species
2.5. Genetic Diversity Analysis of 20 Alfalfa Accessions
3. Materials and Methods
3.1. Plant Material and DNA Isolation
3.2. Identification of SSRs and Primer Design
3.3. Prediction of miRNA Targets and Functional Gene Ontology (GO) Analysis
3.4. PCR Amplification
3.5. Genetic Diversity Analysis
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Urrestarazu, J.; Miranda, C.; Santesteban, L.G.; Royo, J.B. Genetic diversity and structure of local apple cultivars from northeastern spain assessed by microsatellite markers. Tree Genet. Genomes 2012, 8, 1163–1180. [Google Scholar] [CrossRef]
- Jiang, G.-L. Molecular markers and marker-assisted breeding in plants. In Plant Breeding from Laboratories to Fields; Andersen, S.B., Ed.; InTech: Rijeka, Croatia, 2013; pp. 123–126. [Google Scholar]
- Lassois, L.; Denancé, C.; Ravon, E.; Guyader, A.; Guisnel, R.; Hibrand-Saint-Oyant, L.; Poncet, C.; Lasserre-Zuber, P.; Feugey, L.; Durel, C.E. Genetic diversity, population structure, parentage analysis, and construction of core collections in the french apple germplasm based on SSR markers. Plant Mol. Biol. 2016, 34, 827–844. [Google Scholar] [CrossRef]
- Ištvánek, J.; Dluhošová, J.; Dluhoš, P.; Pátková, L.; Nedělník, J.; Řepková, J. Gene classification and mining of molecular markers useful in red clover (Trifolium pratense) breeding. Front. Plant Sci. 2017, 8, 307. [Google Scholar] [CrossRef] [PubMed]
- Yuan, S.; Ge, L.; Liu, C.; Ming, J. The development of EST-SSR markers in Lilium regale and their cross-amplification in related species. Euphytica 2013, 189, 393–419. [Google Scholar] [CrossRef]
- Abbas, G.; Hameed, A.; Rizwan, M.; Ahsan, M.; Asghar, M.J.; Iqbal, N. Genetic confirmation of mungbean (Vigna radiata) and mashbean (Vigna mungo) interspecific recombinants using molecular markers. Front. Plant Sci. 2015, 6, 1107. [Google Scholar] [CrossRef] [PubMed]
- Bruford, M.W.; Wayne, R.K. Microsatellites and their application to population genetic studies. Curr. Opin. Genet. Dev. 1993, 3, 939–943. [Google Scholar] [CrossRef]
- Ni, J.; Colowit, P.M.; Mackill, D.J. Evaluation of genetic diversity in rice subspecies using microsatellite markers. Crop Sci. 2002, 42, 601–607. [Google Scholar] [CrossRef]
- Khatoon, A.; Verma, S.; Wadiye, G.; Zore, A. Molecular markers and their potentials. Int. J. Bioassays 2016, 5, 4706–4714. [Google Scholar] [CrossRef]
- Liu, M.; Xu, Y.; He, J.; Zhang, S.; Wang, Y.; Lu, P. Genetic diversity and population structure of broomcorn millet (panicummiliaceuml.) cultivars and landraces in China based on microsatellite markers. Int. J. Mol. Sci. 2016, 17, 370. [Google Scholar] [PubMed]
- Liu, W.; Liu, W.; Jun, W.; Gao, A.; Li, H.; Li, I. Analysis of genetic diversity in natural populations of Psathyrostachys huashanica keng using microsatellite (SSR) markers. J. Integr. Agric. 2010, 9, 463–471. [Google Scholar] [CrossRef]
- Mondal, T.K.; Ganie, S.A. Identification and characterization of salt responsive miRNA-SSR markers in rice (Oryza sativa). Gene 2014, 535, 204–209. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Min, X.; Wang, Z.; Wang, Y.; Liu, Z.; Liu, W. Genome-wide development and utilization of novel intron-length polymorphic (ILP) markers in Medicago sativa. Mol. Breed. 2017, 37, 87. [Google Scholar] [CrossRef]
- Bartel, D.P. Micrornas: Target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Pan, X.; Cobb, G.P.; Anderson, T.A. Plant microrna: A small regulatory molecule with big impact. Dev. Biol. 2006, 289, 3–16. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. Micrornas: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef]
- Vitsios, D.M.; Davis, M.P.; Dongen, S.V.; Enright, A.J. Large-scale analysis of microRNA expression, epi-transcriptomic features and biogenesis. Nucleic Acids Res. 2016, 45, 1079–1090. [Google Scholar] [CrossRef] [PubMed]
- Lukasik, A.; Zielenkiewicz, P. Plant microRNAs-novel players in natural medicine? Int. J. Mol. Sci. 2016, 18, 9. [Google Scholar] [CrossRef] [PubMed]
- Voinnet, O. Origin, biogenesis, and activity of plant microRNAs. Cell 2009, 136, 669–687. [Google Scholar] [CrossRef] [PubMed]
- Fahlgren, N.; Jogdeo, S.; Kasschau, K.D.; Sullivan, C.M.; Chapman, E.J.; Laubinger, S.; Smith, L.M.; Dasenko, M.; Givan, S.A.; Weigel, D. MicroRNA gene evolution in Arabidopsis lyrata and Arabidopsis thaliana. Plant Cell 2010, 22, 1074–1089. [Google Scholar] [CrossRef] [PubMed]
- Yadav, C.B.; Muthamilarasan, M.; Pandey, G.; Khan, Y.; Prasad, M. Development of novel microRNA-based genetic markers in foxtail millet for genotyping applications in related grass species. Mol. Breed. 2014, 34, 2219–2224. [Google Scholar] [CrossRef]
- Ganie, S.A.; Mondal, T.K. Genome-wide development of novel miRNA-based microsatellite markers of rice (Oryza sativa) for genotyping applications. Mol. Breed. 2015, 35, 51. [Google Scholar] [CrossRef]
- Wang, X.; Gui, S.; Pan, L.; Hu, J.; Ding, Y. Development and characterization of polymorphic microRNA-based microsatellite markers in Nelumbo nucifera (nelumbonaceae). Appl. Plant Sci. 2016, 4, 1500091. [Google Scholar] [CrossRef] [PubMed]
- Fu, D.; Ma, B.; Mason, A.S.; Xiao, M.; Wei, L.; An, Z. MicroRNA-based molecular markers: A novel PCR-based genotyping technique in Brassica species. Plant Breed. 2013, 132, 375–381. [Google Scholar] [CrossRef]
- Wang, T.; Lei, C.; Zhao, M.; Tian, Q.; Zhang, W.H. Identification of drought-responsive microRNAs in Medicago truncatula by genome-wide high-throughput sequencing. BMC Genom. 2011, 12, 367. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Yu, J.; Li, D.; Zhang, Z.; Liu, F.; Zhou, X.; Wang, T.; Ling, Y.; Su, Z. Pmrd: Plant microRNA database. Nucleic Acids Res. 2010, 38, D806–D813. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Han, G.; Shang, C.; Li, J.; Zhang, H.; Liu, F.; Wang, J.; Liu, H.; Zhang, Y. Proteomic analyses reveal differences in cold acclimation mechanisms in freezing-tolerant and freezing-sensitive cultivars of alfalfa. Front Plant Sci. 2015, 6, 105. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Zhang, Z.; Chen, S.; Ma, L.; Wang, H.; Dong, R.; Wang, Y.; Liu, Z. Global transcriptome profiling analysis reveals insight into saliva-responsive genes in alfalfa. Plant Cell Rep. 2016, 35, 561–571. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Khurshid, M.; Sun, Z.M.; Tang, Y.; Zhou, M.L.; Wu, Y.M. Genetic engineering of alfalfa (Medicago sativa L.). Protein Pept. Lett. 2016, 23, 495–502. [Google Scholar] [CrossRef] [PubMed]
- Jonker, A.; Yu, P. The role of proanthocyanidins complex in structure and nutrition interaction in alfalfa forage. Int. J. Mol. Sci. 2016, 17, 793. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.K.; Kim, D.; Uhm, T.; Limpens, E.; Lim, H.; Mun, J.H.; Kalo, P.; Penmetsa, R.V.; Seres, A.; Kulikova, O. A sequence-based genetic map of Medicago truncatula and comparison of marker colinearity with M. Sativa. Genetics 2004, 166, 1463–1502. [Google Scholar] [CrossRef]
- Zhu, H.; Choi, H.K.; Cook, D.R.; Shoemaker, R.C. Bridging model and crop legumes through comparative genomics. Plant Physiol. 2005, 137, 1189–1196. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Prasad, M. Development and characterization of genic ssr markers in Medicago truncatula and their transferability in leguminous and non-leguminous species. Genome 2009, 52, 761–771. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Jia, X.; Liu, Z.; Zhang, Z.; Wang, Y.; Liu, Z.; Xie, W. Development and characterization of transcription factor gene-derived microsatellite (TFGM) markers in Medicago truncatula and their transferability in leguminous and non-leguminous species. Molecules 2015, 20, 8759–8771. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Chu, C. MicroRNAs in crop improvement: Fine-tuners for complex traits. Nat. Plants 2017, 3, 17077. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Wang, T.; Zhao, M.; Tian, Q.; Zhang, W.H. Identification of aluminum-responsive microRNAs in Medicago truncatula by genome-wide high-throughput sequencing. Planta 2012, 235, 375–386. [Google Scholar] [CrossRef] [PubMed]
- Long, R.; Li, M.; Kang, J.; Zhang, T.; Sun, Y.; Yang, Q. Small RNA deep sequencing identifies novel and salt-stress-regulated microRNAs from roots of Medicago sativa and Medicago truncatula. Physiol. Plant. 2015, 154, 13–27. [Google Scholar] [CrossRef] [PubMed]
- Trindade, I.; Capitão, C.; Dalmay, T.; Fevereiro, M.P.; Santos, D.M. miR398 and miR408 are up-regulated in response to water deficit in Medicago truncatula. Planta 2010, 231, 705–716. [Google Scholar] [CrossRef] [PubMed]
- Bandelj, D.; Jakše, J.; Javornik, B. Assessment of genetic variability of olive varieties by microsatellite and AFLP markers. Euphytica 2004, 136, 93–102. [Google Scholar] [CrossRef]
- Wang, Z.; Yan, H.; Fu, X.; Li, X.; Gao, H. Development of simple sequence repeat markers and diversity analysis in alfalfa (Medicago sativa L.). Mol. Biol. Rep. 2013, 40, 3291–3298. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Chen, T.; Wang, Y.; Liu, Z. The development of 204 novel EST-SSRs and their use for genetic diversity analyses in cultivated alfalfa. Biochem. Syst. Ecol. 2014, 57, 227–230. [Google Scholar] [CrossRef]
- Kozomara, A.; Griffithsjones, S. MiRBase: Integrating microRNA annotation and deep-sequencing data. Nucleic Acids Res. 2011, 39, D152–D157. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Niu, B.; Gao, Y.; Fu, L.; Li, W. Cd-hit suite: A web server for clustering and comparing biological sequences. Bioinformatics 2010, 26, 680–682. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Zhao, P.X. Psrnatarget: A plant small RNA target analysis server. Nucleic Acids Res. 2011, 39, W155–W159. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Search Items | Numbers |
|---|---|
| Total number of pri_miRNA sequence examined | 356 |
| Total number of identified SSRs | 189 |
| Number of SSR containing sequences | 137 |
| Number of pri-miRNA containing more than 1 SSR | 46 |
| Number of SSRs present in compound formation | 21 |
| Repeat type | |
| Mononucleotide | 133 |
| Dinucleotide | 27 |
| Trinucleotide | 8 |
| Total length of sequences searched (kb) | 407.9 |
| Frequency of SSRs | One per 0.5 kb |
| Species | Transferability |
|---|---|
| Medicago truncatula | 157 (92.90%) |
| Medicago sativa | 126 (74.56%) |
| Glycine max | 131 (77.51%) |
| Vicia sativa | 151 (89.35%) |
| Melilotus | 150 (88.76%) |
| Lotus corniculatus | 153 (90.53%) |
| Sophora alopecuroides | 151 (89.35%) |
| No. | Primer Name | Primer Sequence (5′–3′) | No. of Alleles | He | PIC Value |
|---|---|---|---|---|---|
| 1 | Mt-miRNA-SSR-7 | F: CGCAACCAACATAGAAGCAA | 11 | 0.84 | 0.82 |
| R: CGCGGTCTTATTAGGGATCA | |||||
| 2 | Mt-miRNA-SSR-10 | F: GCATCGCCGTTATTAACAAAA | 3 | 0.56 | 0.47 |
| R: CGGCTTCATACACAGGGAAT | |||||
| 3 | Mt-miRNA-SSR-26 | F: TTGCAAACCAAACACACACA | 5 | 0.78 | 0.74 |
| R: GCGACATACAATTTGGGCTT | |||||
| 4 | Mt-miRNA-SSR-28 | F: CAAACGTTTTCTCAATTTCTAATCG | 7 | 0.71 | 0.66 |
| R: CCAAGGTTGTTCCAAGGTGT | |||||
| 5 | Mt-miRNA-SSR-29 | F: AGCCTCTCATTTAATTGGTGC | 4 | 0.69 | 0.62 |
| R: GCAGGTGCAAATGCAGATAA | |||||
| 6 | Mt-miRNA-SSR-37 | F: TCCTTTGCTCTTCCAACTCTTT | 18 | 0.90 | 0.89 |
| R: CCCCCTTTGTTAGCAGATGA | |||||
| 7 | Mt-miRNA-SSR-38 | F: AATGTATGGAGAGGATGAGCTTT | 6 | 0.68 | 0.65 |
| R: AACCAGATTACCTTCATCATTCG | |||||
| 8 | Mt-miRNA-SSR-41 | F: TGGTTCAGAAACGGTTAGGG | 5 | 0.76 | 0.72 |
| R: CAGAAAGGTCCAGAAGCCAA | |||||
| 9 | Mt-miRNA-SSR-45 | F: GTGAAGCAATGGTGCCTTTT | 10 | 0.75 | 0.71 |
| R: TCACGGCTCAAAGGTATGTG | |||||
| 10 | Mt-miRNA-SSR-47 | F: TCAATCAGAAAAATTGCACCC | 13 | 0.87 | 0.86 |
| R: AAAGTTTTTGTTGGGAAAGATGA | |||||
| 11 | Mt-miRNA-SSR-52 | F: ATTTTGTGTGCCATCGTGAA | 14 | 0.85 | 0.83 |
| R: GGGACCGGTTATCTTTTGGT | |||||
| 12 | Mt-miRNA-SSR-63 | F: GCCATGTTTTGCATGACTGT | 13 | 0.90 | 0.89 |
| R: TGCAGGTCCAAATTCAACAA | |||||
| 13 | Mt-miRNA-SSR-87 | F: TGAAATGCCTTTTTCTTCCC | 7 | 0.83 | 0.81 |
| R: TTCCCAAACACCATCATCAA | |||||
| 14 | Mt-miRNA-SSR-99 | F: GAGGACGGATCAATAGGCAA | 4 | 0.69 | 0.62 |
| R: TGTCTGGAAAGTGCTTCACAA | |||||
| 15 | Mt-miRNA-SSR-102 | F: CCACGATGCTACACACGTTC | 4 | 0.44 | 0.39 |
| R: CTTCCACGTCCAGACCAACT | |||||
| 16 | Mt-miRNA-SSR-125 | F: TAGCCCTGCCAGCCTATTTA | 15 | 0.81 | 0.79 |
| R: AAGGTGTCATCTCTCCTGCG | |||||
| 17 | Mt-miRNA-SSR-126 | F: TTCTCCAGCAGTGCTATTCTGA | 7 | 0.83 | 0.81 |
| R: TGCTGTTCCTTTGTTTTCAATG | |||||
| 18 | Mt-miRNA-SSR-130 | F: TCCATGTTTTTGGCATCAGA | 4 | 0.60 | 0.52 |
| R: AATTGGGGAAATAAGGGGTG | |||||
| 19 | Mt-miRNA-SSR-142 | F: CCAAAAAGATTTGGCCCTTT | 6 | 0.81 | 0.78 |
| F: GCATGGTTGTCCCTTGCTAT | |||||
| 20 | Mt-miRNA-SSR-144 | R: TGCTTGTTCAATTTCGAATG | 4 | 0.69 | 0.62 |
| F: CTTAAGTTACCTGTCCGGCG | |||||
| 21 | Mt-miRNA-SSR-148 | R: TGCTCATGTTGATTCCCAGA | 5 | 0.58 | 0.50 |
| F: AGCATTAGTTGTCATGCCCC | |||||
| 22 | Mt-miRNA-SSR-151 | R: AAACATGTGGGGTTTGGTGT | 10 | 0.89 | 0.88 |
| F: AGCCAAGTTTGGACCATCAG | |||||
| 23 | Mt-miRNA-SSR-163 | R: GGGGAGGAAAGGTTGAATTT | 12 | 0.87 | 0.86 |
| F: GCTGCAGTTAACTACCGAGGA | |||||
| 24 | Mt-miRNA-SSR-164 | R: GGGGAGGAAAGGTTGAATTT | 6 | 0.78 | 0.74 |
| F: GCTGCAGTTAACTACCGAGGA | |||||
| 25 | Mt-miRNA-SSR-166 | R: GCACCATTAGTGTGGTGTGAG | 4 | 0.70 | 0.64 |
| F: GCCAACATTCCCCTCAAATA | |||||
| Average | 7.88 | 0.75 | 0.71 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Min, X.; Zhang, Z.; Liu, Y.; Wei, X.; Liu, Z.; Wang, Y.; Liu, W. Genome-Wide Development of MicroRNA-Based SSR Markers in Medicago truncatula with Their Transferability Analysis and Utilization in Related Legume Species. Int. J. Mol. Sci. 2017, 18, 2440. https://doi.org/10.3390/ijms18112440
Min X, Zhang Z, Liu Y, Wei X, Liu Z, Wang Y, Liu W. Genome-Wide Development of MicroRNA-Based SSR Markers in Medicago truncatula with Their Transferability Analysis and Utilization in Related Legume Species. International Journal of Molecular Sciences. 2017; 18(11):2440. https://doi.org/10.3390/ijms18112440
Chicago/Turabian StyleMin, Xueyang, Zhengshe Zhang, Yisong Liu, Xingyi Wei, Zhipeng Liu, Yanrong Wang, and Wenxian Liu. 2017. "Genome-Wide Development of MicroRNA-Based SSR Markers in Medicago truncatula with Their Transferability Analysis and Utilization in Related Legume Species" International Journal of Molecular Sciences 18, no. 11: 2440. https://doi.org/10.3390/ijms18112440