1. Introduction
Increasing the complexity of in vitro systems to mimic three-dimensional tissues and the cellular interactions within them will increase the reliability of data that were previously collected with in vitro systems. The development of these in vitro systems requires models that display maximum resemblance to the situations found in tissues in vivo. Adequate morphogenesis and cell functions depend on close interactions between cells as well as between cells and their matrix, because both types of interaction are critical for the organization of a microenvironment in vitro that is as close to the in vivo situation as possible. Without the latter, cells lose their organotypical characteristics and transdifferentiate [
1,
2].
In vivo, capillaries are embedded in a microenvironment that consists of the extracellular matrix (ECM) and of cellular components including fibroblasts (FBs) as well as immune cells. The ECM is a complex, noncellular network composed of distinct components that is found in two different locations, i.e., in the interstitium, as interstitial ECM and in association with all epithelia and endothelia, as the basement membrane (BM) [
3]. In vivo, the ECM components are mainly synthesized by FBs, which, among others, secrete various collagens, fibronectin, and heparin sulfate proteoglycans [
4,
5]. The development of blood vessels, which is a requirement for growth and regeneration, depends on a highly structured communication of endothelial cells (ECs) with their surrounding tissue. In vivo vascularization is based on complex cell–matrix and cell–cell interactions, in which the ECM and the FBs seem to play a very important role [
6,
7]. Blood vessels either develop de novo by the process of vasculogenesis, or they arise by angiogenesis, where new capillaries grow from already existing ones. Vasculogenesis is the determination and differentiation of endothelial precursor cells that arrange themselves into aggregates of cells and then create a simple network of tubes having a lumen [
8]. Angiogenesis appears to occur by two mechanisms, namely non-sprouting (intussusceptive) and sprouting angiogenesis [
9]. During intussusception, endothelial protrusions of opposing capillary walls extend towards each other and fuse creating an interendothelial contact [
8].
Sprouting angiogenesis has been a recent focus of intense research. It is a process having many sequential hierarchical steps that require the close interaction of ECs with both acellular and cellular components of the surrounding tissue, including FBs [
4]. In fact, FBs exert a significant role in angiogenesis [
5]. These cells synthesize angiogenic growth factors that trigger sprouting angiogenesis. These factors include vascular endothelial growth factor (VEGF-A), transforming growth factor-β (TGF-β), and platelet-derived growth factor (PDGF) [
4,
5,
10]. Angiogenic stimuli activate the ECs to migrate into the avascular tissue [
11]. ECs express VEGF-receptor 2 that responds to the VEGF-A gradient and binds to the growth factor. Once an angiogenic stimulus occurs, metalloproteinases (MMPs) break down the basement membrane (BM) of the blood vessel, mainly near the trigger sites [
12]. During sprouting, ECs are triggered by the VEGF-R2–VEGF-A reaction to temporarily transform into migrating tip cells. These cells are polarized and have well-developed filopodia that enable them to interact with the ECM via integrins. Integrins (primarily alphaVß3) on the ECs’ filopodia surface have an adhesive function during endothelial migration [
11]. ECM proteins, mainly synthesized by FBs, are important for adhesion and migratory processes of the endothelial tip cells and therefore promote angiogenesis [
3,
6,
13,
14]. The endothelial tip cells traverse into the surrounding avascular extracellular matrix towards the angiogenic stimulus [
15]. To enable this process, the MMPs form tunnels in the ECM to facilitate endothelial migration [
16]. Membrane-type 1 matrix metalloproteinases (MT1-MMP), selectively expressed by the endothelial tip cells, are responsible for most of the proteolysis of the ECM [
15,
17]. Endothelial stalk cells follow the tip cells into the ECM where they proliferate and elongate the developing capillary sprout, as well as establish its internal lumen. The tubular lumen is formed by intraendothelial vacuoles that fuse. For the development of the vacuoles, MT1-MMP and the integrins alphaVß3 and alpha5ß1 play important roles [
18]. In addition, FB-derived matrix proteins, i.e., collagen I and fibronectin, are necessary for tube formation [
5,
6]. Tight and adherens cell junctions are established between the stalk cells of the newly built tube, and, consequently, a new vessel arises.
Due to their roles in cell–matrix interactions and especially in matrix remodeling, FBs are crucial in vascular development through transmitting biochemical signals and mechanical forces that affect endothelial cell survival, cell shape, and cell orientation [
19]. In cooperation with endothelial stalk cells, surrounding FBs synthesize basement membrane proteins, namely laminin, collagen IV, perlecan, nidogen, collagen XVIII, and fibronectin. The BM envelops and stabilizes the newly developing capillary sprout, serving as an acellular barrier against the capillaries microenvironment and ensuring the correct polarity of ECs [
12]. It typically consists of an electron-dense layer (lamina densa) separated from the cellular plasma membrane by an electron-lucent layer (lamina lucida). The outermost third sheet of the BM is a less dense but thicker layer of diffuse ECM that constitutes the lamina fibroreticularis [
20]. Maturation, stabilization, and remodeling of the dynamic capillary structures follow initial angiogenesis [
21]. As tubules mature, their ECs transform into quiescent phalanx cells [
22].
While the molecular aspects of angiogenesis, including signal molecules and their specific receptors, have been studied in great detail, hardly any attention has been given to the physical interaction, communication, and exchange of information between cells or between cells and their immediate surroundings.
The aim of this study was to monitor and visualize the cellular events and interactions between ECs, FBs, and their surrounding microenvironment during vascular morphogenesis in a three-dimensional coculture model. Moreover, the ECM proteins laminin, fibronectin, and collagen III, which are known to be significant at different steps of angiogenesis, were examined morphometrically.
3. Discussion
The multifaceted composition and interplay of cells and molecules within a tissue or organ play an essential role and can only be mirrored in cocultures of at least two different cell species [
13,
23,
24,
25]. To optimize the existing in vitro models of angiogenesis [
26,
27], it is essential to take into account the interaction of the ECs with the cells and molecules present in their physiologic environment, and to include further cell types, especially stromal FB [
13,
24,
25,
28].
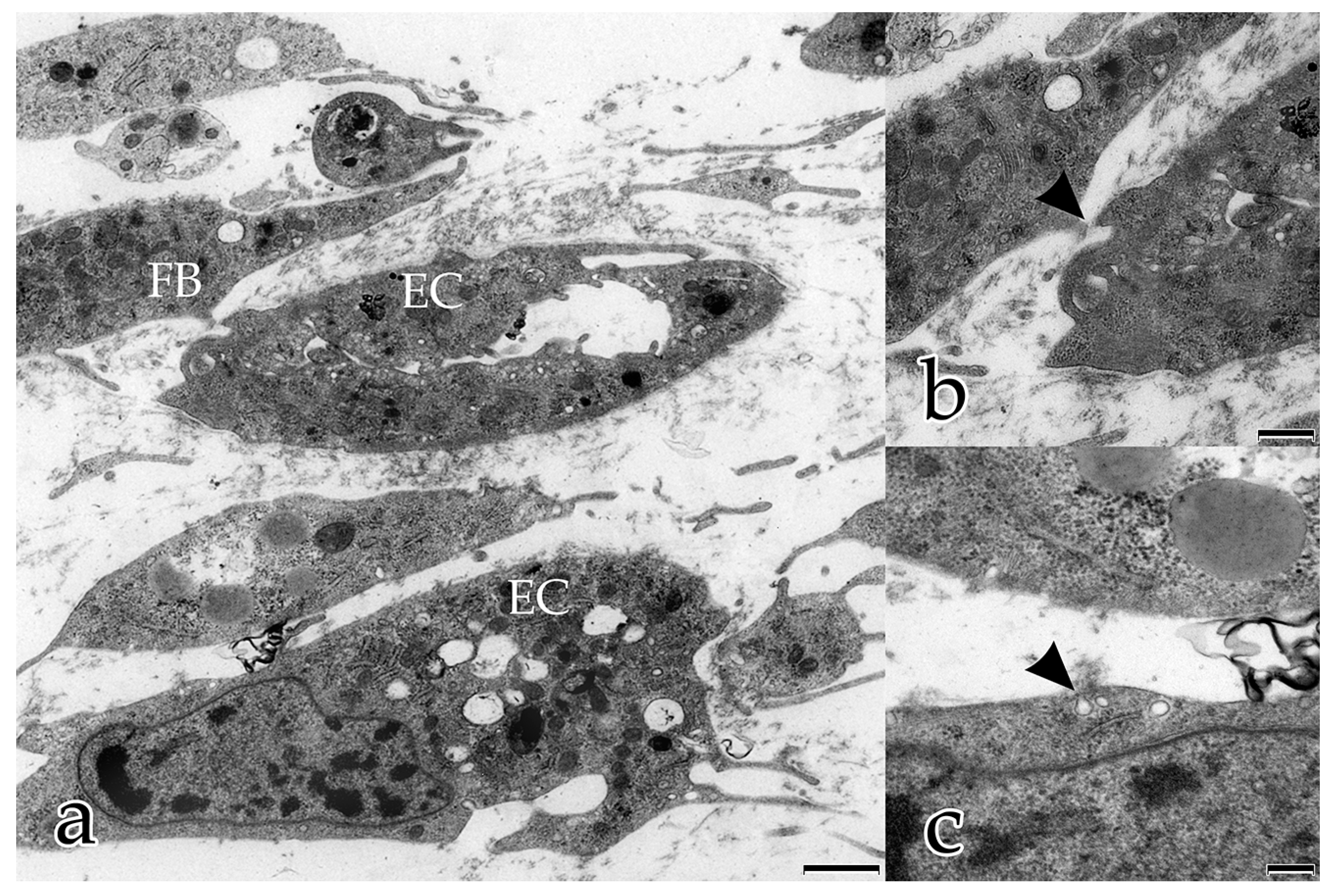
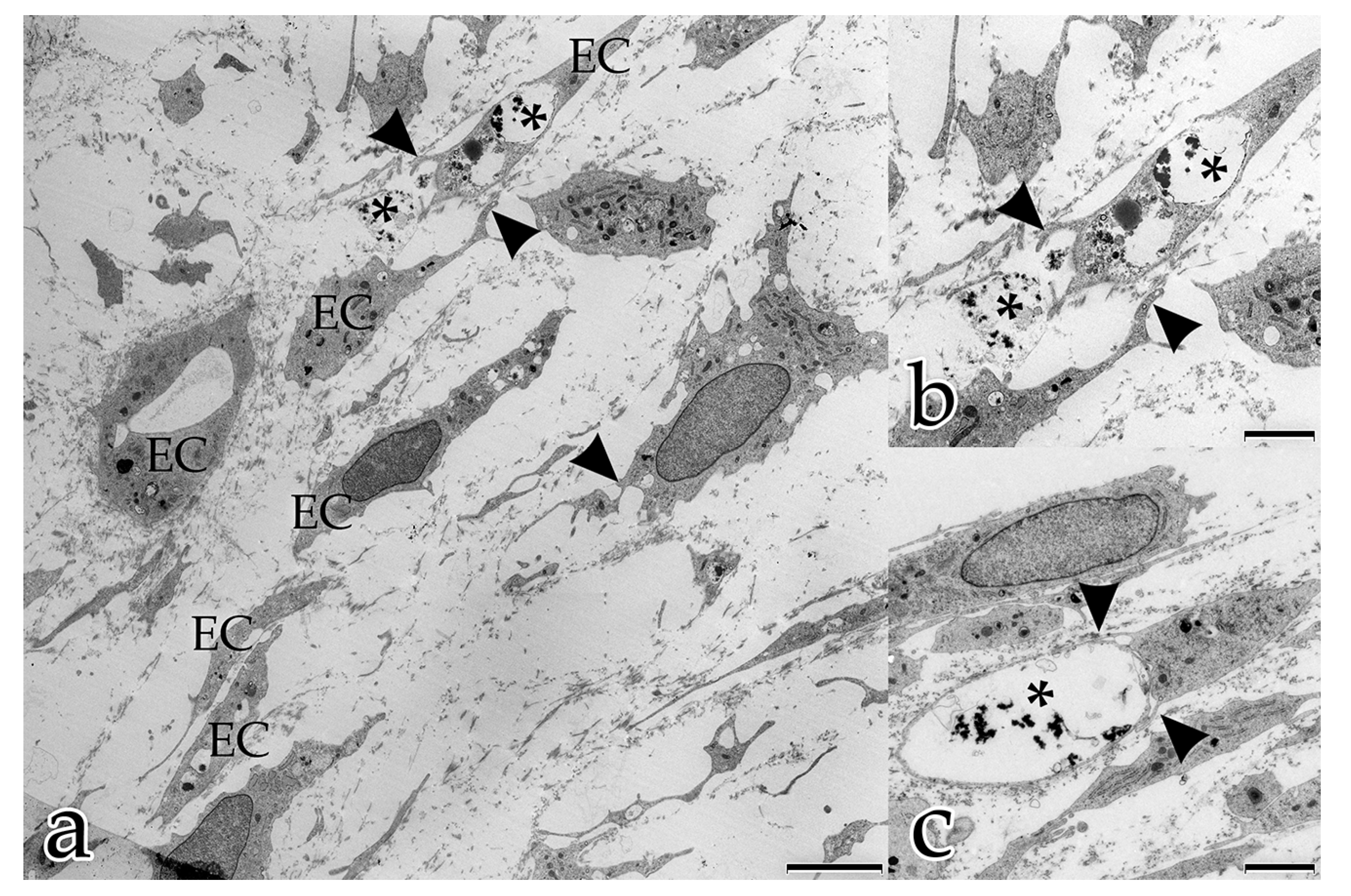
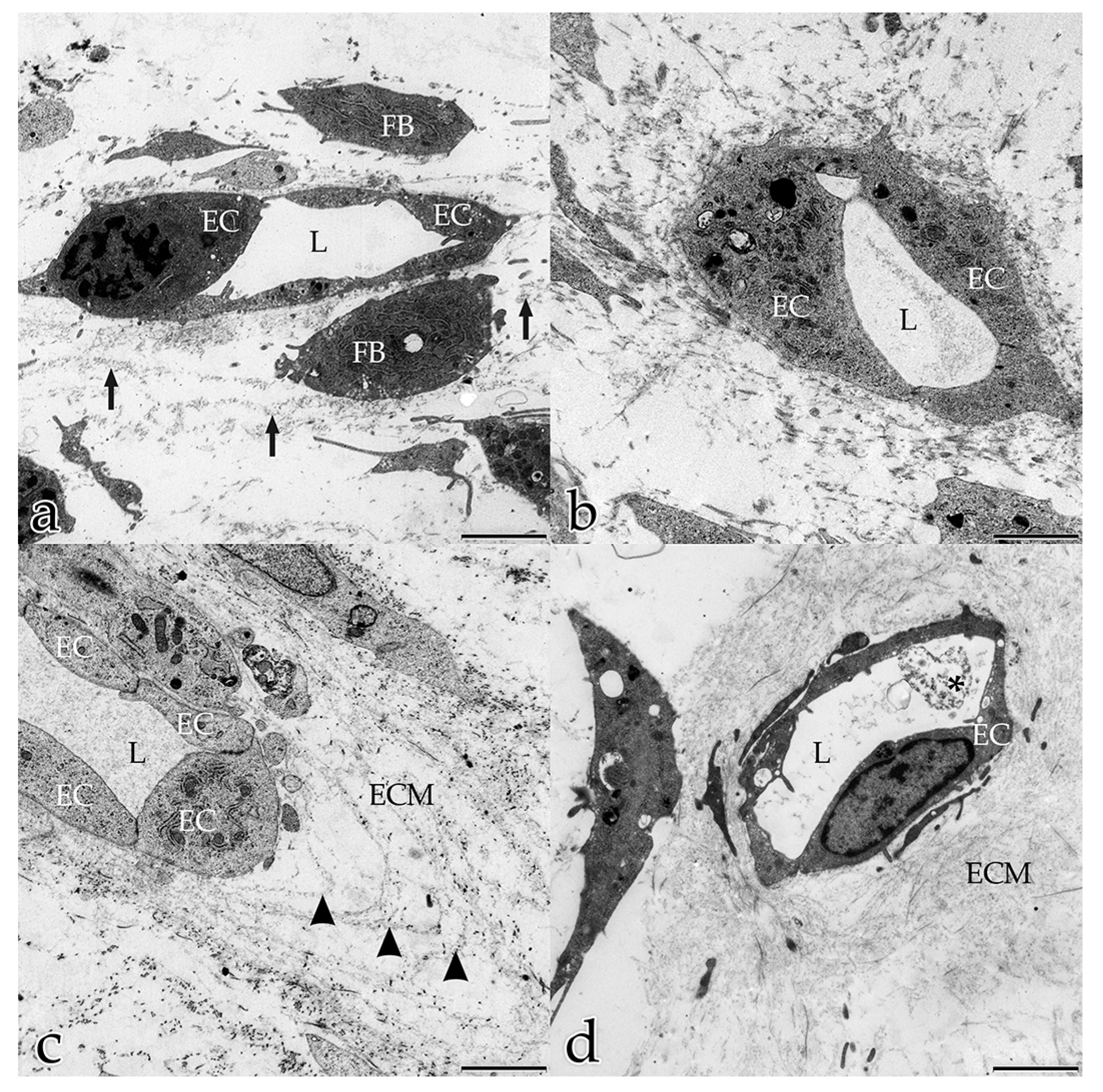
3.1. FBs Take an Active and Physical Part in the Angiogenesis Process of ECs
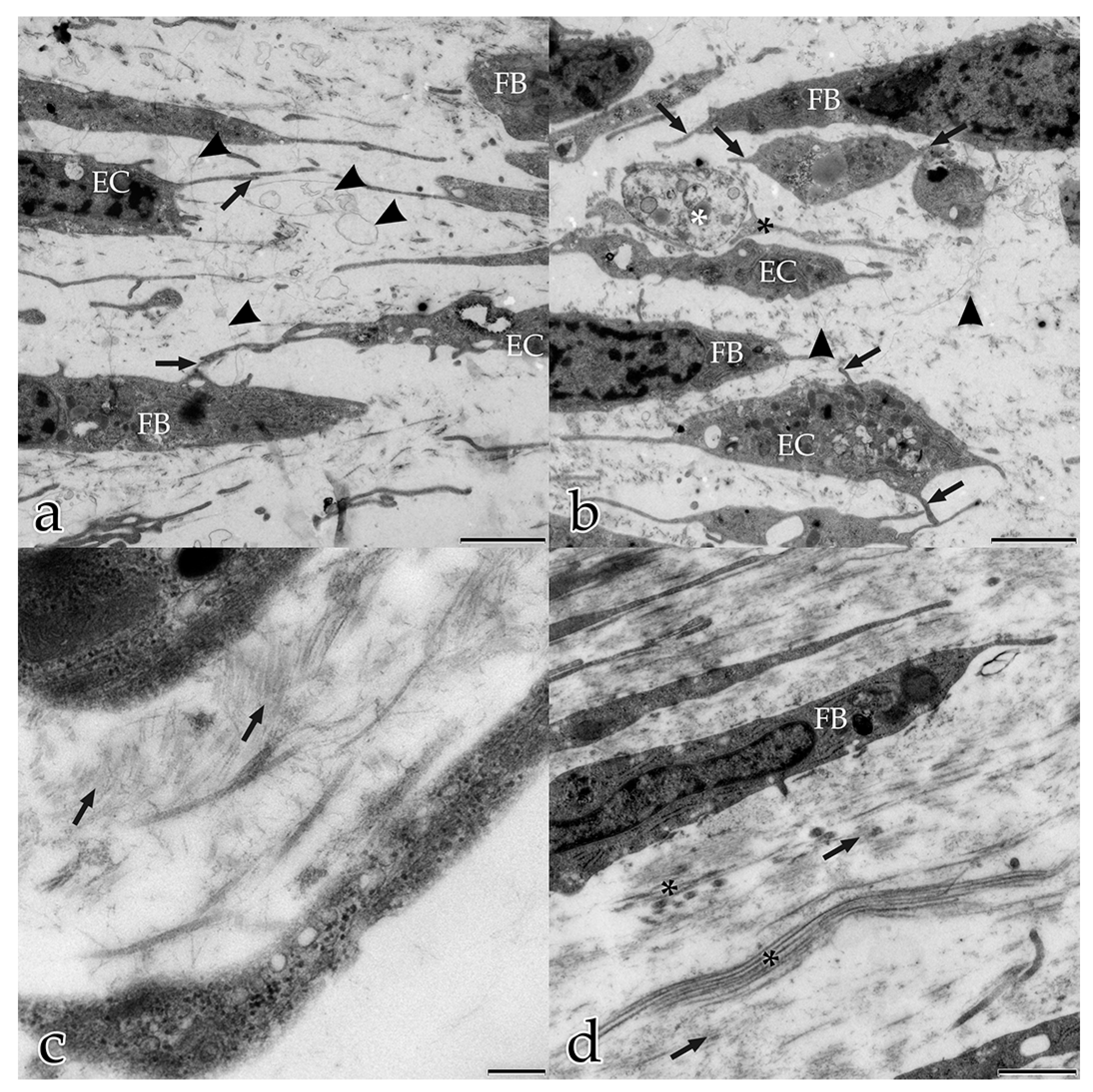
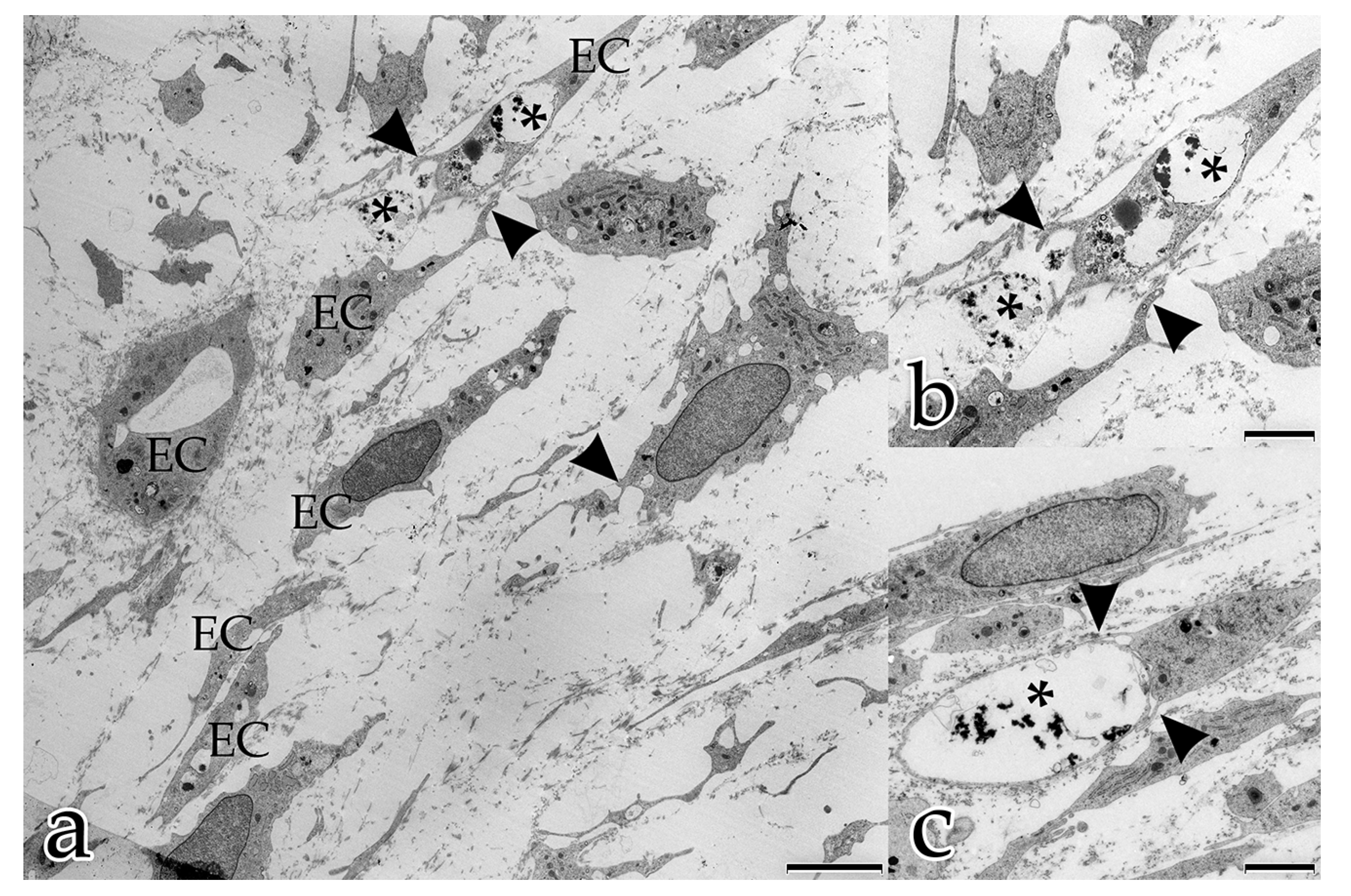
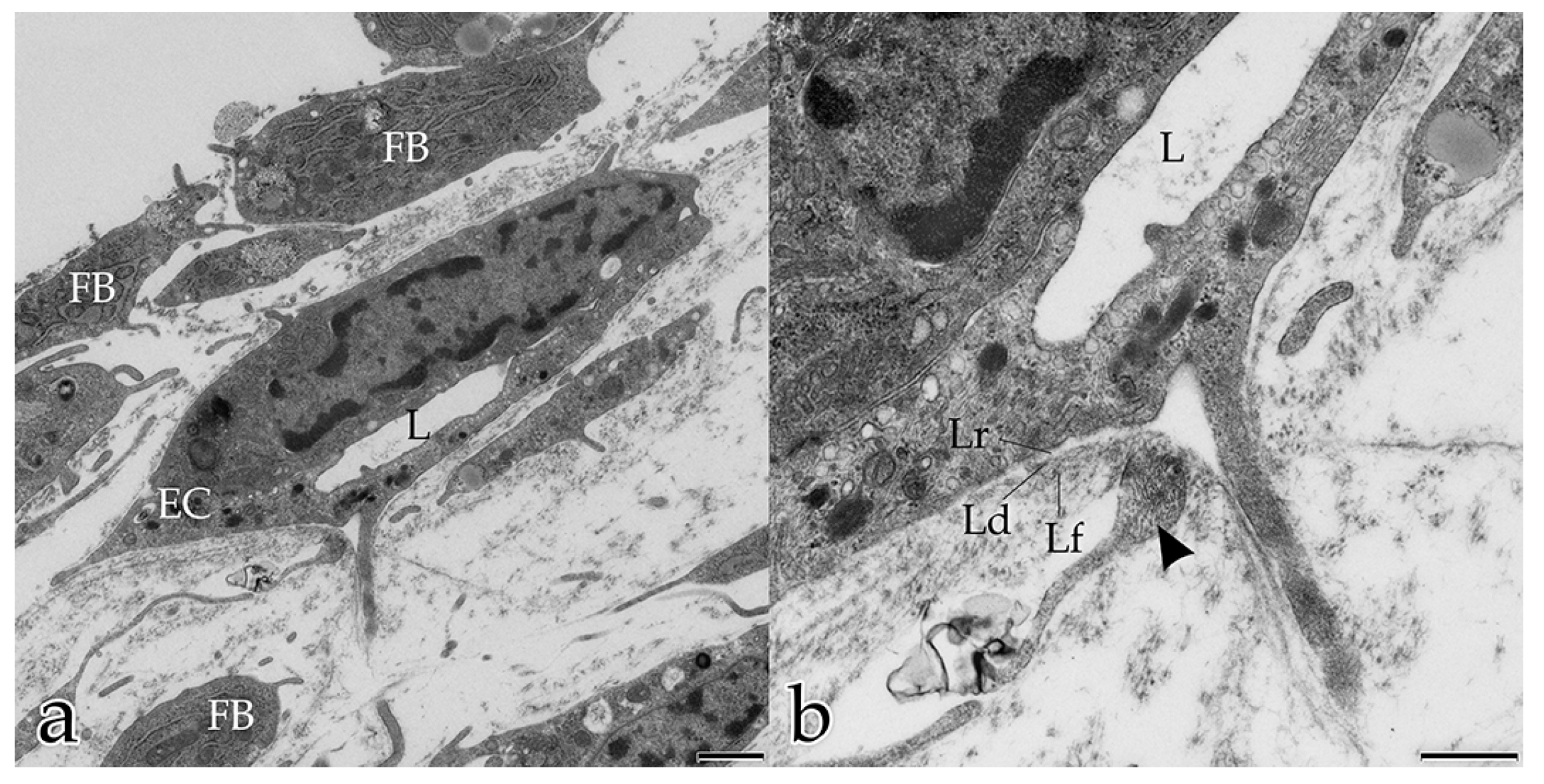
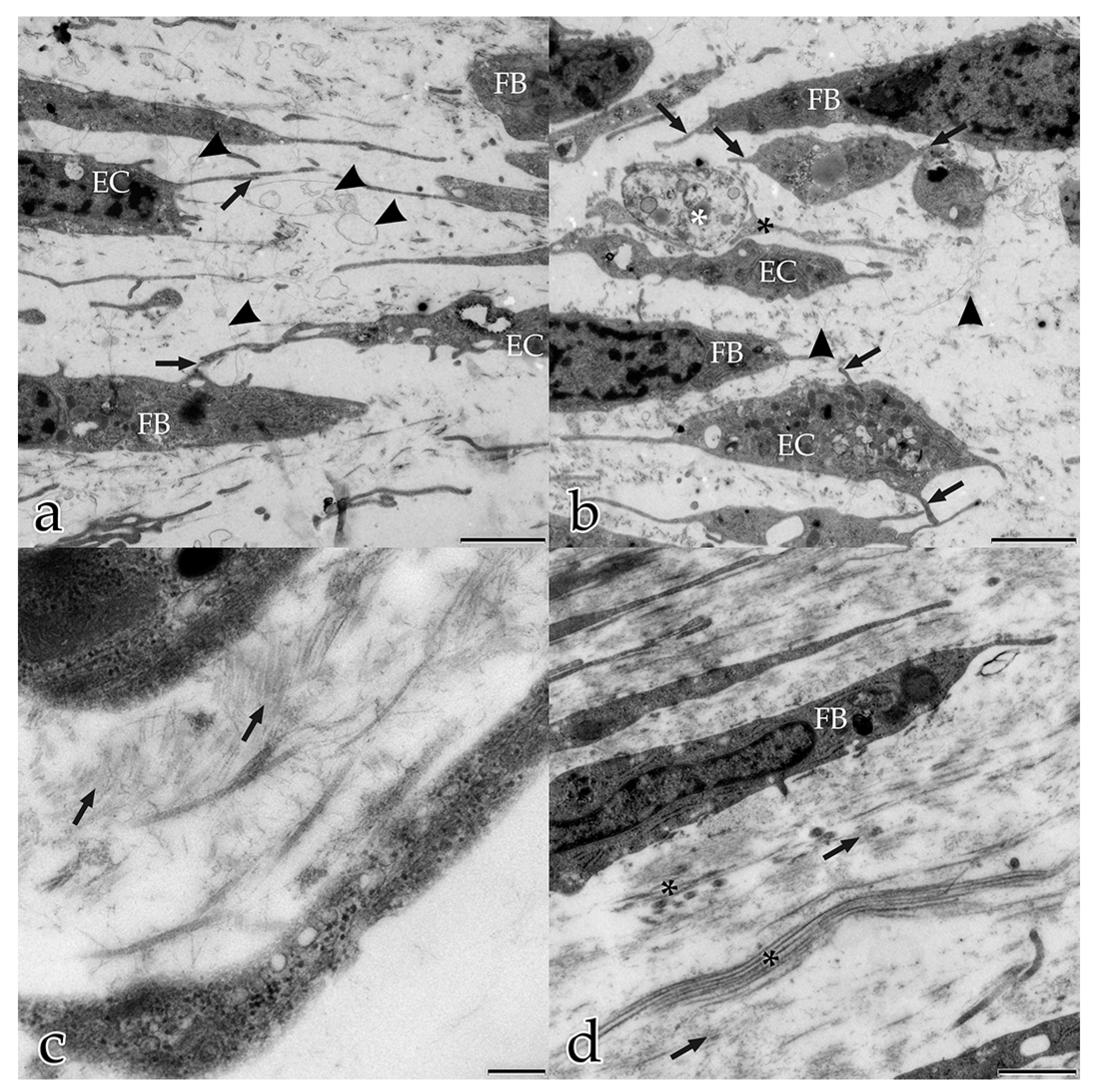
In our study, during the coculture of ECs with FBs, a microenvironment was generated that allowed and furthered cell–cell and cell–matrix interactions. In the early stages, both FBs and ECs possessed long filopodial processes that interconnected FBs with FBs, FBs with ECs, and ECs with ECs. FBs were seen frequently adjacent to the endothelial tubular structures, and physically contacted endothelial tubes at specific sites thereby initiating the degradation of the BM and the sprouting of a new vessel. These observations demonstrate that FBs take an active and physical part in the angiogenesis process of ECs. Fibroblasts are the main cells that secrete ECM components and growth factors, including VEGF-A, FGF, PDGF [
6]. Our results suggest that FBs are not only passive bystanders that secret scaffold material and proangiogenic factors, but they also influence the activation of endothelial angiogenesis by their physical contact with ECs. Our observations of the dynamic involvement of FBs in angiogenesis suggest that angiogenesis models based on endothelial monocultures do not adequately explain in vivo intercellular communication [
13,
23]. The obvious influence of FBs must be considered when collecting and interpreting in vitro results.
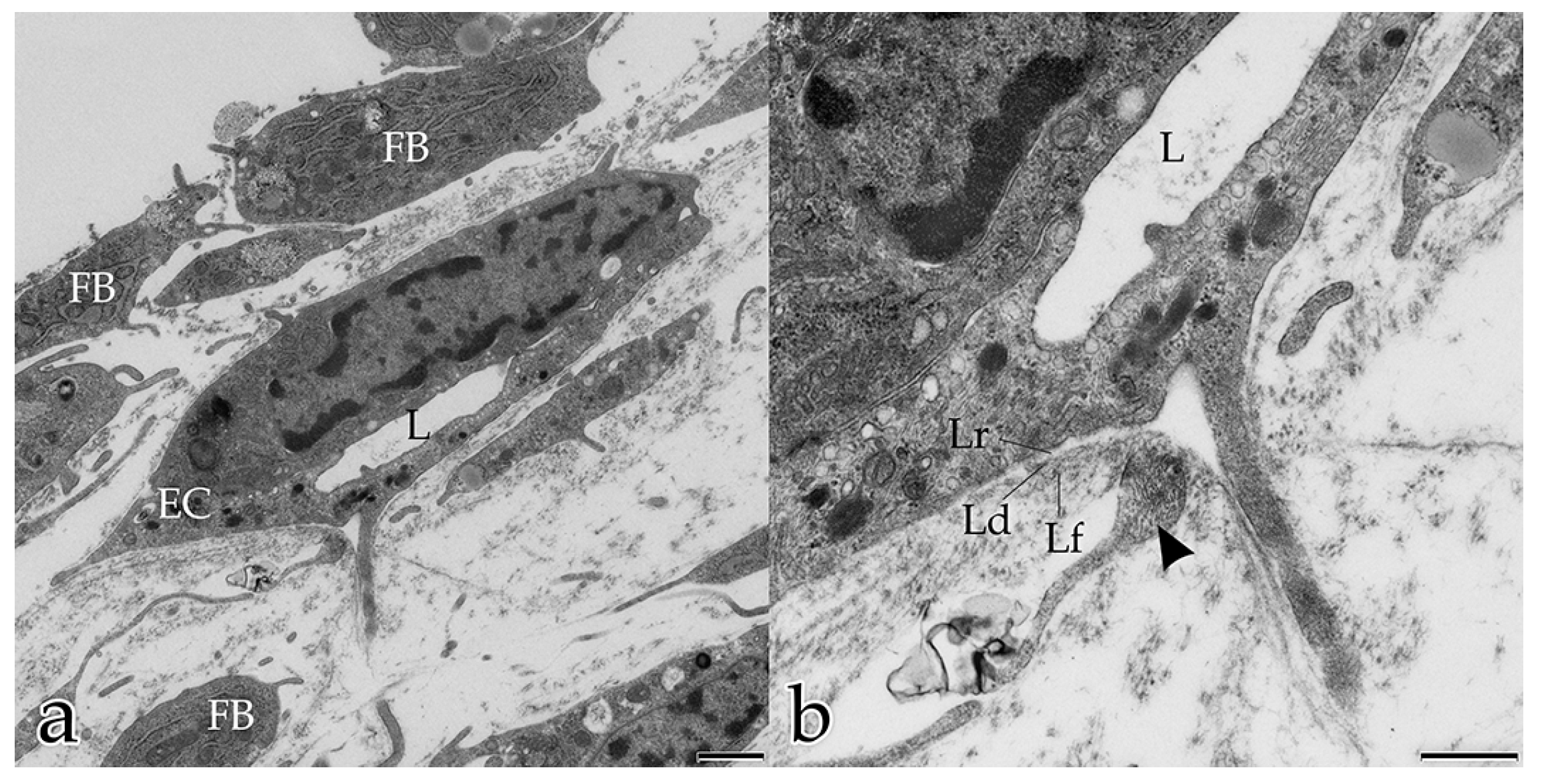
3.2. Microvesicles in Intercellular Information Transfer
In the present study, several different mechanisms of intercellular communication were observed in the cocultured cells. To bridge short distances, cellular projections of adjacent ECs and FBs contacted each other. In addition, cellular material that was released by specific cells into the extracellular space appeared to be taken up by their neighboring cells.
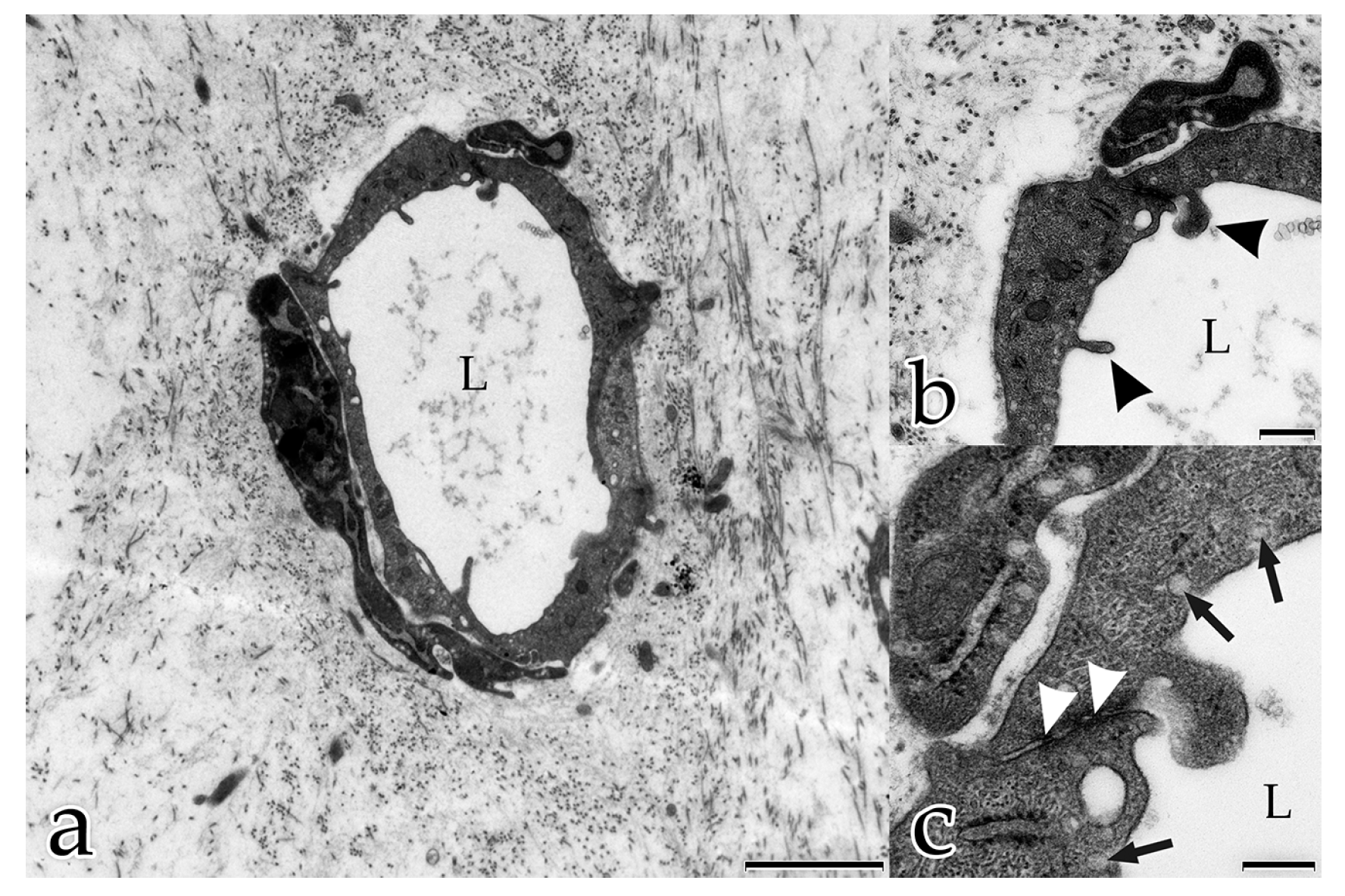
This form of intercellular interaction was seen frequently in close proximity to tubular structures during the early stages of the morphogenesis of the in vitro constructs. As our morphological study presents no evidence for the exchange of material between the different cell species, further studies are planned to investigate this largely unknown phenomenon. In addition, in each developmental stage examined, extracellular vesicles surrounded by membranes were found to be scattered amongst the interstitial matrix fibrils, and cellular filopodia from both cell types were in contact with these extracellular vesicles. We hypothesize that these structures are microvesicles that were released by the cells, serving as vehicles for the transfer of angiogenic factors over longer distances [
29]. Microparticles were found to be released in monocultures of ECs in an earlier study at the time of their integration from the monolayer into three-dimensional capillary-like structures [
30]. It is only recently that the role of microvesicles in intercellular interactions has become a focus of research interest. Today, it is generally accepted that microvesicles mediate communication between cells in vivo [
29,
31,
32]. After their synthesis, microvesicles are released via exocytosis and float within the extracellular space to interact with an adjacent cell in a paracrine manner. Vesicles deliver information to their recipients by binding to specific receptors [
33]. Recent studies report that extracellular vesicles play a role as proangiogenic mediators [
33,
34,
35]. It has been suggested that angiogenic extracellular vesicles are released from different types of cells, and that their content ranges from lipids, to proteins, to mRNA [
33,
34,
35]. Interestingly, a cargo of microvesicles is also involved in the early steps of the angiogenic cascade. Soucy and Romer [
36] described endothelial vesicles that carry MT1-MMP, i.e., the MMP responsible for the degradation of the BM in the beginning of sprouting angiogenesis. Using transmission electron microscopy, we observed that the ECs took up extracellular vesicles and that the capillary lumen formed by fusion of those vesicles. Our results mirror the dynamics of lumen formation in vivo and are supported by the observations from Kamei et al. [
18], who reported lumen formation by intracellular and intercellular fusion of endothelial vacuoles in live animals. They used time-lapse imaging to examine the dynamics of endothelial vacuoles and their contribution to vascular lumen formation [
18].
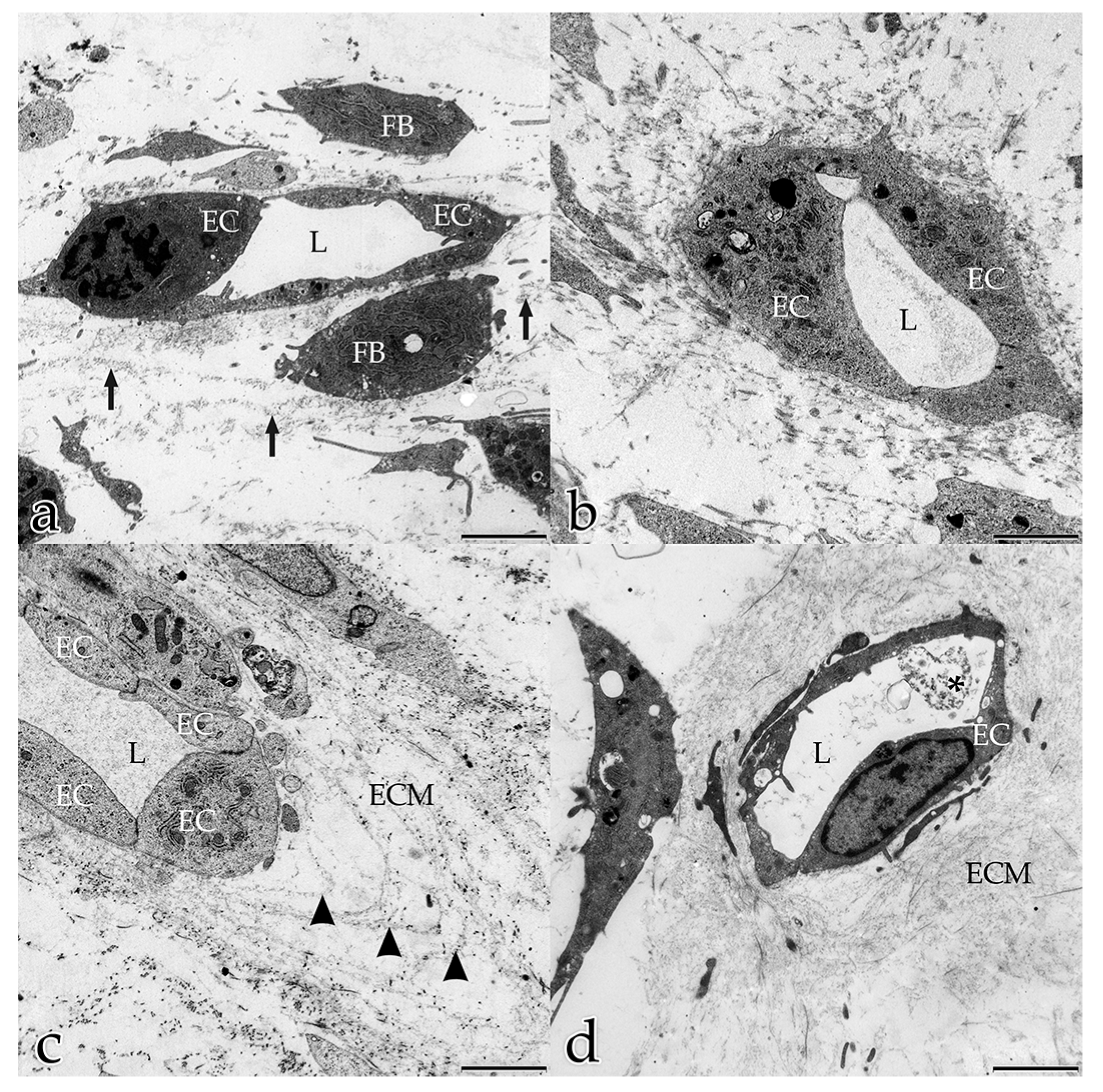
3.3. The Vascular Structures and the ECM of Cocultures Mirror the Topography of Histological Tissue Sections
In vivo, the fibrillary and nonfibrillary interstitial ECM milieu, in which blood capillaries are embedded, is rich in fibrillar and nonfibrillar collagens, elastic fibers, glycosaminoglycans, and glycoproteins, such as fibronectin, laminin, and other molecules [
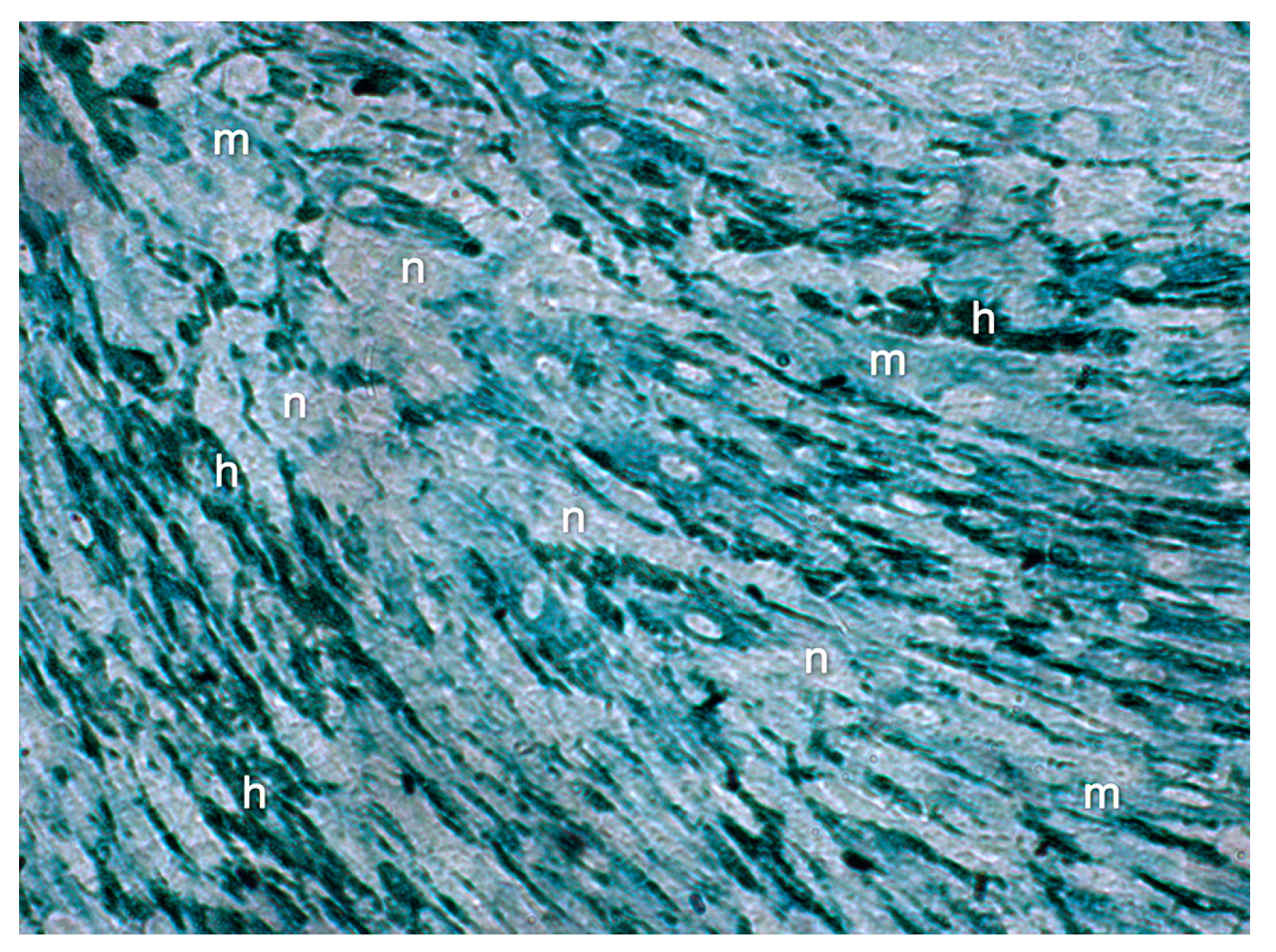
3]. Our ultrastructural observations revealed that the interstitial ECM progressively filled the spaces between the cells over the investigation timeline (day 5–day 20), until the in vitro construct consisted of a densely organized ECM that formed a framework for the organization of the cells and displayed a close resemblance to histological sections of loose connective tissue.
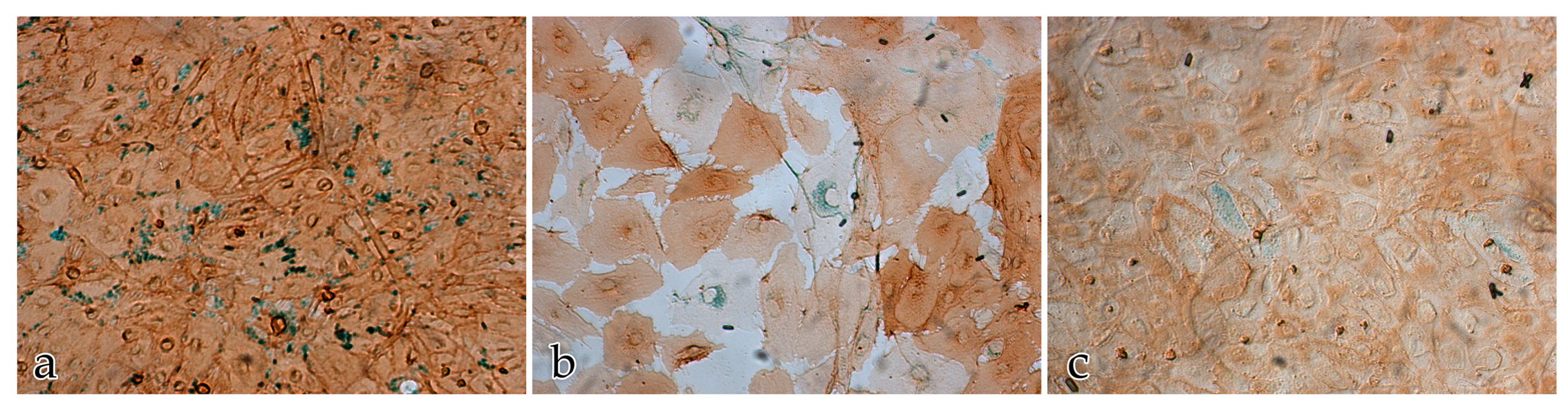
We examined the expression pattern of the three different ECM components collagen III, fibronectin, and laminin, which are known to be relevant in different steps of angiogenesis [
6,
7]. In our study, the amount of immunolocalized ECM differed significantly over the observation timeline as it followed an undulating pattern with the total ECM being 36.21 ± 5.92% at day 5, followed by a decrease to 23.40 ± 19.64% by day 10, then an increase to 58.86 ± 0.97% by day 14, and a final decrease to 11.01 ± 2.73% (
p = 0.010) by day 20. This pattern mirrors ECM development and maturation over time as analyzed by the pixel intensity profile. Ultrastructurally, its configuration and accumulation started from a randomly organized structure finally maturing into a homogenously distributed matrix consisting of smaller but more condensed molecules over the cultivation timeline. Our results corroborate the possibility that, similar to the in vivo situation, the ECM produced in the coculture assays is actively remodeled by FBs [
25] and changes its composition in order to adjust to the individual developmental demands of the tissue. Notably, matrix remodeling FBs are crucial in vascular development through transmitting biochemical signals and mechanical forces affecting cell survival, cell shape, and cell orientation [
19]. Therefore, it is possible that the ECM that we immunolocalized was replaced by other ECM components during the maturation process. In addition, our studies, comparing the cocultures with the monocultures of FBs and ECs, support that the ECM is mainly synthesized by the FBs.
In our cocultures of ECs with FBs, the three-dimensional ECM was synthesized by the cells themselves. This ECM not only served as a scaffold for the vascular structures, but it also played an active role in the communication between cells involved in angiogenesis. This confirms observations by Neve et al. [
3], who reported that the ECM consists of and stores a multitude of proteins, glycoproteins, and polysaccharides that act as proangiogenic or antiangiogenic factors controlling vascular development and remodeling [
3].
3.4. Molecules and Fibers of the ECM Guide the ECs during Angiogenesis
At the onset of coculturing, FBs and ECs were distributed randomly. The ECM had built up a connecting network of very fine fibrils between these cells. We assume that these fine fibers, running from cell to cell and connecting them with each other, may be fibronectin. Soucy and Romer [
36] described that a characteristic of fibronectin is its configuration into fine fibrillar structures, and that fibronectin initiates the matrix organization into its typical architecture [
36]. This is supported by our observations that the fine fiber net not only dictated the position of the cells within the cocultures, but also constituted a framework for other ECM components, including collagen fibrils. We also observed that the ECs lined up with other ECs using the fine ECM fiber net to guide their migratory activities. In this context, it is important to know that ECs selectively adhere to fibronectin [
36].
In our study, the expression of fibronectin was found to decrease significantly after 10 days of cocultivation. In vivo, fibronectin is strongly associated with the periphery of developing vessels, but it is only weakly expressed in the mature vasculature [
6,
37]. The decrease of fibronectin found in this study can be interpreted as an indication of the maturation of the capillary-like structures. Hetheridge et al. [
25], who also cocultured ECs with FBs, reported that the growth of tubes in vitro ceased after 14 days. At the endothelial intercellular junctions, they measured an increase in vascular endothelial cadherin, which is known to suppress the sprouting of new tubes [
25].
Collagen III is a fibrillary protein that plays an important role in stabilizing blood vessels and affects fibroblast function [
7]. Our observations showed an increasing amount of immunolabeled collagen III from day 10 to day 20 in parallel to the appearance of differentiated fibrillar structures within the ECM.
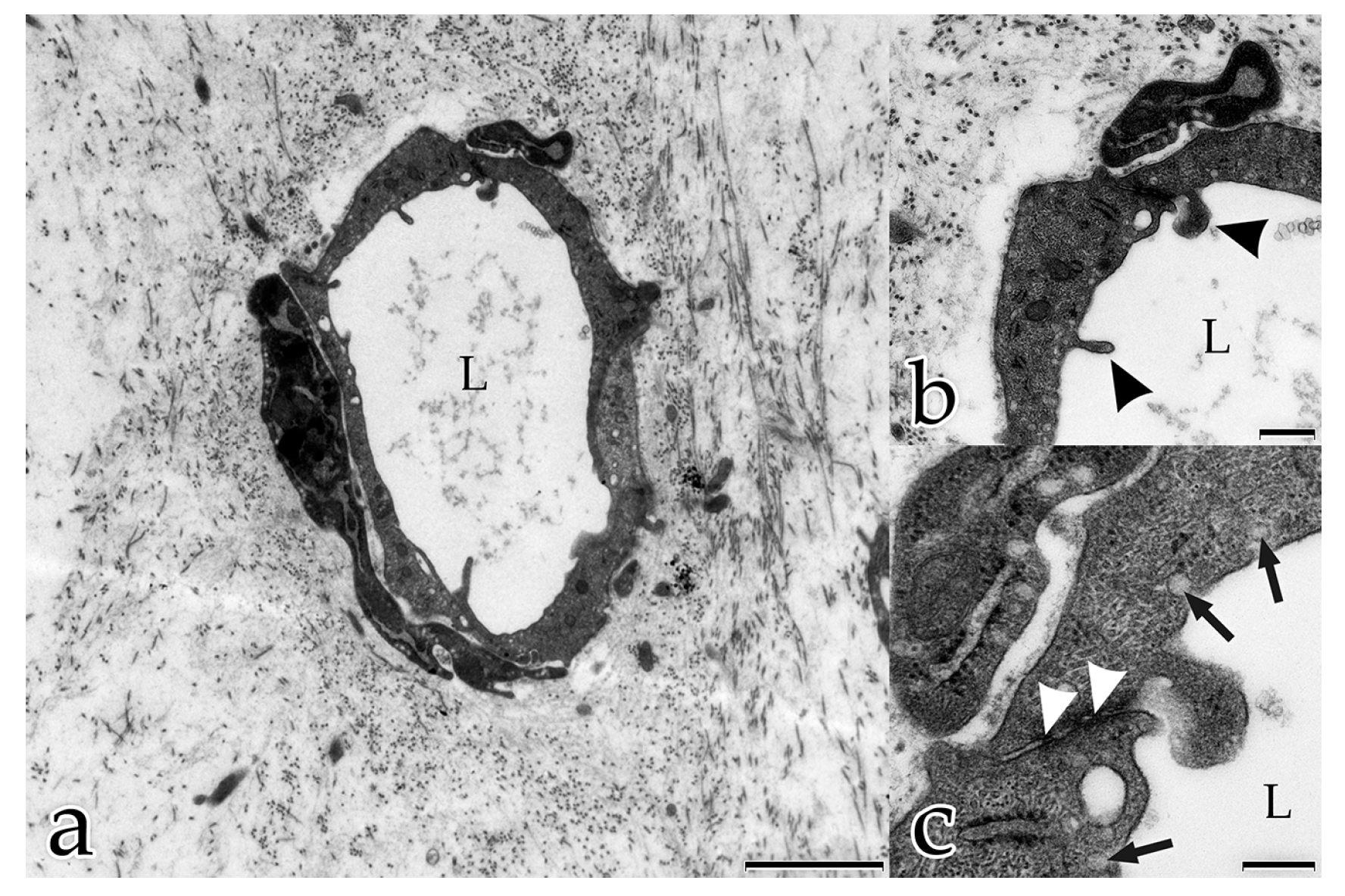
3.5. Formation of A Three-Layered BM In Vitro
Sheet-like organized ECM components surrounded the tubular structures on their external surface and resembled the BM. The formation of a BM synthesized by both ECs and FBs proceeded with the ongoing maturation and stabilization of a new vessel [
9]. The BM typically consisted of an electron-dense layer (lamina densa) separated from the plasma membrane by an electron-lucent layer (lamina rara). In monocultures of ECs, a BM consisting of several layers mimicking the in vivo situation did not develop [
38]. The BM is multifunctional and important for endothelial luminal polarization. The ECs in the capillary-like structures that developed in our cocultures clearly showed a correct polarity with nuclei bulging towards the lumen. EC nuclei orientated towards the “wrong” abluminal side are an artefact that occurs frequently in monocultures of angiogenic ECs [
38].
Laminin is an important molecule of the BM [
3,
7] and, together with fibronectin, forms networks within the BM contributing to BM stability [
3,
7]. In our study, laminin was evenly expressed over the period of observation. In monocultures of ECs the amount of laminin was significantly higher than in the cocultures. Whether this is an effect of the absence of FBs can only be speculated.
3.6. Signs of Maturation of Capillary-Like Tubular Structures In Vitro by Day 20
In summary, the following signs indicate maturation of capillary-like tubular structures in the in vitro constructs by day 20. Firstly, the interstitial ECM presented a dense, well-differentiated meshwork of collagen bundles, fibers, and fibrils that fully enmeshed all cells and tubular structures. Secondly, tubular structures were build up by ECs that were connected to each other by tight and adherens junctions. Thirdly, the endothelial tubes were surrounded by a BM that had differentiated into three clearly separated layers. Fourthly, individual fibroblastic cells with small amounts of rER were detected, indicating their differentiation into fibrocytes. Lastly, the morphometric analysis revealed a decrease in the number of tubes and an increase of multibranched endothelial tubes, as well as an increase in the length of endothelial branches, indicating that remodeling and maturation of the vascular network in vitro had occurred.
In conclusion, our study underlines the importance of morphological and ultrastructural investigations that should proceed hand in hand with molecular analyses.
4. Materials and Methods
4.1. Cells and Culture Conditions
Human dermal microvascular ECs from neonatal foreskin (HMVEC-D; Lonza, Walkersville Inc., Walkersville, MD, USA) were cultured according to the manufacturer’s instructions in basic EC growth medium 2 (EGM-2-MV, Lonza, Switzerland) supplemented with fetal bovine serum (FBS), fibroblast growth factor (rhFGF), vascular endothelial growth factor (VEGF), vitamin C, GA-1000 (gentamicin/amphotericin B), and hydrocortisone. Human juvenile foreskin fibroblasts (FB) were isolated from residual tissue of circumcision surgery (with patient’s ethical consent and permission from Charité, Berlin, Germany EA1/081/13) and cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) containing 10% FBS, 1% l-glutamin, and 1% penicillin/streptomycin (10,000 U/mL; all from Sigma-Aldrich, Taufkirchen, Germany). All cells were maintained at 37 °C in a humidified atmosphere (37 °C, 5% CO2). The cell culture medium was changed every 2–3 days. For each experiment, HMVEC-D were used at passage 5, and FBs were used at passages 9–10. The bottom of the culture wells was not coated with gelatin or other substances.
4.2. Priming of Fibroblasts
Adaptation (priming) of the FB monocell cultures to the EC medium was achieved in 48 h steps. The fresh FB medium was first mixed with 25% of EC medium, after 48 h with 50%, after further 48 h with 75%, and finally with 100% of EC medium.
4.3. Direct Cocultures of ECs with FBs and Monocultures of ECs or FBs
For cocultures, adapted FBs (5.000 cells per well) were seeded on a 24-well plate and cultured for 10 days. After this period of time, 20.000 ECs were seeded on top of the FBs for direct cellular contact and incubated for another 5, 10, 14, and 20 days in EC medium, which was replaced every 2–3 days (modified assay according to Richards and Mellor, 2016). After 5, 10, 14, and 20 days of culturing, the cells were fixed and processed as described in
Section 4.4 and
Section 4.5.
The monocultures of ECs or FBs were cultivated separately to assess and compare their cell behavior, morphometric features of angiogenesis, and extracellular matrix production. The monocultures were fixed and processed after 5, 10, 14, and 20 days of culturing, as described
Section 4.4 and
Section 4.5.
4.4. Transmission Electron Microscopy of the Cocultures
Cocultures for transmission electron microscopy (TEM) were grown on transwell membranes with a pore size of 0.4 µm (Corning, Tewksbury, MA, USA) for 5, 10, 14, and 20 days. The cultures were washed with 0.1 M cacodylate buffer (cacodylic acid sodium salt trihydrate, Roth, Karlsruhe, Germany) and fixed for 1 h at 4 °C in Karnovsky’s fixative (Merck Eurolab, Darmstadt, Germany). After fixation, the cells were postfixed in 1% osmium tetroxide (Chempur, Karlsruhe, Germany) in 0.1 M sodium cacodylate buffer for 1 h. Dehydration followed through a graded series of ethanol. The specimens were embedded in a mixture of Agar 100 (epoxy resin), DDSA (softener), MNA (hardener), and DMP 30 (catalyst) (all: Agar Scientific, Stansted, Great Britain, UK). The polymerization was done at 45 °C and 55 °C, each for 24 h. Semithin sections (0.5 µm) were cut with an ultramicrotome Reichert UltracutS (Leica Microsystems, Wetzlar, Germany) and stained with modified Richardson solution [
39] for 45 s on an electric hotplate at 80 °C. The examination of semithin sections was carried out using a light microscope (Olympus CX21, Olympus, Stuttgart, Germany). Next, ultrathin sections were cut with the UltracutS. Sections of 70 nm were mounted on nickel grids (Agar Scientific, Stansted, Great Britain, UK), stained with 2% uranyl acetate, stabilized by lead citrate Ultrostain 2 (Leica, Wetzlar, Germany), and examined to detect the occurrence of capillary structures, cell–cell contacts, cell organelles, and cell shapes as well as to analyze ECM organization, distribution, and differentiation using an EM109 electron microscope (Zeiss, Jena, Germany). Photographs were taken and processed using an Adobe Photoshop Program (Adobe System, Unterschleissheim, Germany).
4.5. Immunocytochemistry
Cells were washed with PBS, fixed with methanol/acetone (1:1) at −20 °C, and air-dried for at least 1 h. Afterwards, the cells were fixed for a second time with 4% formalin diluted in PBS overnight at 4 °C and then washed briefly with tap water and TBS. After antigen demasking with Target Retrieval Solution pH 9.0 (DAKO, Glostrup, Denmark) for 30 min at 86 °C and immersion in Protein Block Serum-Free (DAKO Diagnostika, Hamburg, Germany), the cultures were immunolabelled. For specific EC identification, mouse anti-CD31 (1:50, clone JC70A, DAKO, Glostrup, Denmark) was employed. To assess ECM protein distribution, mouse anti-human fibronectin (1:50, clone A-11, Santa Cruz, Heidelberg, Germany), rabbit anti-human collagen III (1:50, Abcam, GBR, Cambridge, UK), and rabbit anti-human laminin (1:50, DAKO, Glostrup, Denmark) were used. After 2 h incubation with CD31 at room temperature, cells were washed with TBS and incubated with anti-mouse Polymer EnVision System: HRP (DAKO, Glostrup, Denmark) for 30 min at 37 °C. After PBS wash, HRP was visualized by diaminobenzidine (Sigma-Aldrich, Taufkirchen, Germany) for 20 min at 37 °C. To assess ECM proteins, an overnight incubation with the different antibodies was performed at 4 °C. The antibodies had been diluted earlier in 3% bovine serum albumin (BSA) (Roth, Karlsruhe, Germany), 2% goat normal serum (GNS) (DAKO, Glostrup, Denmark), and incubation buffer (with 1% milk solely for rabbit antibodies) for 30 min at room temperature. Antibody incubation was followed by a TBS wash and the addition of anti-mouse Polymer EnVision System-HRP (DAKO, Glostrup, Denmark) and donkey-anti-rabbit-Ig-HRP (Linaris, Dossenheim, Germany), respectively, for 30 min at 37 °C. Negative controls were performed, in which the primary antibody was replaced by buffered anti-rabbit IgG (1:500, DAKO, Glostrup, Denmark) or anti-mouse IgG1 and IgG2b (1:25, both DAKO, Glostrup, Denmark). Subsequently, the cells were washed with PBS and treated with HistoGreen (Linaris, Dossenheim, Germany) for 10 min at room temperature for HRP detection.
4.6. Morphometric Analysis and Quantification of Angiogenesis
Four wells from each day (5, 10, 14, and 20) and culture type (cocultures of ECs with FBs, monocultures of ECs or FBs, respectively) were examined with a light microscope (Zeiss MicroImaging GmbH, Jena, Germany) and photographed with a digital camera (ColorView, Olympus, Münster, Germany). PC-based laboratory imaging programs (analysis docu, Version 5.2, Olympus, Münster, Germany and NIS-Elements AR, Version 4.5, Nikon, Düsseldorf, Germany) were used for processing. For measurements of capillary-like structures, visual fields were standardized by taking 7 images of each cell culture at the same magnification (5×) with a total area of inspection of 40.6 mm2. The morphometric features of capillary-like structures that were evaluated included: average number of tubes per mm2, average length of tubes (10 tubes per field of view), average diameter of tubes (10 tubes per field of view), average number of tubes with branches, including the percentage proportion of branching and the average length of branches (10 branches per field of view), and branching distribution pattern (single-, double-, triple- and multibranched). Branches were defined as projections of capillary-like structures, which were detectable at a magnification of 5×. The percentage proportion of branching was defined by the number of branched tubes in relation to the total number of tubes that were counted.
4.7. Quantitative Area Percentage Measurement for the Assessment of ECM Immunolocalization
The assessment of immunolabeled ECM components including the three proteins laminin, collagen III, and fibronectin, was conducted in cocultures and in monocultures after 5, 10, 14, and 20 days in vitro. Ten images were taken with a 20× objective from each cell culture type (Zeiss MicroImaging GmbH, Jena, Germany and ColorView, Olympus, Münster, Germany). The quantification of the fixed, immunostained ECM proteins was evaluated using a quantitative multiphase analysis for determining phase composition (analySIS docu, Version 5.2, Olympus, Münster, Germany) with a corresponding pixel intensity profile that was calibrated manually. We defined the color intensity ranges (negative, moderate, and high) for the separate phases. The proportion of positively labeled ECM (regions that displayed a distinct color change) was expressed as a percentage of the total area. The total amount of ECM was defined as the percentage of all positively labeled areas of one slide (moderate and high intensity). The amount of the individual ECM proteins (laminin, collagen III, and fibronectin) was determined as the proportion of the total response. The ECM production was measured through the pixel intensity profile, and the percentage of positively labeled pixels mirrored the quantity of immunolabeled ECM.
4.8. Statistical Analysis
Statistical analysis was performed by using IBM SPSS 24 (IBM Deutschland GmbH, Ehningen, Germany). Linear mixed regression models were conducted to determine the effect of the different wells and plates on the morphometric features of angiogenesis. In these models, (incubation) time was the fixed effect and the plate the random effect. Separate models were adapted for the following dependent variables: the average number, the length or diameter of the tubes respectively, the length of the branches, and the percentage proportion of branching over a set period of time. The Intraclass Correlation Coefficient (ICC) was defined as the percentage of variance due to differences between plates. A high ICC meant that there was a large variance between the plates, and thus the plate had a large influence on the result. In contrast to a fixed factor, the ICC generally refers to all plates and not distinctly to the plates that were used. Although the ICC values were high (between 22% and 65%), they were not statistically significant. Considering this, we utilized linear mixed regression models with Bonferroni correction for further uni- and multifactorial morphometric analyses. All the morphometric features of angiogenesis listed in 2.6 were tested for significant differences between the time points after 5, 10, 14, and 20 days of culturing, with particular focus placed on the time dependency and the association of morphometric variables. Bonferroni pairwise comparisons were conducted between each time of investigation.
All ECM parameters quantitatively analyzed were investigated by One Way ANOVA. All dependent variables (total amount of labeled ECM proteins, laminin, collagen III, or fibronectin, respectively) were tested with the independent variables time and cell culture type (cocultures of ECs with ECs, monoculture of ECs or FBs), the remaining ECM parameters, as well as the interactions between time and cell culture types. As the values of total ECM amounts were not normally distributed, we used logarithmic values for the regression models. Tukey’s (all pairwise) and Dunnett’s (all against the reference group day 20) post hoc test as well as Bonferroni correction adjustment in multiple comparisons were used in this model. To find out if there was a positive or negative feedback between the total ECM amount and specific ECM components, the influence of determining factors, collagen III, fibronectin, and laminin was tested against the total amount of ECM that had been immunolocalized.
The results were considered statistically significant at p < 0.05. All data were reported as the mean values and standard deviation.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}