1. Introduction
Membrane fusion plays an important role in multiple biological processes, such as exocytosis, fertilization, synaptic transmission etc. [
1]. Besides these processes normal for a living cell, it also constitutes a crucial stage in viral infection [
2]. A commonly adopted paradigm is that any physiological processes involving the remodeling of a biological membrane must not violate its primary function as a physical barrier separating the cell and its organelles from the exterior. Therefore, the stalk-fusion diaphragm model was proposed for description of the rearrangement of the interacting lipid bilayers in the course of fusion [
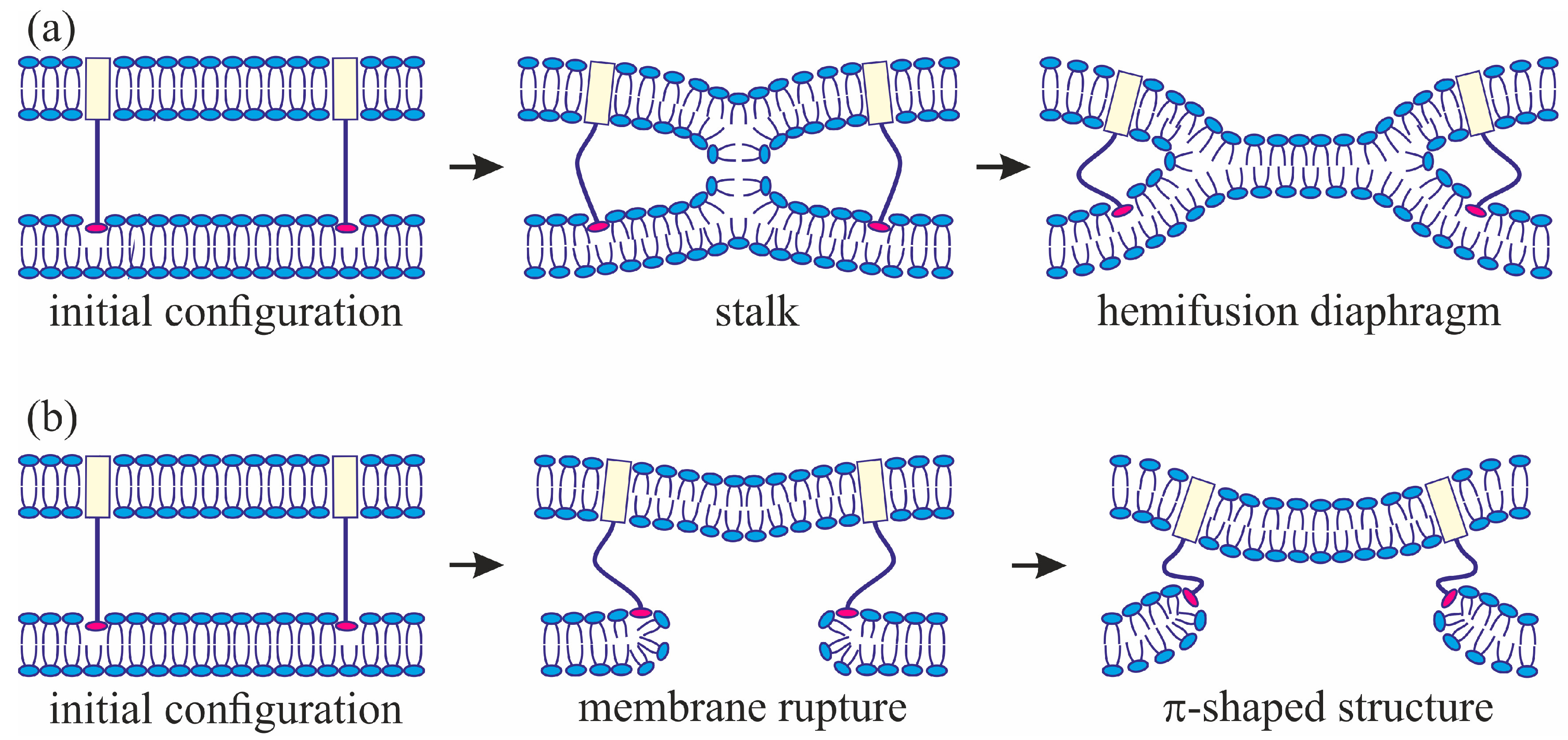
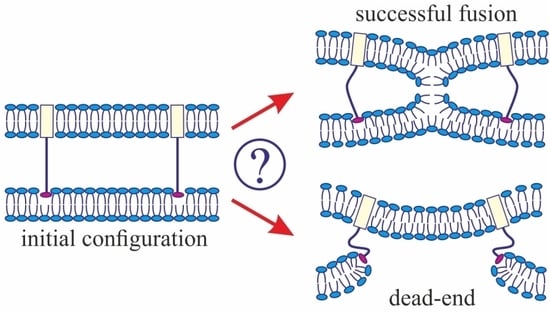
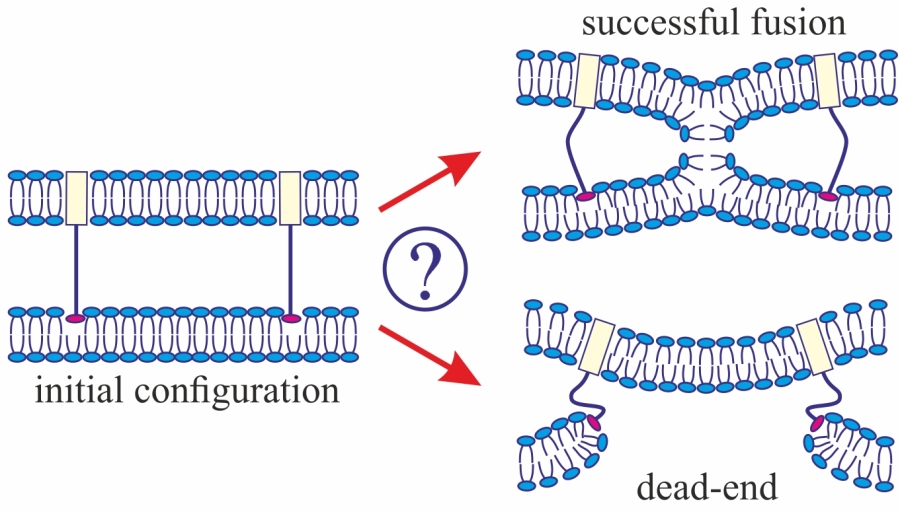
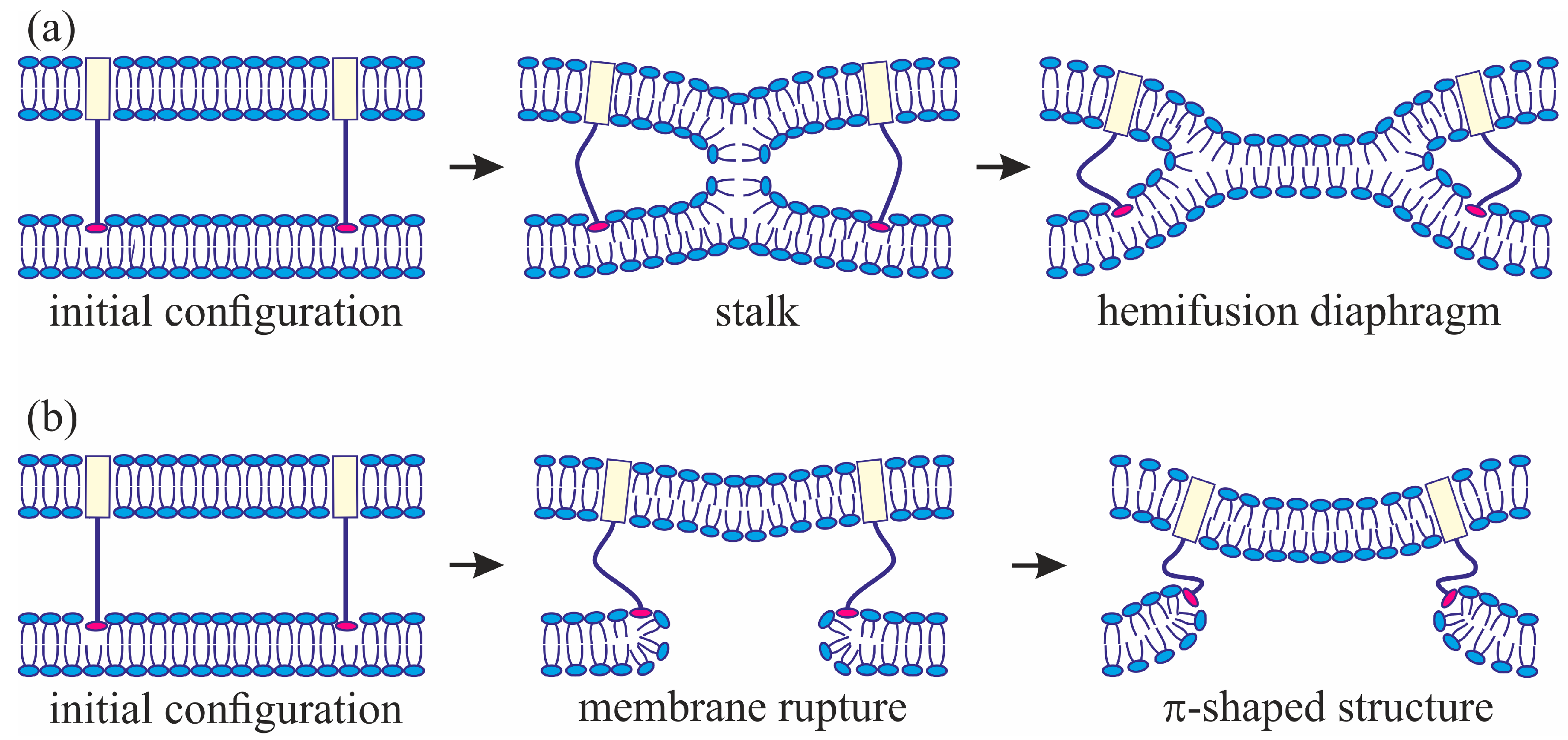
3]. The model assumes that, at the initial stage of fusion, the contact monolayers of the membranes locally merge to form an hourglass-shaped structure (stalk) (
Figure 1a), which then expands to bring the distal monolayers into contact and form the so-called trilamellar structure, or hemifusion diaphragm [
4] (
Figure 1a). Formation of a pore in the hemifusion diaphragm constitutes the final state of fusion. According to theoretical estimates made in a number of publications [
5,
6,
7], the energy barrier to be crossed for stalk formation is estimated at about 40
kBT (
kBT ~ 4 × 10
−21 J). This means that, without an additional impact on the system, the membrane would not merge within a physically reasonable time ranging from a few seconds to a few tens of seconds. In biological systems, special proteins known as fusion proteins are responsible for this additional intervention into the system [
2,
8]. The most thoroughly characterized fusion proteins are those of the synaptic system, participating in the process of Ca
2+-dependent exocytosis [
9,
10], and fusion proteins of various envelope viruses, such as influenza virus [
11,
12,
13], HIV [
14,
15], vesicular stomatitis virus [
16], etc.
By contrast with the fusion process associated with synaptic transmission, where the fusion process is mediated by a relatively large ensemble of different proteins, viral fusion is often mediated by a single protein [
17], e.g., hemagglutinin (HA) in the case of influenza A virus. The protein forms trimers and consists of a transmembrane (TM) domain and an ectodomain protruding out of the lipid membrane of the virus. Besides this, a fusion peptide is accommodated in a hydrophobic pocket of the ectodomain. The peptide incorporates into the target membrane during a pH-dependent conformational transition of hemagglutinin occurring in the process of viral infection in the cellular endosomes. Subsequent rearrangement of the HA conformation results in close apposition of the viral membrane and the target membrane, bringing the TM domains and fusion peptides into close proximity [
18,
19]. Coordinated action of several HA proteins is known to be needed for successful fusion in order to form the so-called fusion rosette [
20,
21,
22]. Three major classes of fusion proteins are usually discriminated [
23,
24] based on their ectodomain structure, and mostly the structure and mechanism of insertion of the fusion peptides into the host cell membrane [
25]. However, whether the fusion subunit has a prominent central α-helical coiled-coil (Class I viral fusion proteins), whether it primarily or exclusively consists β-structure (Class II viral fusion proteins), or whether it displays a combination of α-helical and β-structures (Class III viral fusion proteins), the fusion subunits ultimately fold back into a trimer-of-hairpins, in which three C-terminal regions pack on the outside of a central N-terminal trimeric core. When the fold-back is complete, the fusion peptide and the transmembrane anchor are brought together, thereby pulling the two attached membranes (target cell and viral, respectively) together and facilitating their merger [
23]. The information on the phenomenology of the viral fusion we used in this work is related to influenza and HIV fusion proteins belonging to class I, since this class is especially well investigated. However, the fusion peptide model we use in our analysis is that of a generalized inclusion into the host membrane, irrespective of the secondary structure details. The only implied assumption is the mechanism whereby the viral and host cell membranes are forced into close proximity by the fusion protein complexes, which is common for all the three classes. As a result, in the approximation of our model, the three classes are essentially equivalent; the only variable being the depth of insertion of the fusion peptide into the host cell membrane and the lateral dimensions of the inserted fragment.
The change of the membrane topology associated with fusion is believed to be impossible achieve by merely breaking the bilayers and then reclosing the system with a topology. The fusion is rather assumed to occur without contact of the cytoplasm with the external environment. Any long-term deterioration of barrier function is a manifestation of a pathological process [
26]. However, experimental evidence suggests that a pore piercing the membrane can sometimes be formed at the early stages of viral fusion in one of the membranes [
27]. Formation of the through pores at the early stages of membrane fusion was also observed during synaptic vesicle exocytosis or yeast mating [
28]. Molecular dynamic simulations also corroborate the possibility of fusion accompanied by leak at the early stage [
29,
30], as well as the calculations based on the self-consistent field model [
31].
The geometry of the possible leaky structure had been unknown until very recently. However, the recent experiments with the use of cryoelectronic microscopy to visualize the process of membrane fusion mediated by the HA [
32,
33,
34] revealed a structure notably different from stalk: the so-called π-shaped structure. In this configuration, a through pore is likely to exist in one of the membranes, with the HA fusion peptides aligned along the edge of the pore (
Figure 1b). The membrane is strongly convex, bulging towards the other membrane, into which the HA transmembrane domains are anchored. The pore is assumed to be formed due to membrane deformation caused by interaction with the fusion proteins. In particular, the structure of the fusion protein domains was hypothesized to impose considerable elastic stress on the relatively flat membrane surface, whereas in the strongly bent edge of the pore the elastic tensions can be relieved [
29]. It seems reasonable to assume that the structure of fusion proteins would determine the choice of the fusion scenario. In the present work, we investigate this correlation and try to identify the parameters of the fusion proteins that play a definitive role in the selection of one of the alternative trajectories of the membrane fusion process. We also explore the ability of the membranes to fuse in case a π-shaped structure is formed after the conformational transition of the fusion proteins.
Ostensibly, the diversity of the fusion process pathways cannot be related to the structure of the transmembrane domains of the fusion proteins anchored into the viral membrane. First of all, their structure is rather simple and conservative. As a rule, the TM domains are formed by a single α-helix, the length of the hydrophobic part of which roughly coincides with the hydrophobic thickness of the membrane (or slightly exceeds it) [
2]. The viral or virus-like particle membrane containing the TM domains of fusion proteins remains almost completely undeformed in the experiments. This should largely suppress (inhibit) the responsiveness of the fusion process to any changes of structure or conformation of the TM domains. The fact that the domains are usually separated from the point of initial contact of the fusing membranes by a considerable distance makes the influence of their precise structure and physical state on the trajectory and fine regulation of the fusion process even less plausible.
It is the interaction of the peptides with the target membrane that appears to play a primary role in this context. Point mutations of the fusion peptide are known to be capable of causing the appearance of pores in the membrane or complete inhibition of fusion [
35,
36,
37,
38]. The fusion peptides of different proteins can differ in the mechanisms of their incorporation into the membrane. The HA fusion peptides are known to form boomerang-shaped α-helices, incorporating into the host cell membrane at a small depth and small angle [
37,
39]. It is also experimentally verified that they can be incorporated into the membrane approximately at the right angle and pierce an entire monolayer [
40]. HIV fusion peptides can form either α-helices or β-sheets in the bilayer lipid membranes, depending on the membrane composition [
41]. According to [
42], the depth of insertion of the HIV fusion peptide into the membrane correlates positively with the effectiveness of the fusion process. In recognition of the available information about interaction of different fusion peptides with the target membrane, we consider peptides of different sizes and take into account the effects of different depths of peptide insertion into the membrane.
The fusion event usually involves several fusion proteins ensuring tight contact between the membranes by cooperatively applying mechanical force to them. A cluster of fusion proteins is termed a fusion rosette. In case of the influenza virus, the fusion rosette consists of three to eight HA proteins [
20,
21,
22]. It can be deduced from the results reported in [
28] that the appearance of the leaky structures can be caused by an improperly assembled fusion rosette, and that special proteins exist that can surround the leak location to prevent expansion of the pore and death of the cell. It also implies that the size of the protein rosette can influence the process of pore formation in the target membrane.
We consider two possible scenarios for the system behavior: membrane fusion through stalk formation, and the occurrence of a pore in one of the membranes with formation of a π-shaped structure. For each of the scenarios, we calculate the total energy of the system consisting of the two membranes. In order to calculate the deformational component of the energy, we employ an adaptation of the liquid crystal elasticity theory to lipid membranes (for detailed description of the methodology see, e.g., [
43,
44,
45]). The membrane is treated as a continuous medium subject to elastic deformations. The sources of deformations are the fusion protein domains incorporated into the membrane. The membranes are subdivided into several parts so that deformations in each of them can be deemed small. It is assumed that, for each fixed configuration of proteins, the membrane adopts the state with the minimal energy for the given boundary conditions. The steady state configurations are found by means of minimization of the membrane elastic energy functional. The functional variation yields the Euler–Lagrange differential equations, solution of which with the appropriate boundary conditions determines the energy of each part of the membrane. The boundary conditions are determined by the topology of the membrane parts and the geometry of incorporation of the fusion proteins into the membrane. Thus, the membrane elastic energy is determined as a function of the membrane parameters and boundary conditions. Besides the energy of the membrane deformations, we take into account the energy of hydration and hydrophobic interactions between the membranes, the contribution of which becomes significant at small distances. Thus, we obtain the dependence of the total membrane energy on the geometric parameters—the distance between the membranes and the radius of pore in one of them— while the energy is minimized with respect to all other degrees of freedom. This allows the determination of the height of the energy barrier that has to be crossed by the system to form a stalk or a pore. The geometric characteristics of the fusion peptide complexes are varied in order to evaluate the sensitivity of the energy barrier height to their changes. Comparison of the resultant heights of the energy barriers for each of the scenarios allows making judgement about the relative likelihood of each of them.
2. Results
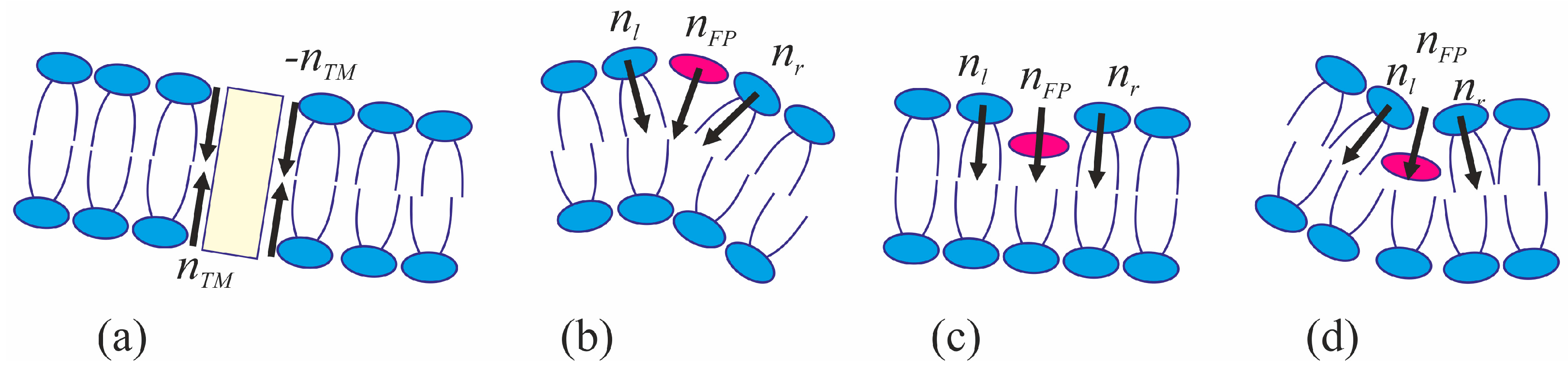
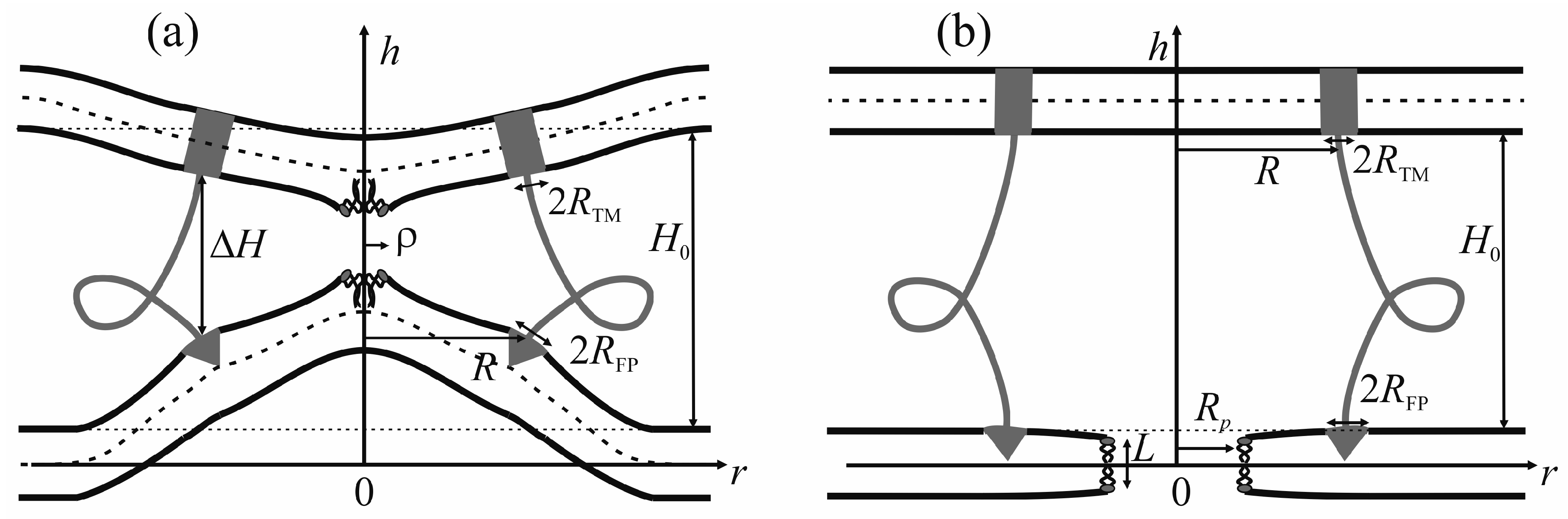
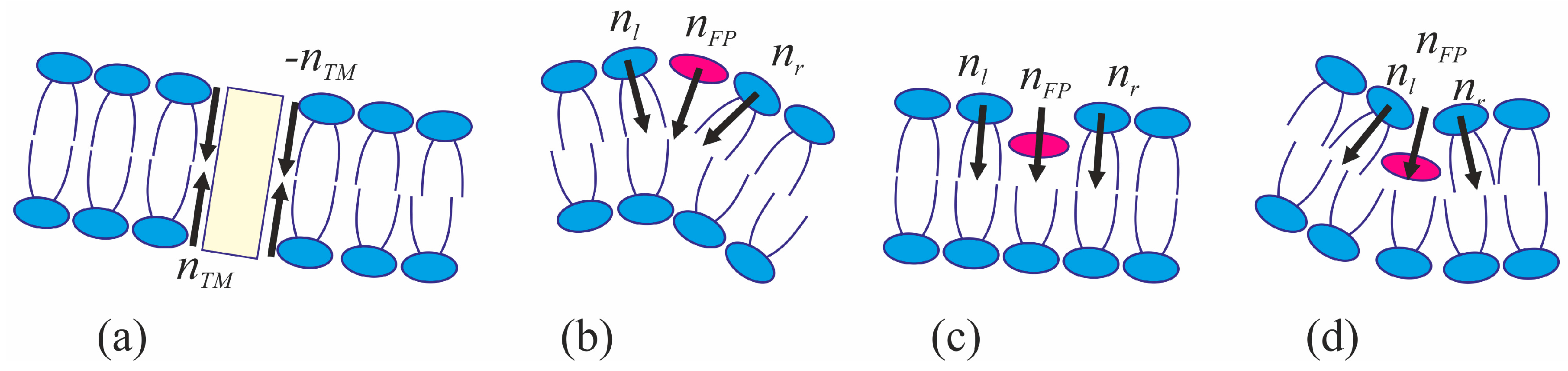
The viral particle and host cell membrane curvature (~50 nm) greatly exceeds the characteristic length of decay of deformations (~1 nm), and hence herein we assume the membranes to be planar away from the incorporated fusion proteins. We consider two planar parallel bilayers with incorporated fusion proteins. The membrane housing the TM domains of the fusion proteins will be termed the viral membrane, and the membrane into which the fusion peptides are incorporated will be referred to as the target membrane. The membranes are assumed to be horizontal, with the viral membrane on the top (
Figure 2). The fusion protein rosette is considered as a continuous annular structure. The TM domains and fusion peptides are modeled by two coaxial rings with the radius of
R and the half-widths of
RTM and
RFP, respectively. The TM domains are modeled as annular inclusions piercing the entire depth of the bilayer of the viral membrane, whereas fusion peptides—as annular inclusions incorporated into one of the monolayers to a certain depth depending on the specifics of the peptide structure (see
Figure 2). For trimeric configuration of fusion proteins, the annular structure of fusion peptides can be formed by aligning two peptides of each trimer along the fusion rosette circle, and directing the third peptide outward the circle center. Let us introduce a cylindrical coordinate system
Ohr, with the origin
O, the axis
Or lying in the plane of the inter-monolayer surface of the bottom membrane, and the axis
Oh along the rotational symmetry axis of the system. Due to cylindrical symmetry, the system is effectively unidimensional, i.e., all the values only depend on
r.
We assume that the fusion proteins bring the membranes to close apposition at a certain distance
H0, at which the attraction force imposed by the proteins is equilibrated by the repulsion forces induced by membrane hydration [
46]. Further evolution of the system can only be driven by thermal fluctuations of the lipid bilayers. Two possible trajectories of the system evolution corresponding to different modes of interaction of proteins with the membrane are considered. In the first case, the membranes are brought into tight contact at the expense of conformational transitions of the fusion proteins causing juxtaposition of their transmembrane domains with the fusion peptides. It is assumed that, in the course of fusion, the distance is ∆
H between the ring of the transmembrane domains in the viral membrane and the ring of the fusion peptides in the target membrane, while the distance between the membranes away from the fusion rosette remains equal to
H0 (see
Figure 2a). An energy barrier associated with hydration-induced repulsion has to be crossed in order to bring the membranes in close proximity [
47]. It is assumed that, under the conditions when fusion proteins attempt to bring the membranes in juxtaposition, strong hydration repulsion results in lateral displacement of the lipid head groups from the area of contact of the membranes [
6]. Thus, hydrophobic defects are generated in the contacting monolayers of the merging membranes [
48]; the radius of the hydrophobic defect is designated as
ρ on
Figure 2a. Such defects can serve as monolayer fusion nucleation centers, since their formation induces local loss of order of the hydration layers and appearance of hydrophobic attraction between them [
49]. The attraction between hydrophobic defects in the contact monolayers of the merging membranes leads to stalk formation (
Figure 1a).
An alternative theory of the fusion process is associated with spontaneous formation of a transversal pore. The fusion peptides are assumed to partition preferentially into the vicinity of the pore (
Figure 2b), because incorporation of the peptides generally leads to the target membrane deformation in relation to the unperturbed planar initial state. The membrane at the edge of the pore is also strongly deformed, and preferential partitioning of the peptides into this area alleviates the elastic stress associated with the both the deformations around the peptide and the deformations in the vicinity of the pore. This possibility is conditional to the peptide lateral mobility allowing them to choose the preferred localization. Occurrence of the hydrophobic zones capable of providing the attraction needed to juxtapose the merging membranes is believed to be hindered in the π-shaped structure configuration (
Figure 2b). As opposed to the stalk configuration, formation of hydrophobic zones in the vicinity of the center of the fusion rosette is impossible in the π-shaped structure case as there is a pore in the bottom membrane. Hydrophobic regions can only occur if the fusion peptides remain localized away from the edge; in this case, formation of radially symmetrical hydrophobic belts is likely to be possible. However, the energy associated with formation of such belts would be much higher than the energy of formation of a compact circular defect in the area of tight contact between the membranes because the perimeter of the annular belt has to exceed the perimeter of the hydrophilic pore, which cannot be smaller than ~15 nm (corresponding to the pore radius of ~2 nm) for purely geometric reasons. In principle, a compact hydrophobic region can be formed at the edge of the pore, with a violation of the rotational symmetry of the system. However, in this case, the mutual approach of the membranes in the area of the hydrophobic region is likely to be hindered more than in the case of radially symmetrical stalk due to proximity of the fusion peptides to the region. We therefore contend that, in the π-shaped structure configuration, apposition of the membranes cannot end up in their fusion, and this trajectory of the process leads to a dead end state. As we demonstrated earlier, a hydrophilic pore in the membrane can be formed through an intermediate state of a hydrophobic defect [
44,
45,
50]. In this state, lipid molecules shift radially in the plane of the membrane to form a water-filled cylinder piercing the entire membrane (
Figure 2b). The side surface of the cylinder is formed by the carbohydrate chains of lipid molecules. As the hydrophobic defect expands, polar headgroups of lipids “slip” into the lumen of the cylinder, ultimately forming a hydrophilic pore [
44,
45,
50]. It takes crossing an energy barrier associated with the formation of a cylindrical hydrophobic defect, reorientation of the lipid molecules required to form the hydrophilic pore, and possibly a redistribution of the fusion peptides towards the pore edge.
For each of the two scenarios, we calculated the dependence of the total energy
WT of the membranes upon the process coordinate. In the case of stalk, the coordinate is the change of distance ∆
H between the fusion peptides and the transmembrane domains of the fusion proteins located in the target membrane and in the viral membrane, respectively (
Figure 2a), while the radius of the hydrophobic patch
ρ can vary freely. As we demonstrated earlier, the process of formation of a pore through the membrane is described by at least two coordinates [
44,
45,
50,
51]. In the case of the π-shaped structure, we selected the height of the hydrophobic part of the pore
L, and the pore radius
Rp (
Figure 2b) as the system coordinates. The energy contributors vary significantly between the two scenarios.
In the calculations, we assume the values of the elastic moduli typical for “common” lipids; based on the experimental and theoretical results these are
B = 10
kBT and
K = 10
kBT/nm
2 [
52,
53] for leaflet splay and tilt moduli, respectively. The monolayer surface tension
σ is assumed equal to 0.01
kBT/nm
2. The equilibrium thickness of the monolayer
h0 is assumed at 1.5 nm, which is consistent with the values observed for 1,2-dioleoyl-sn-glycero-3-phosphocholine, 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine, 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine etc. For hydration-induced repulsion parameters, we used the following values from the work [
54]: disjoining pressure
P0 = 60
kBT/nm
3, characteristic length of hydration repulsion
ξh = 0.35 nm. The characteristic length of hydrophobic attraction
ξf is considered equal to 1 nm [
49]. The equilibrium distance between the fusing membranes
H0 is assumed at 5 nm. This distance reflects steric interactions associated with the fusion protein ectodomains that have to be accommodated between the membranes. As the membranes are brought together, the HA protein undergoes a conformational rearrangement [
18], in the course of which one of the parts of the HA ectodomain, namely HA1 subdomain, moves away thereby decreasing the effective diameter of the ectodomain just to the diameter of HA2 subdomain, which is about 5 nm [
18]. Note that the energy of the π-shaped structure is independent on the intermembrane distance, while the stalk and hemifusion diaphragm energy grows up for increasing distance [
55]. The half-width of the TM domain
RTM is assumed at 1 nm.
We vary the parameters determining the geometry of fusion proteins, and for each set of parameters calculate the energy barriers corresponding to the transitions of the membrane with the incorporated peptides to the stalk and π-shaped structure configurations.
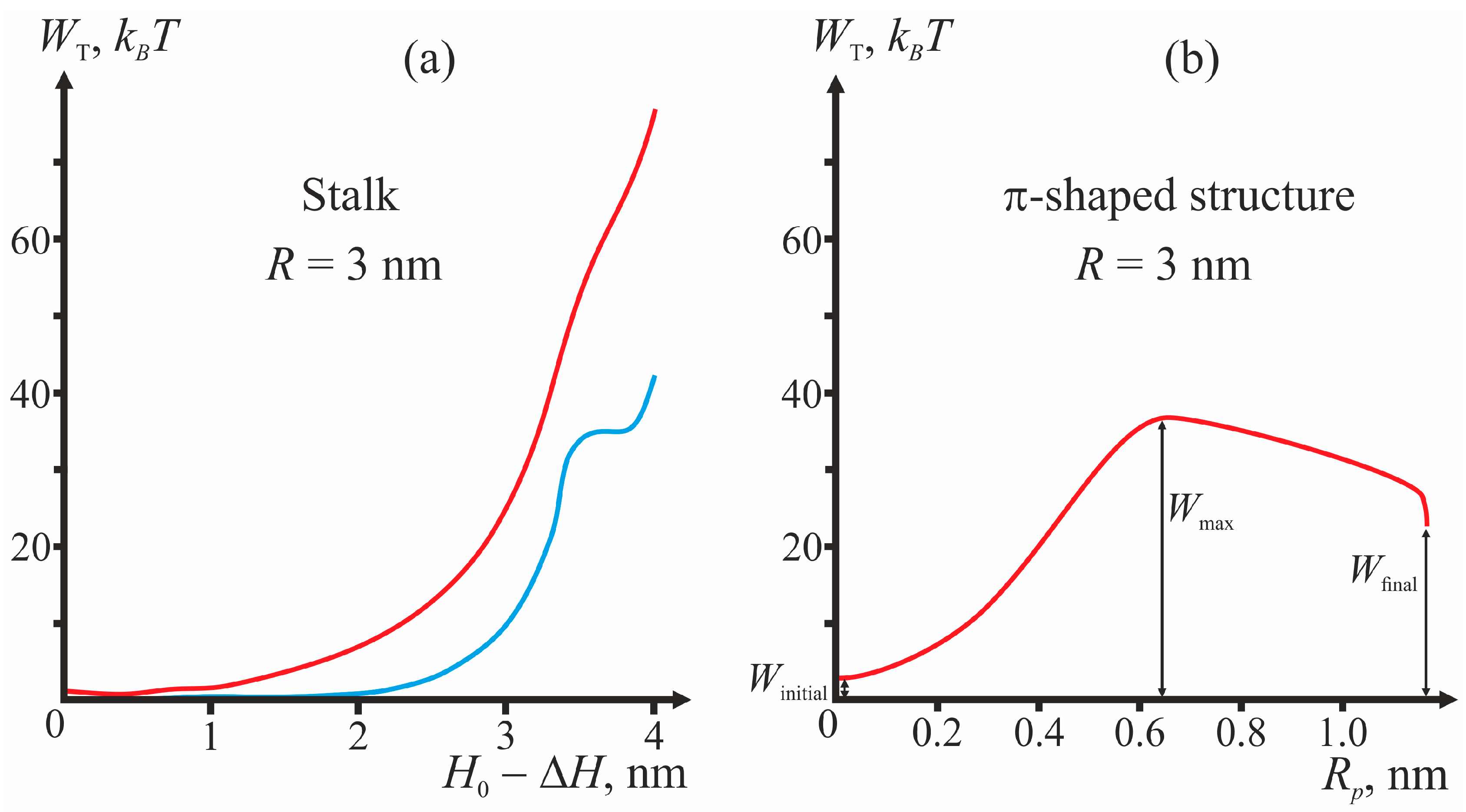
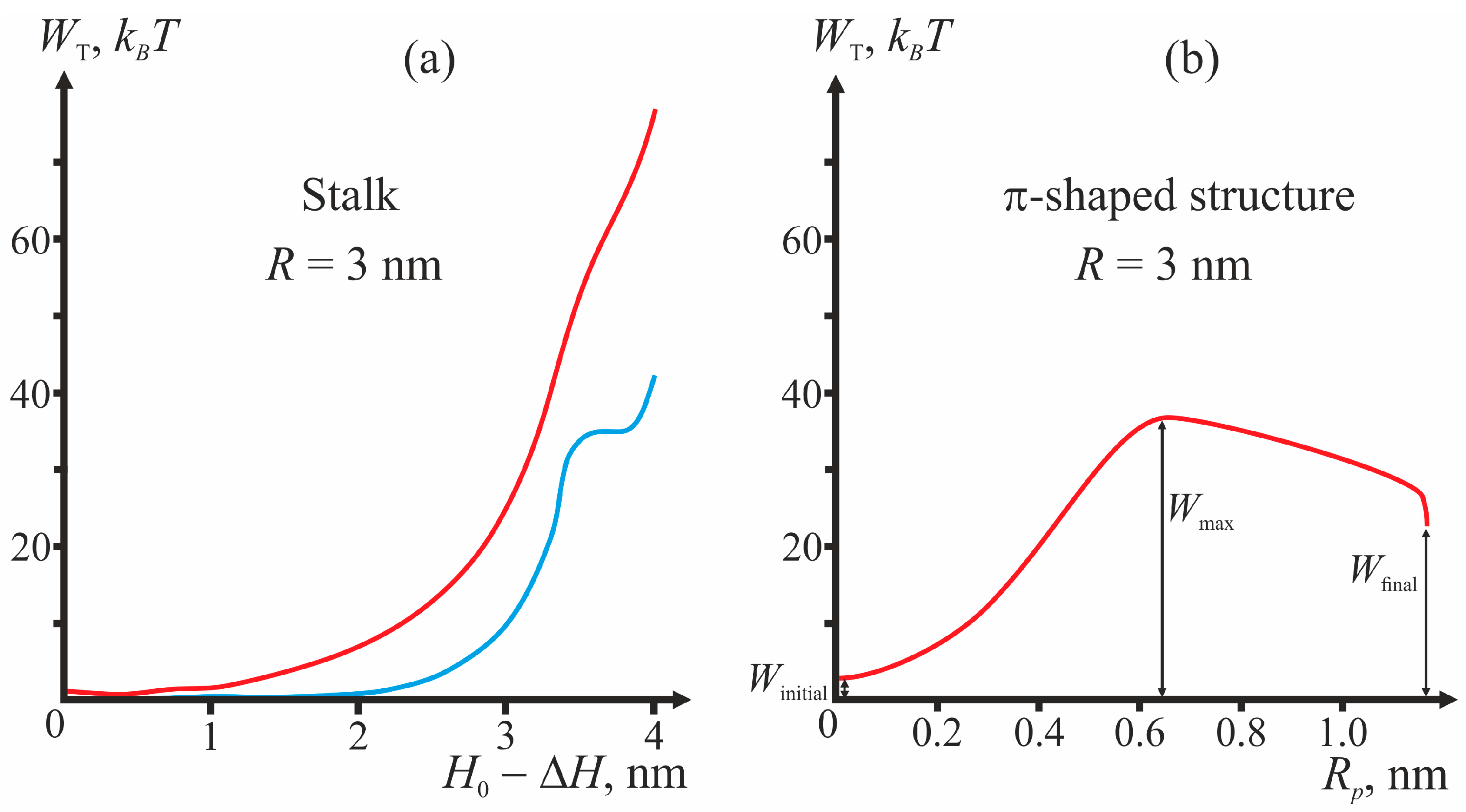
Figure 3 presents the plots of dependencies of the total energy on the coordinates of the process illustrating the algorithm of calculations of the energy barrier for fixed geometry of fusion proteins. For the case of the π-shaped structure, dependence upon only one coordinate, pore radius
Rp, is presented. The height of the hydrophobic part of the pore
L also varies in this process from
L = 3 nm corresponding to the equilibrium thickness of the bilayer 2
h0, to zero. The vertical drop of energy at the point
Rp = 1.1 nm of the plot corresponds to change of the height of the hydrophobic belt
L at constant pore radius
Rp.
As illustrated by
Figure 3, in the initial state of the system, the energy is not equal to zero, since the incorporation of TM domains and fusion peptides causes deformation of membranes. For stalk, the end state of the system (∆
H = 0) can correspond to a local minimum or a maximum of total energy (see, e.g.,
Figure 3a). In the latter case, the calculated height of the energy barrier is only a lower-bound estimate, since further expansion of stalk towards formation of hemifusion diaphragm cannot be considered in the framework of our model and can cost additional energy. In
Figure 3b, the points corresponding to the system’s initial state (
Winitial), final state (
Wfinal), and energy maximum (
Wmax) are shown.
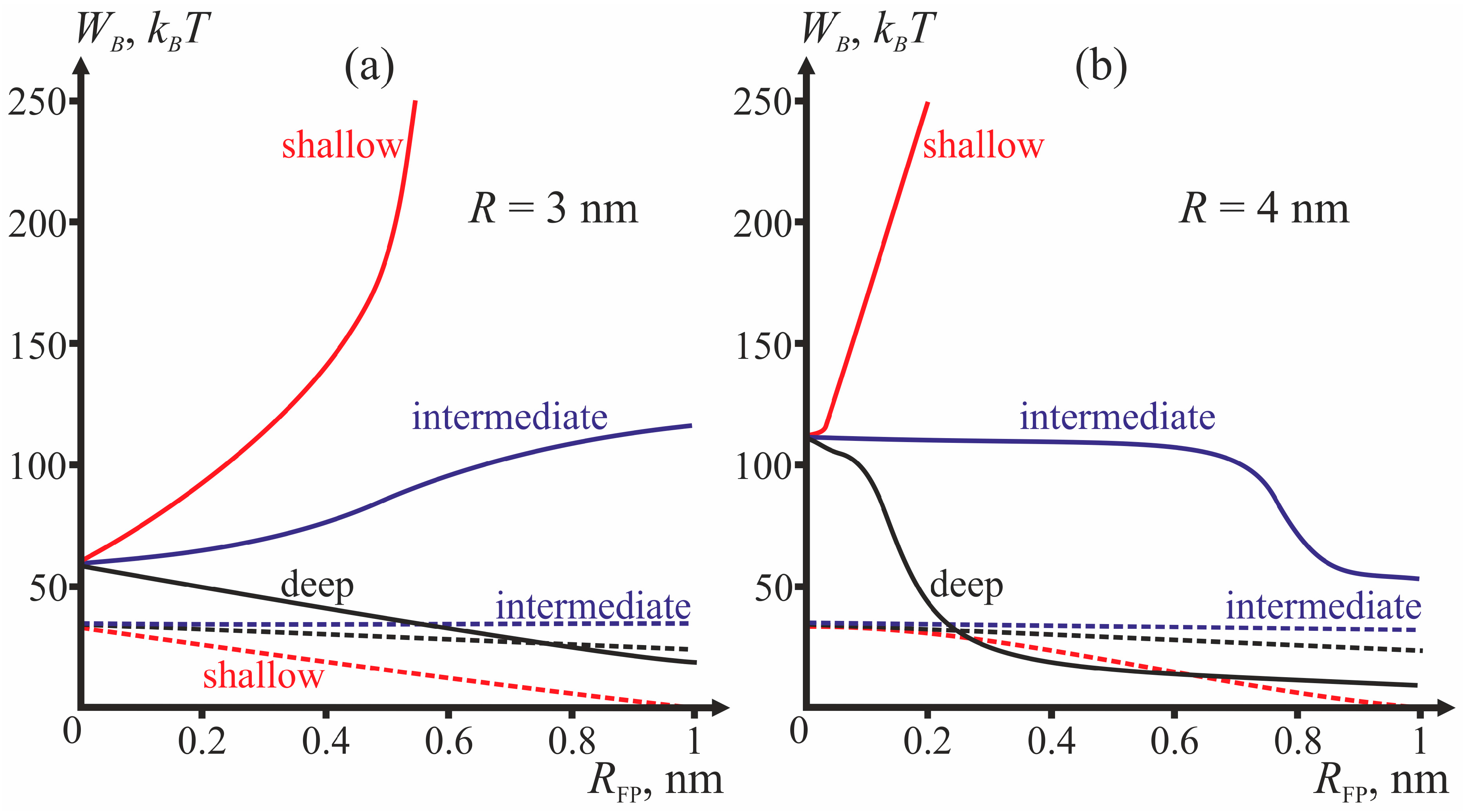
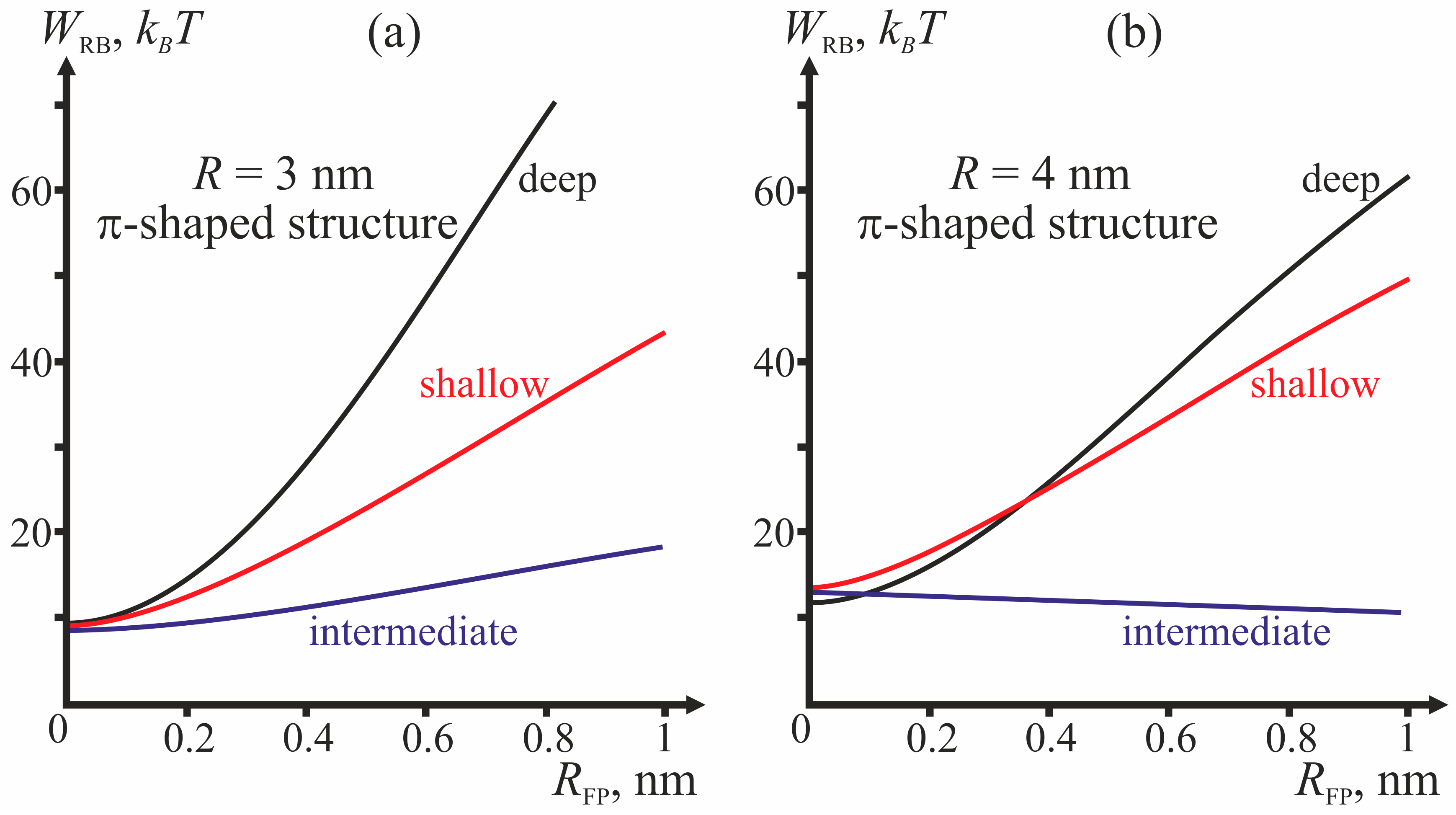
Comparing the energy barriers for system transitions into the stalk and π-shaped structure configurations, we determine for each geometry of fusion proteins the possibility of membrane fusion (stalk formation). We vary three geometric parameters: fusion rosette radius
R, fusion peptide half-width
RFP, and the depth of incorporation of the fusion peptide. The cases of
R = 3 and
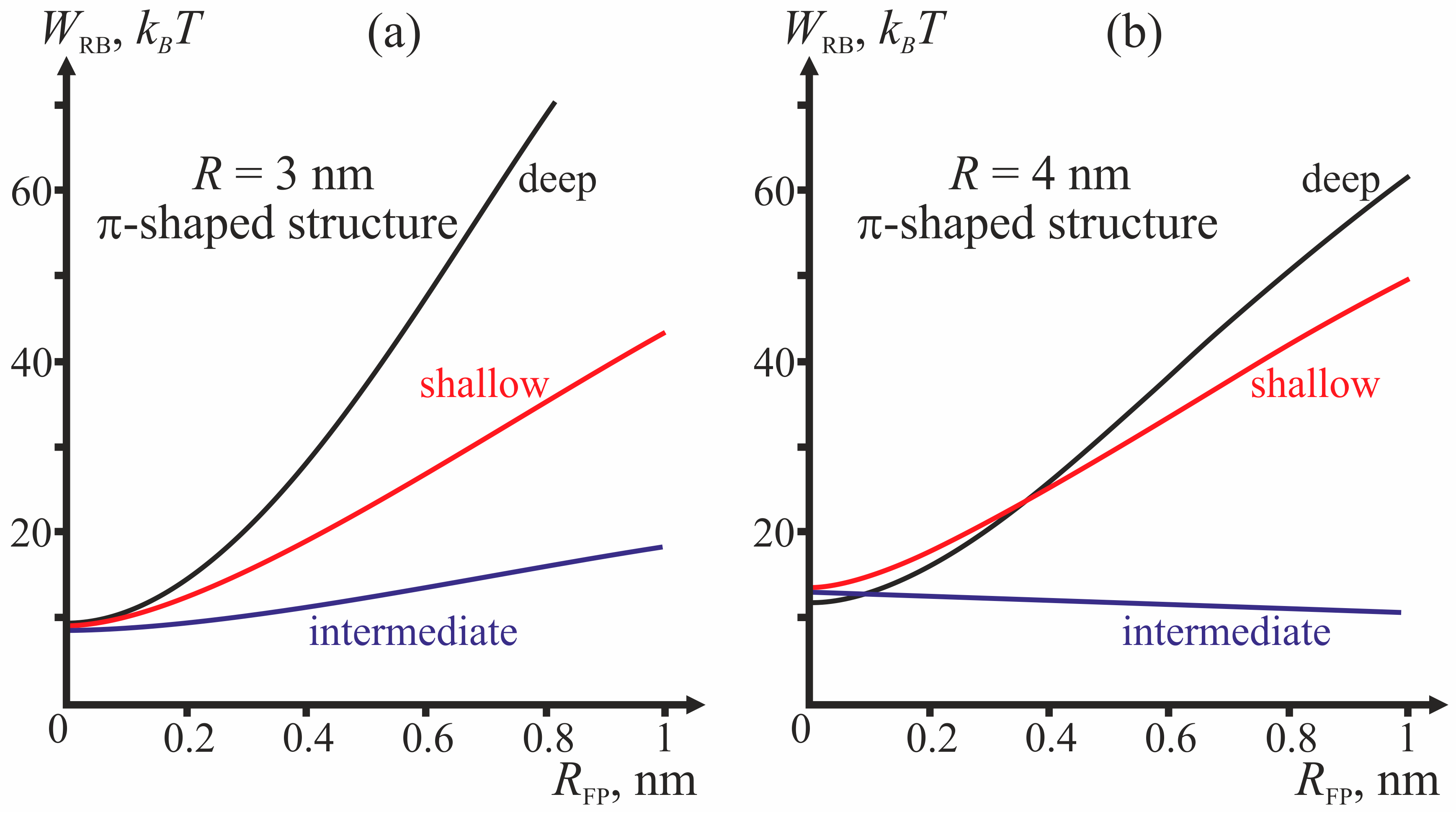
R = 4 nm are analyzed for three different depths of incorporation of the fusion peptide (shallow, intermediate and deep insertion); the half-width of the fusion peptide is varied from 0 to 1 nm.
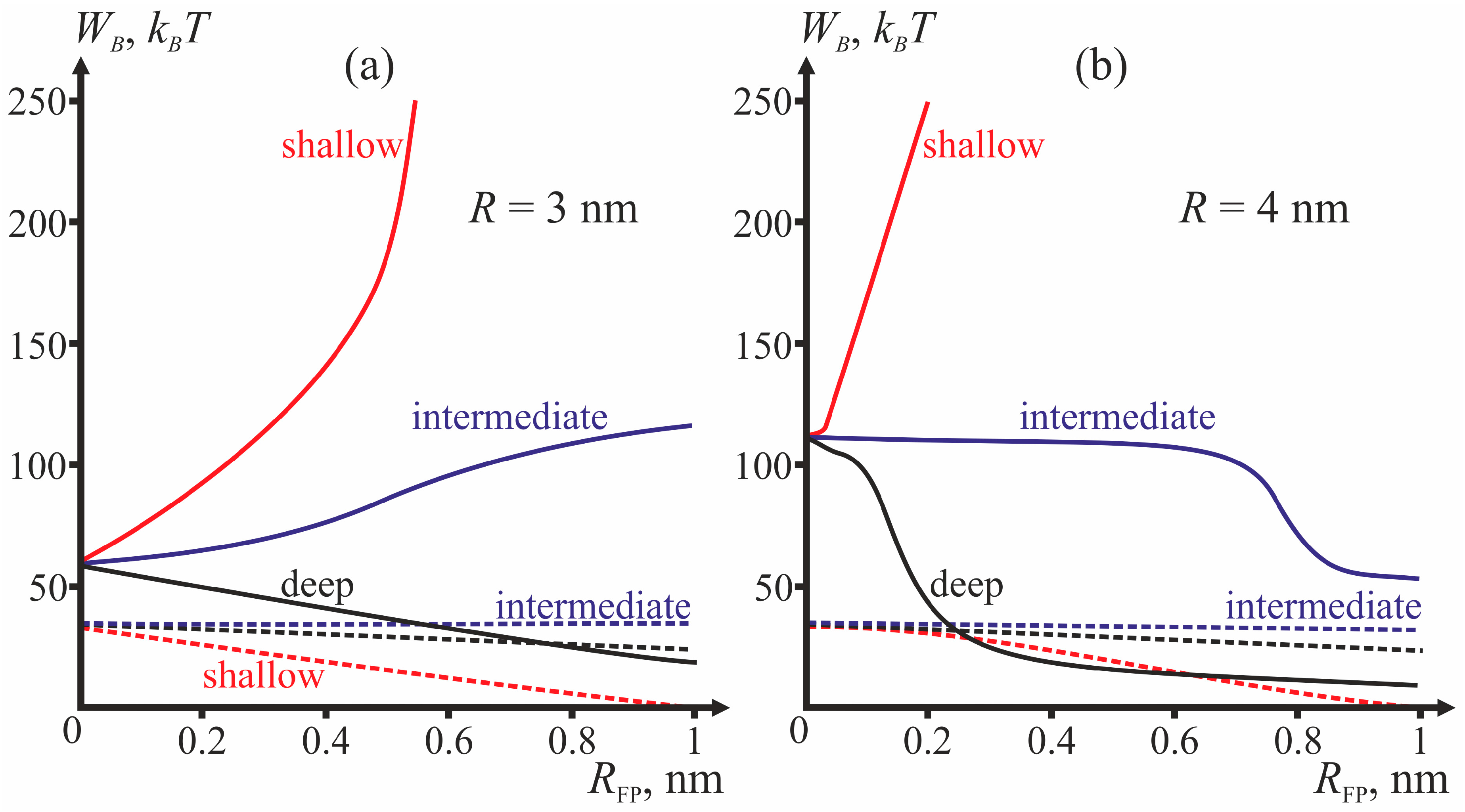
Figure 4 presents dependencies of energy barriers on
RFP at fixed
R for the three depths of insertion.
Figure 4a corresponds to
R = 3 nm,
Figure 4b—to
R = 4 nm.
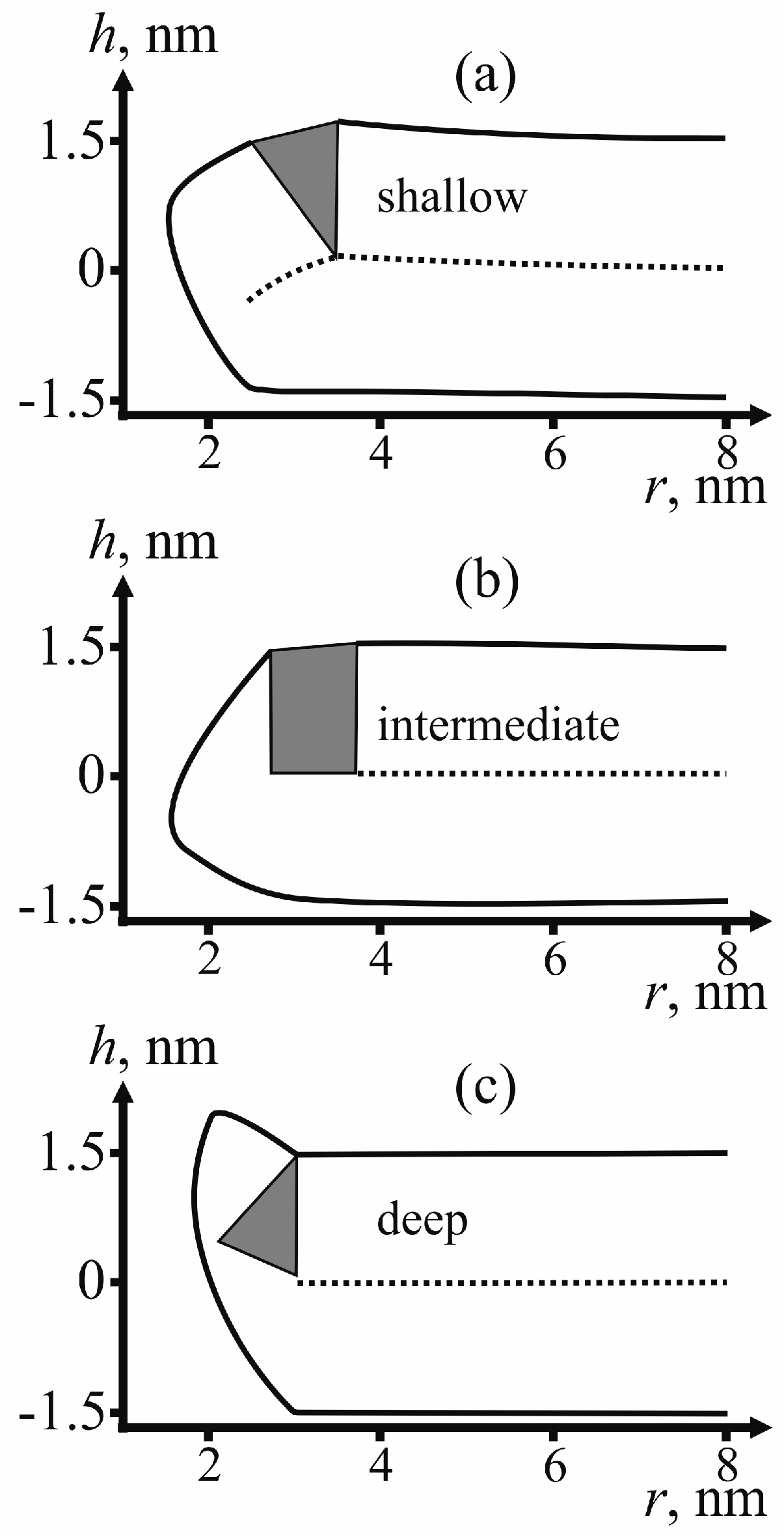
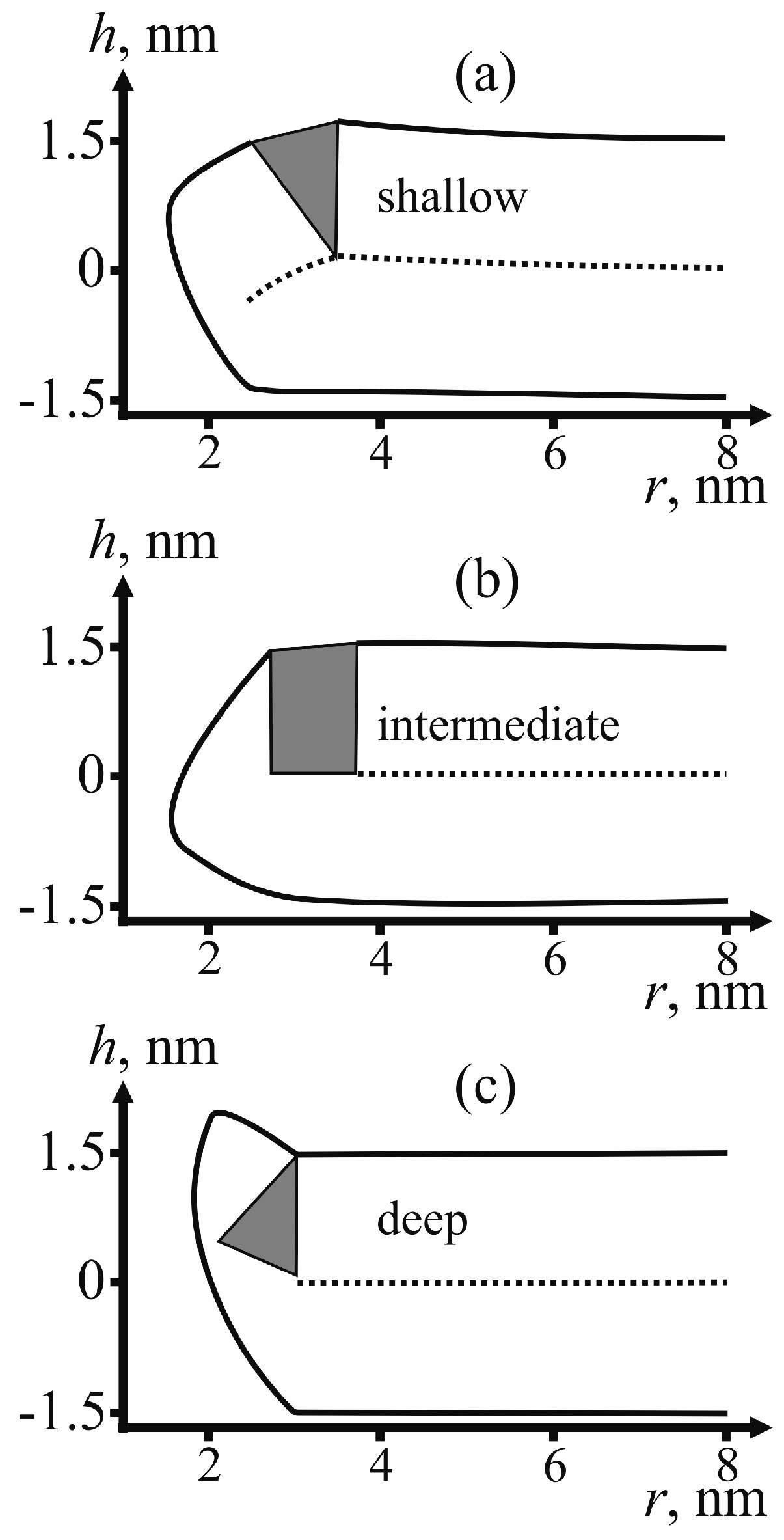
For determining the state of fusion peptides in the configuration of π-shaped structure, we calculate the membrane surface shape for each type of insertion: shallow, intermediate and deep.
Figure 5 provides examples of the membrane surface shape corresponding to the fusion rosette radius
R = 3 nm and the half-width of the fusion peptide
RFP = 0.5 nm.
According to
Figure 5, no displacement of peptides to the equator of the pore edge occurs for any depth of their insertion. To verify that the structure is indeed a dead end, we have to analyze possibilities for its further evolution. One option is closure of the pore with return of the system to the initial state. To achieve it, the system has to go back across the energy barrier. The energy barrier to reach the pore state is defined as the difference of the maximal and the initial energy on the trajectory (see
Figure 3b):
The reverse transition energy barrier is the difference of the maximal and the final energy:
Thus, the reverse barrier can be found from the direct transition barrier as follows:
The dependencies of the reverse barrier on the half-width of the fusion peptide for the cases of
R = 3 and
R = 4 nm are shown on
Figure 6.
Besides the pore “healing” option, a possibility of fusion of the membrane containing the pore with the virus membrane also needs to be considered. For the membrane fusion to take place, hydrophobic defects need to form in the membranes. Due to the presence of the pore in the bottom (target) membrane, the hydrophobic region in it can only be formed along the annulus in the vicinity of the fusion peptide ring. However, the energy barrier associated with the formation and evolution of an annular hydrophobic region is too high to be crossed at the expense of thermal fluctuations. We evaluate it based on the energy barrier calculated for the case of stalk-mediated fusion. As shown in
Figure 3a, the energy not associated with membrane deformations peaks at 40
kBT (blue curve in
Figure 3a). The hydrophobic patch radius at that point is ~1 nm. This means that, in order to create a hydrophobic defect with the area of 1 nm
2, the system has to cross the energy barrier exceeding 12
kBT. According to
Figure 5, the inner radius of the hydrophilic pore is ~1 nm. We consider that for fusion, the width of the hydrophobic belt must also be no smaller than ~1 nm. Its area then exceeds 18 nm
2. It means that the energy barrier associated with formation of such a belt is larger than 100
kBT and cannot be crossed at the expense of thermal fluctuations within the characteristic time of fusion (~1 min) [
6].
3. Discussion
It is presently believed that, in most cases, membrane fusion occurs through no-leak mechanisms, implying formation of a stalk [
9]. Pore formation in the course of fusion was believed to be a pathological process and was not systematically investigated. However, in a number of recent experimental works [
27,
34,
35], the existence of the fusion structure with pores was demonstrated. Molecular dynamics simulations confirm that fusion proteins can facilitate formation of pores in the membrane [
29]. Fusion proteins are also known to play a crucial role in crossing of the energy barrier associated with stalk formation [
9,
56]. It implies that, in certain cases, fusion proteins facilitate membrane fusion, whereas in other cases the formation of the structures does not lead to such fusion, and the latter option is not exceptionally rare. We analyzed such structures and the characteristics of the fusion proteins essential for evolution of the fusing membrane.
The most important parameter for the choice between the stalk and the π-shaped structure scenario is the structure of fusion peptides and geometry of their incorporation into the host cell membrane. In our model, this structure is described by two parameters: half-width
RFP and depth of insertion, i.e., the value of the boundary director. According to
Figure 4, the behavior of the system is determined by the depth of incorporation of the peptide: decreased depth of incorporation hinders fusion due to increase of the energy barrier to stalk formation. Pore formation proves the least favorable in the case of intermediate depth of insertion and the most favorable in case of shallow insertion. The larger the half-width of the fusion peptide, the more favorable is pore formation. For the biologically relevant sizes of the fusion peptide (
RFP ≥ 0.5 nm), shallow incorporation of the fusion peptide facilitates formation of the π-shaped structure, and deep insertion facilitates membrane fusion. In the case of intermediate incorporation, the fusion rosette radius
R plays a definitive role: the larger the fusion rosette, the more favorable stalk formation becomes.
As can be seen from the calculated shape of the membrane (see
Figure 5), the peptides do not relocate towards the equator of the pore. The pore radius in these conditions is about 1 nm. This result is consistent with the previously obtained data on dependence of lipid pore energy on its radius [
44,
45,
50]. According to these calculations, the energy of lipid pore has a minimum corresponding to the radius of ~1 nm; further increase of the radius results in abrupt increase of the deformation energy of the membrane. Due to the presence of hydrophilic pore in a membrane, fusion of the membranes is hindered by the need to form a large hydrophobic area with energy of about ~100
kBT. The reverse barrier corresponding to pore closure depends on the depth of incorporation of the fusion peptide (see
Figure 6). In the case of intermediate depth, it is almost insensitive to the changes of half-width
RFP and amounts to about 10–15
kBT, i.e., it can be relatively rapidly crossed at the expense of thermal fluctuations. In other cases, for biologically adequate sizes of the fusion peptide (
RFP ≥ 0.5 nm) the energy barrier would exceed 50
kBT, i.e., too high to be crossed at the expense of thermal fluctuations in reasonable time. Therefore, π-shaped structure turns out to be a dead end final state in the case of shallow and deep insertion: both fusion and return into the initial state are hindered. In the case of intermediate depth of incorporation, a return to the initial state proves possible. Thus, in the case of intermediate depth a scenario with periodic opening and closure of a pore in the target membrane preceding stalk-mediated fusion is possible. These reversible changes, as well as a significant height of the stalk formation energy barrier (~50
kBT) must substantially slow down fusion of membranes in case of the intermediate depth of insertion of fusion peptides.
Our results offer explanations for a number of experimental findings. Thus, it was demonstrated in the work [
42] that there is a correlation between the depth of incorporation of fusion peptide of HIV and the effectiveness of fusion mediated by the peptide: the deeper the insertion, the more effective the membrane fusion. Similar correlations were observed in a number of works dedicated to analysis of mutants of influenza virus.
It is broadly assumed that the boomerang-shaped HA fusion peptide incorporates deeply into the contact monolayer of the target membrane [
38]. It was demonstrated that a mutation transforming the fusion peptide into α-helix lying on the surface of the membrane completely inhibits fusion [
37]. A mutation causing an increase of the angle between parts of the boomerang-shaped peptide, and hence partial decrease of the depth of its insertion, results in a decrease of the fusion effectiveness [
35], with a leak observed immediately before the fusion. Our results explain both the decreased fusion effectiveness and the observed leak. Insignificant decrease of the depth of incorporation in our terms means that the fusion peptide is now incorporated to an intermediate depth. According to our results (see
Figure 4), intermediate insertion can facilitate opening of the pore with subsequent rapid closure. It means that in the case of intermediate insertion, occurrence of leak is possible before the fusion. In case of shallow insertion, no fusion occurs.
The obtained results are indicative of a generic nature of the correlation between the depth of insertion of the fusion peptide and effectiveness of fusion, and allow concluding that the deeper fusion peptide inserts into the membrane of the target cell membrane, the more effectively fusion occurs. We failed to find any credible correlation between the hydrophobic to hydrophilic residue ratio and the depth of insertion. One of the reasons for that is very low variability of this ratio associated with the known mutations of fusion peptides. More generally, the hydrophobicity/hydrophilicity of individual amino acids, often quantified through “hydrophobic (or lipophilic) potential” or other similar measures, is a predominantly entropic phenomenon determined by the ability of the molecule to form hydrogen bonds and shift electronic density within its moieties to minimize the overall electrostatic energy. Intramolecular H-bonding reduces the apparent hydrophilicity of the molecule by leaving fewer H-bond donor/acceptor groups for interactions with water [
57]. Likewise, an amino acid residue as a part of a peptide or protein molecule can form hydrogen bonds or enter hydrophobic interactions with its neighbors or other proximal residues, thereby reducing its effective hydrophilicity or hydrophobicity score. Thus, a residue of an amino acid behaving as hydrophilic in isolation can be a part of a highly hydrophobic domain by virtue of sequestration of the residue’s polar moieties via intramolecular interactions. As a side note, the occurrence of polar residues within transmembrane domains often drives their oligomerization, the residues serving as oligomerization interfaces, in which case the interactions are intermolecular. Thus, membrane-embedded domains, including fusion peptides, can and do accommodate multiple “hydrophilic” residues. The energy cost of bringing a charged residue into the hydrophobic core is usually prohibitively high, but even such examples, though rare, are not totally unheard of. Accordingly, as stated in the review [
23], it is the fusion peptide primary and secondary structure rather than a sum of properties of individual amino acid residues that determines the depth of insertion of the peptide. Their variance thus provides an instrument for fine regulation of the fusion effectiveness.
For the purpose of this analysis, we assumed homogeneous distribution of lipids. Cellular membranes are known to consist of multiple lipid species with highly heterogeneous lateral distribution, including formation of microdomains of liquid ordered (L
o) phase (“rafts”) with the local lipid composition different from that of the bulk lipid [
58]. The liquid ordered domains are also known to be thicker than the surrounding membrane [
59], which results in line tension of the domain boundary [
43]. According to recent experimental studies, the raft boundary structure plays an important role in the process of viral fusion [
60]. In particular, the HIV fusion peptide tends to be preferentially inserted along the raft boundaries, and phase separation with formation of such boundaries substantially enhances fusion efficiency. By contrast, no enhancement of fusion efficiency was observed in the membranes consisting exclusively of the L
o phase without any phase separation boundaries. These trends, however, do not apply to the influenza virus. This difference in the behavior of the HIV and influenza virus fusion peptides can be attributable to different depths of insertion of the corresponding fusion peptides into the host cell membranes. A correlation can be found between the difference in the insertion depths and the difference in the spontaneous curvatures induced by the insertion: deeper penetration (as in case of the HIV) is similar to induction of negative spontaneous curvature, whereas more shallow incorporation (e.g., influenza virus) induces nearly zero spontaneous curvature. According to our prior results [
59], membrane components with non-zero spontaneous curvature (regardless of the sign) preferentially distribute into the phase separation boundaries, so that such boundaries can serve as local concentrators of the fusion peptides with deep insertion and define their orientation in the membrane. Besides that, regardless of the preferences of the fusion peptides, the phase separation boundary has excess energy that can be characterized by the line tension of the boundary and is proportional to its total length. One of the viable options for minimizing the boundary energy is to bend the domain surface to form a hemispherical bulge protruding from the membrane plane. If it happens in the area between the viral membrane and the host cell membrane, minimal distance between the membranes would decrease, facilitating the fusion process through stalk intermediate. Thus, formation of a domain with a high line tension of the boundary should further facilitate fusion. We suppose that incorporation into the target membrane of the fusion peptides with preference towards the phase separation boundary can serve to nucleate domain formation in the target membrane. In other words, under appropriate conditions the fusion peptides can induce formation of phase separation boundaries rather than preferentially partitioning into the pre-existing boundaries. A detailed theoretical model of the process of fusion of phase-separating membranes is an intended topic of our further investigations.

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}