Rapid Non-Enzymatic Glycation of the Insulin Receptor under Hyperglycemic Conditions Inhibits Insulin Binding In Vitro: Implications for Insulin Resistance



Abstract
:1. Introduction
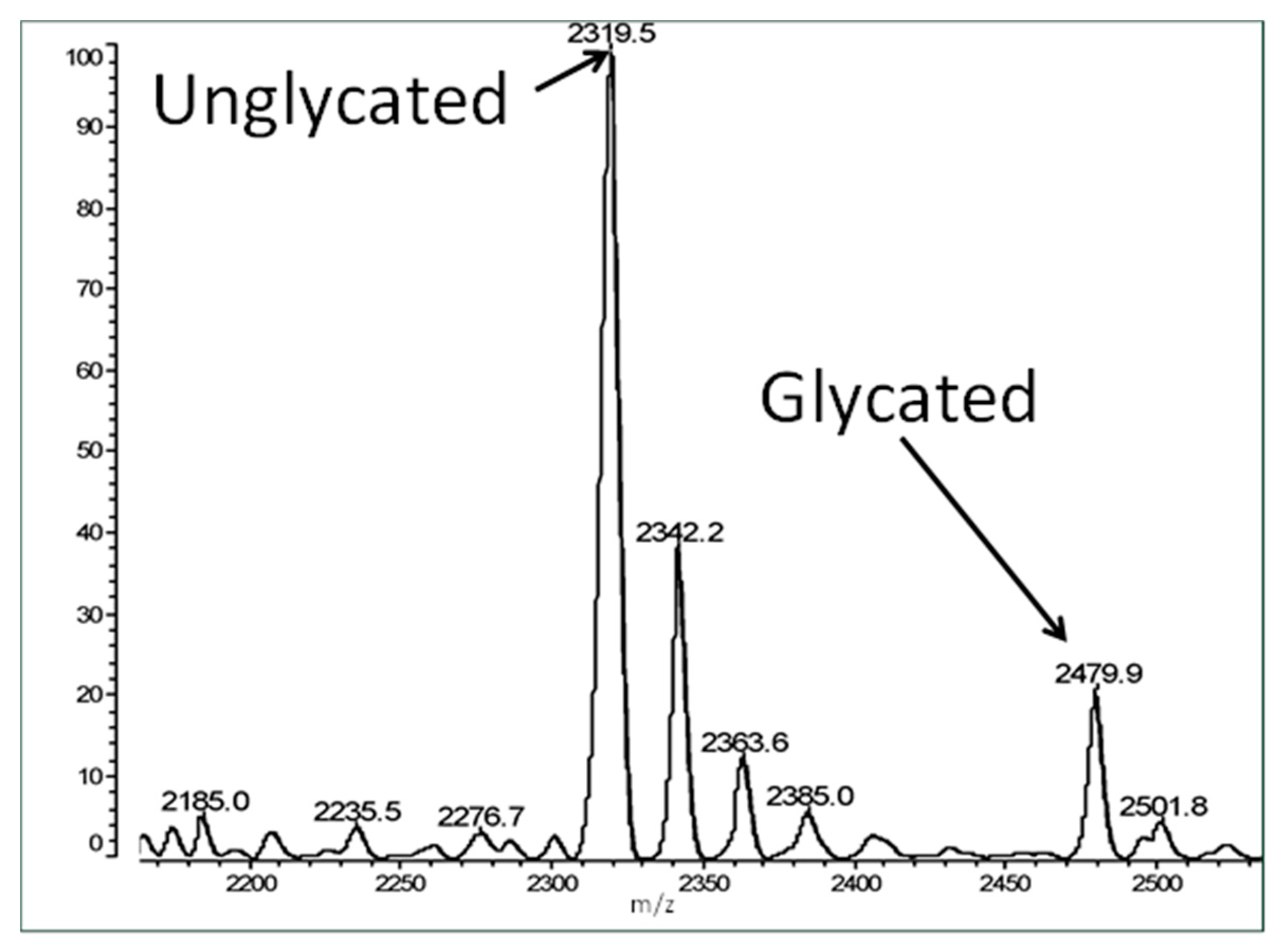
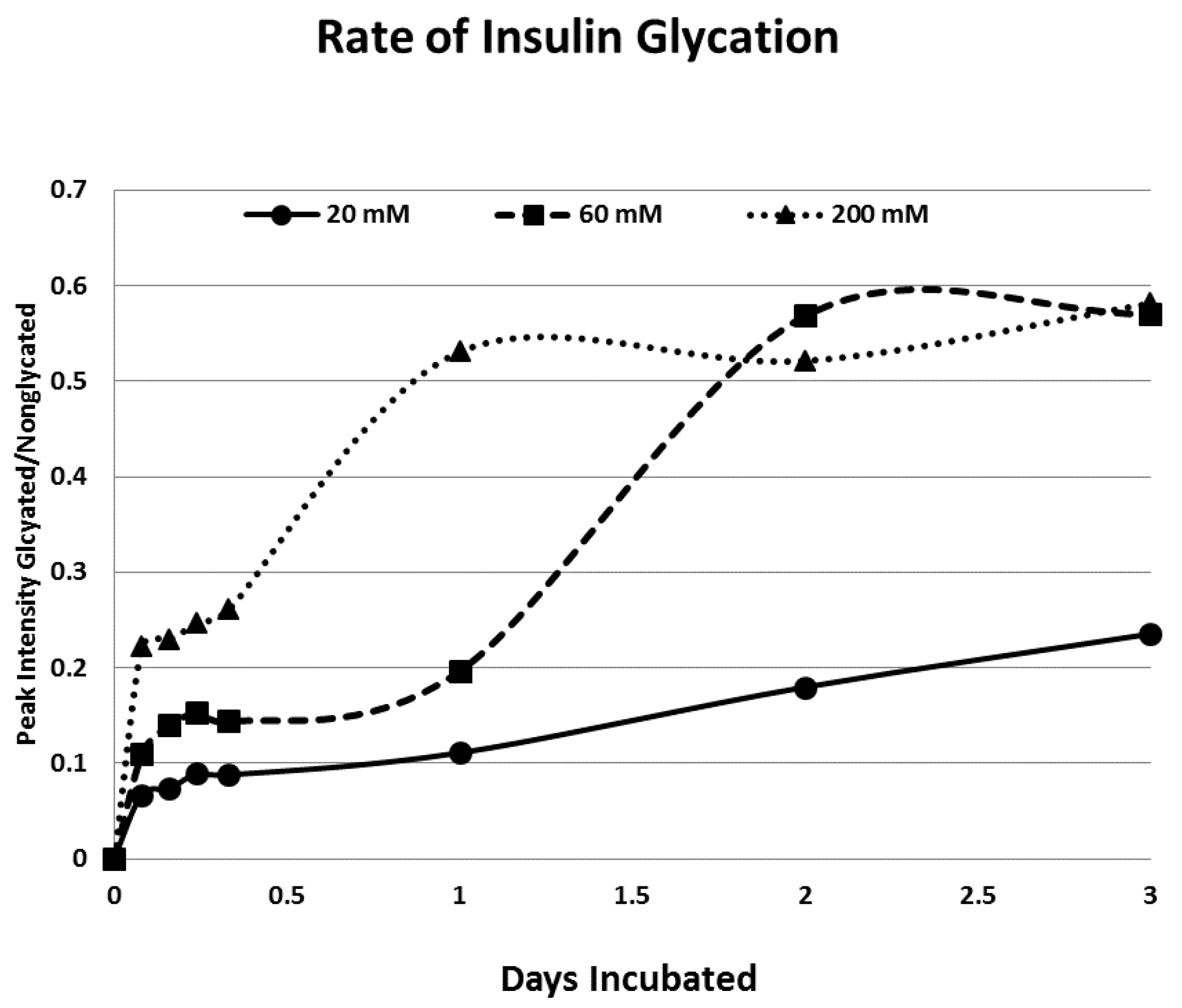
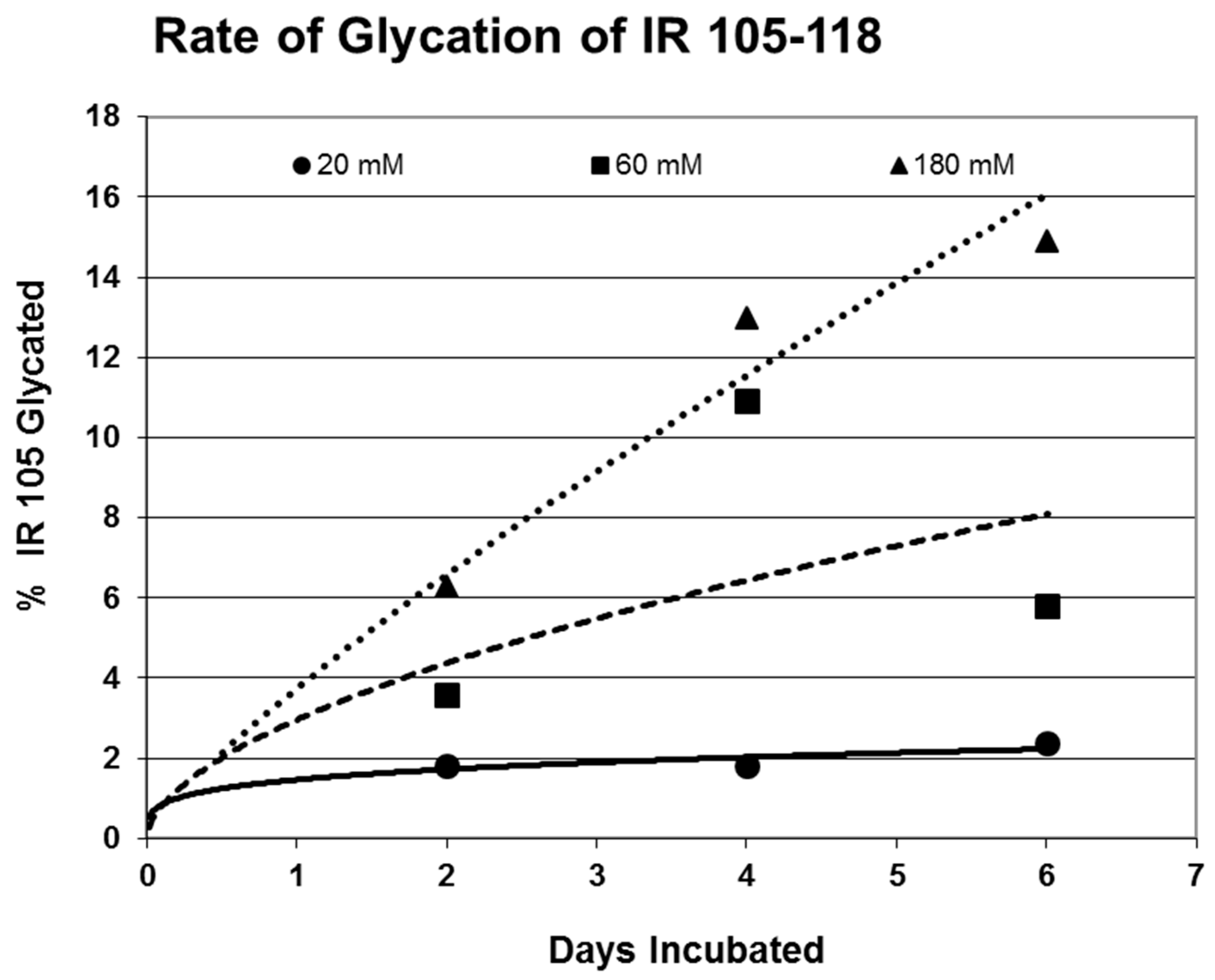
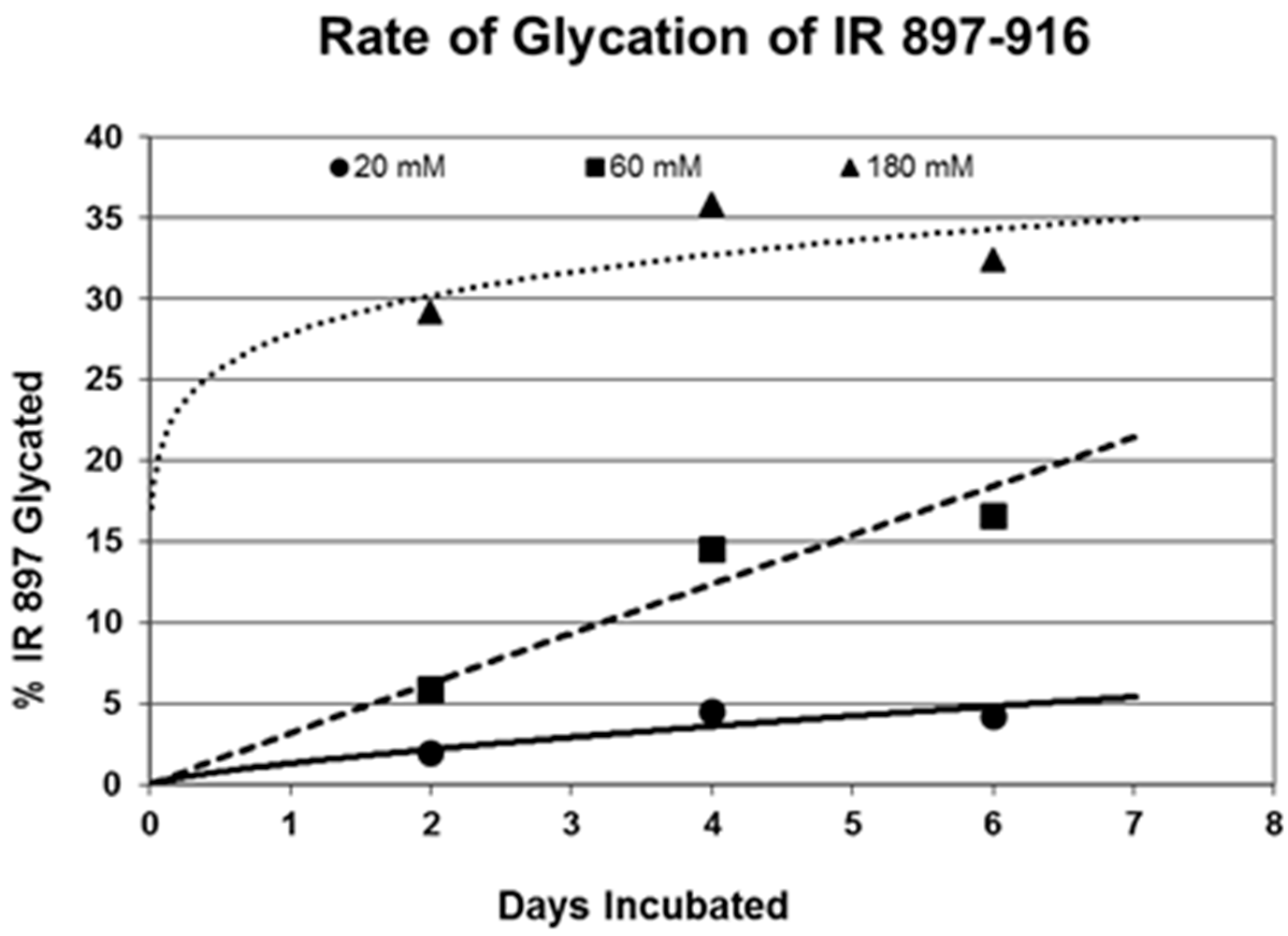
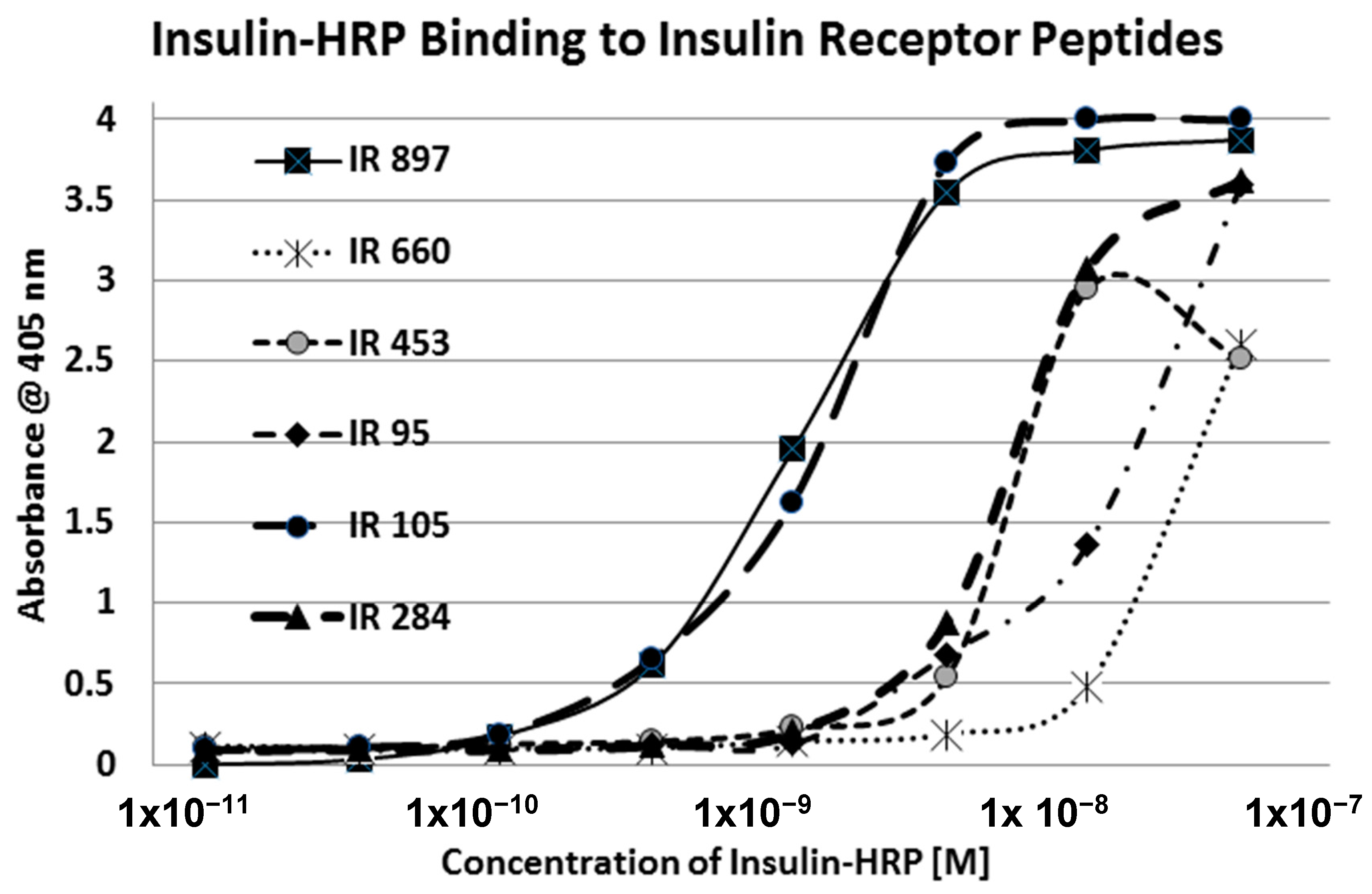
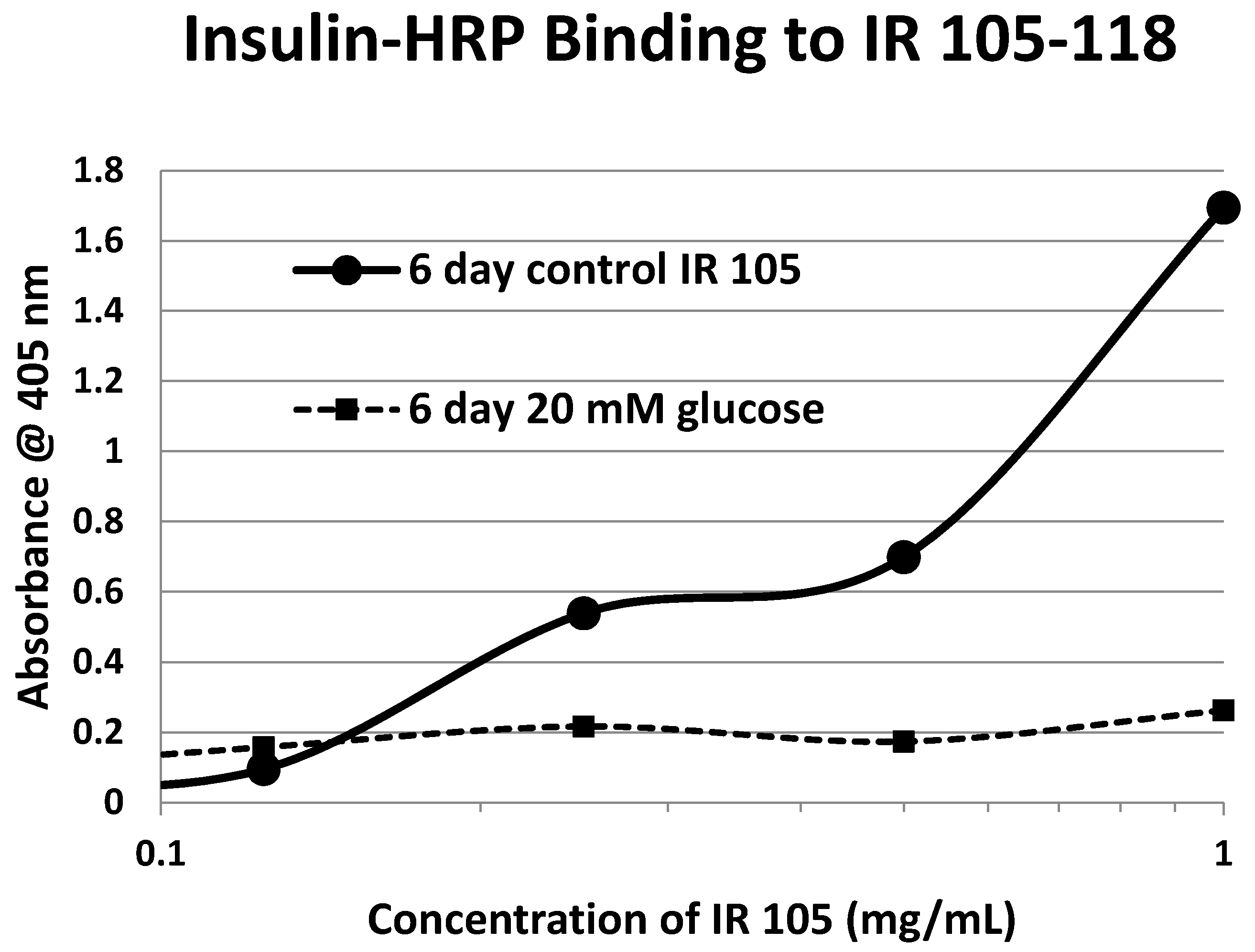
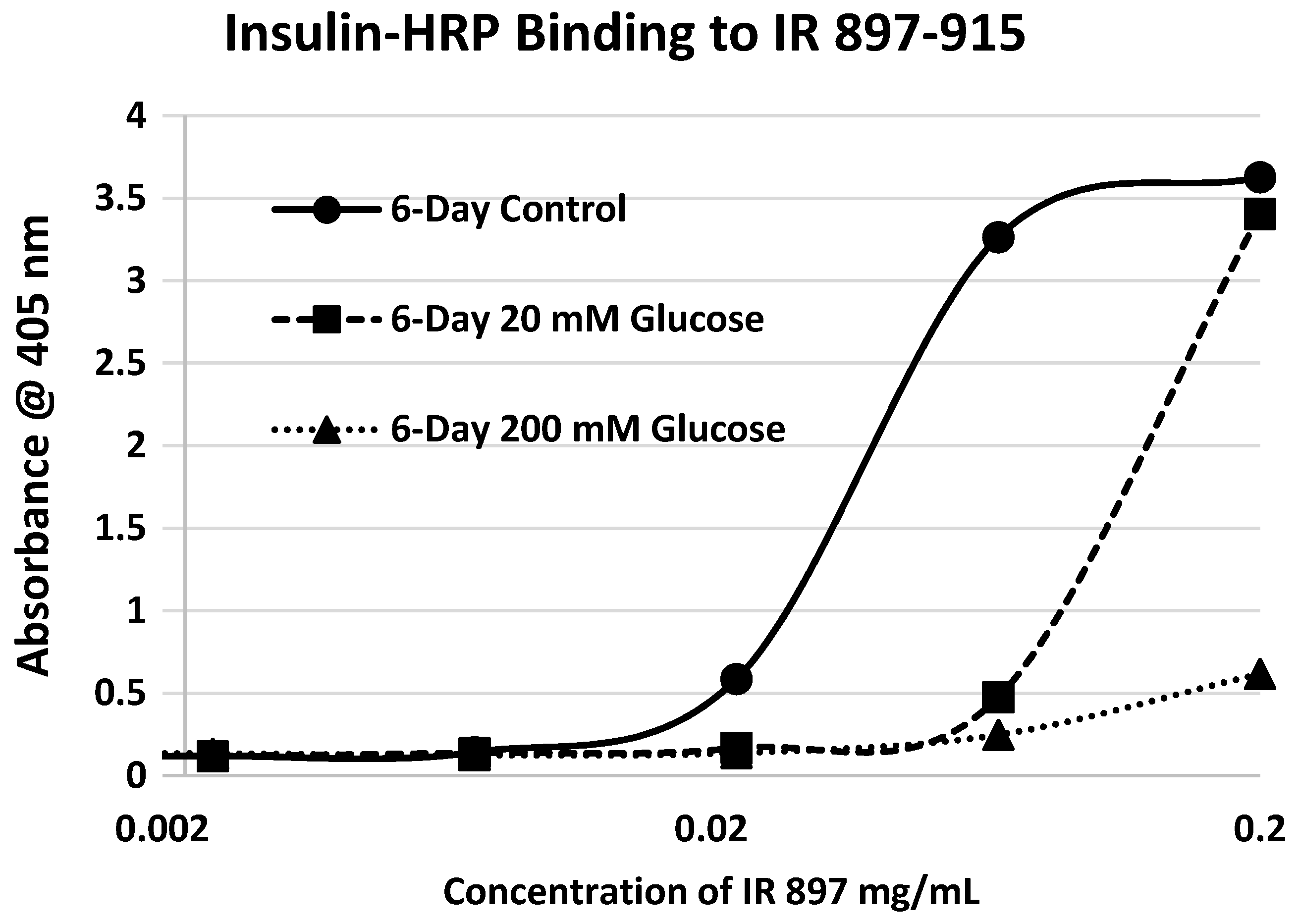
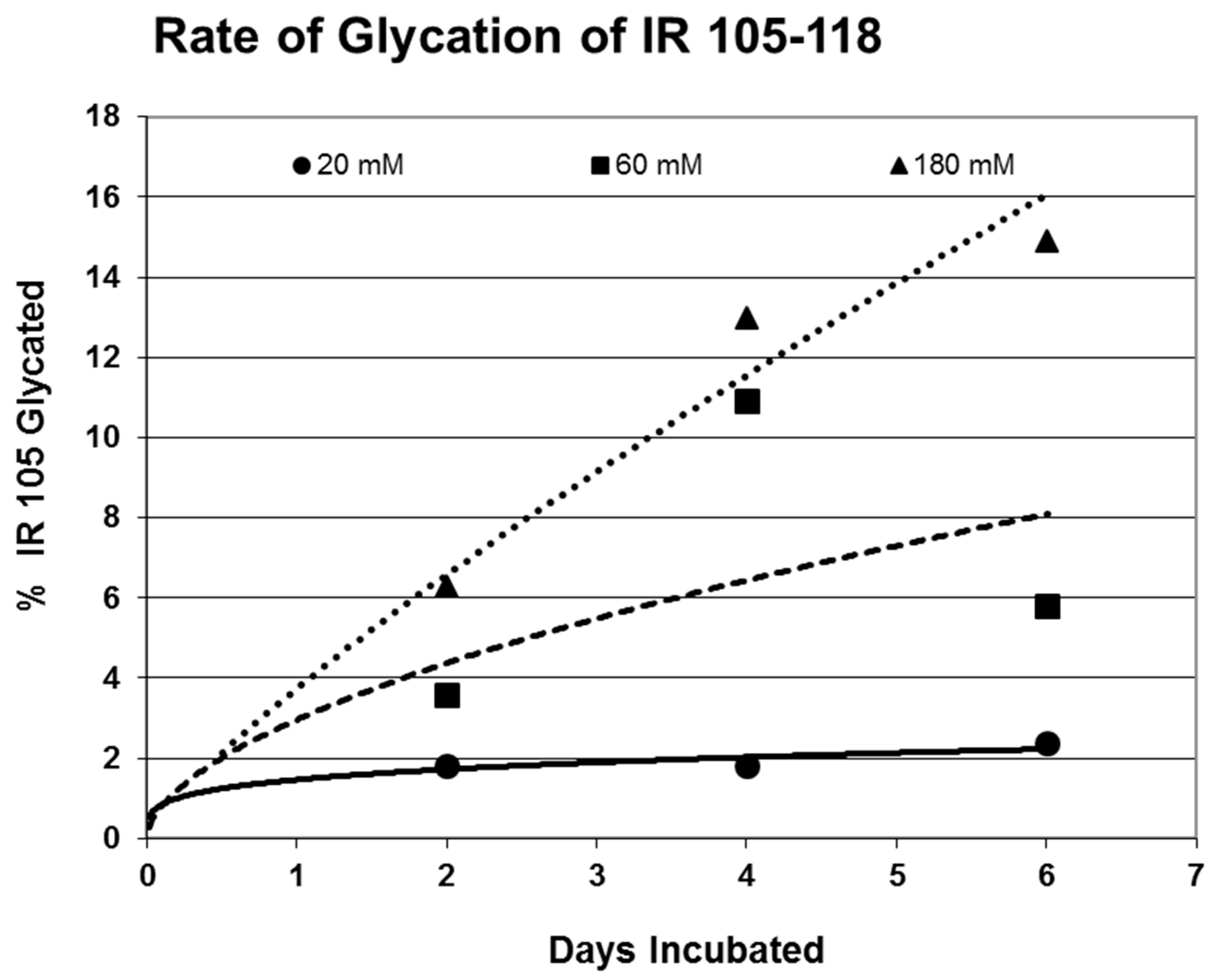
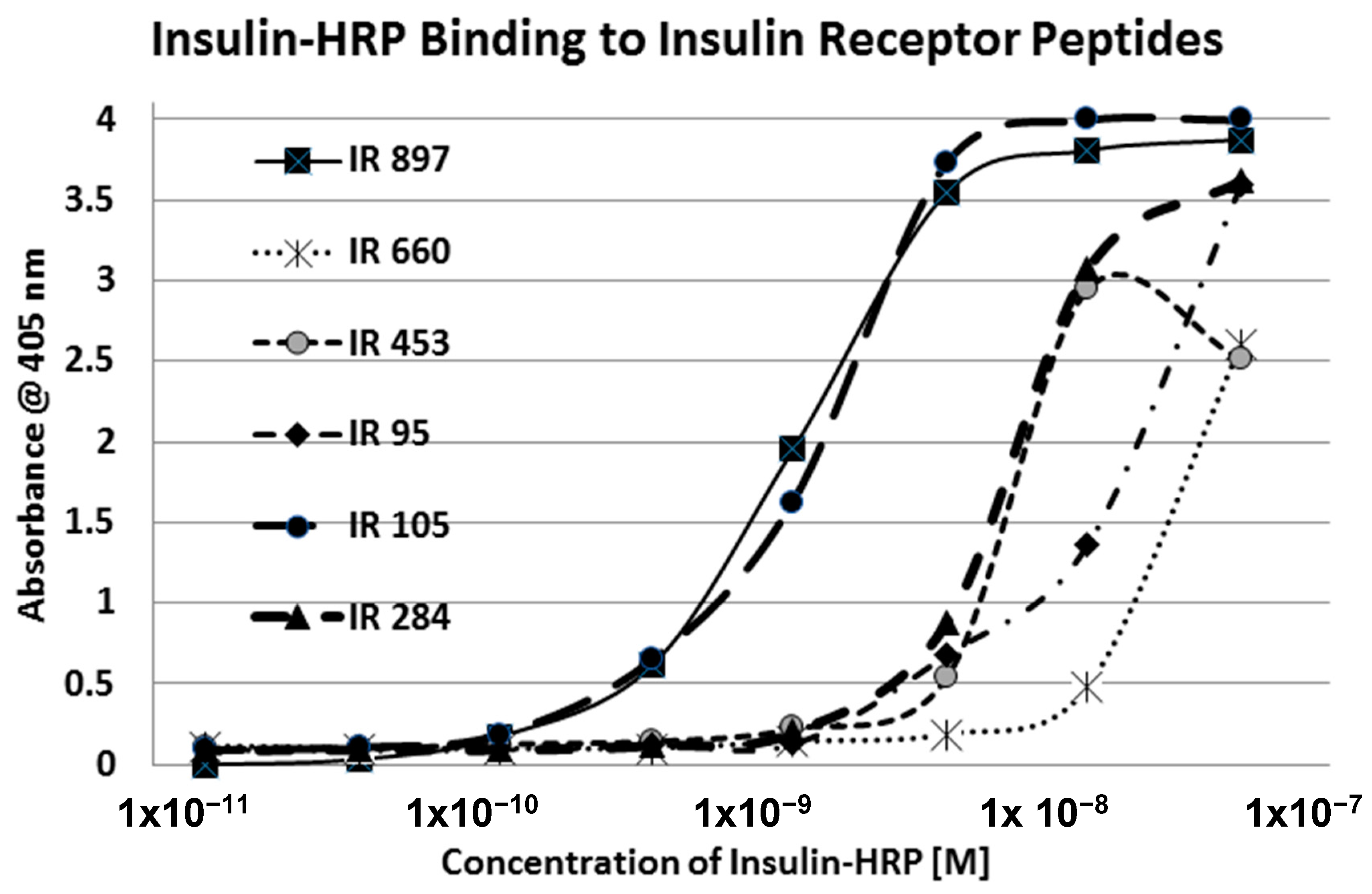
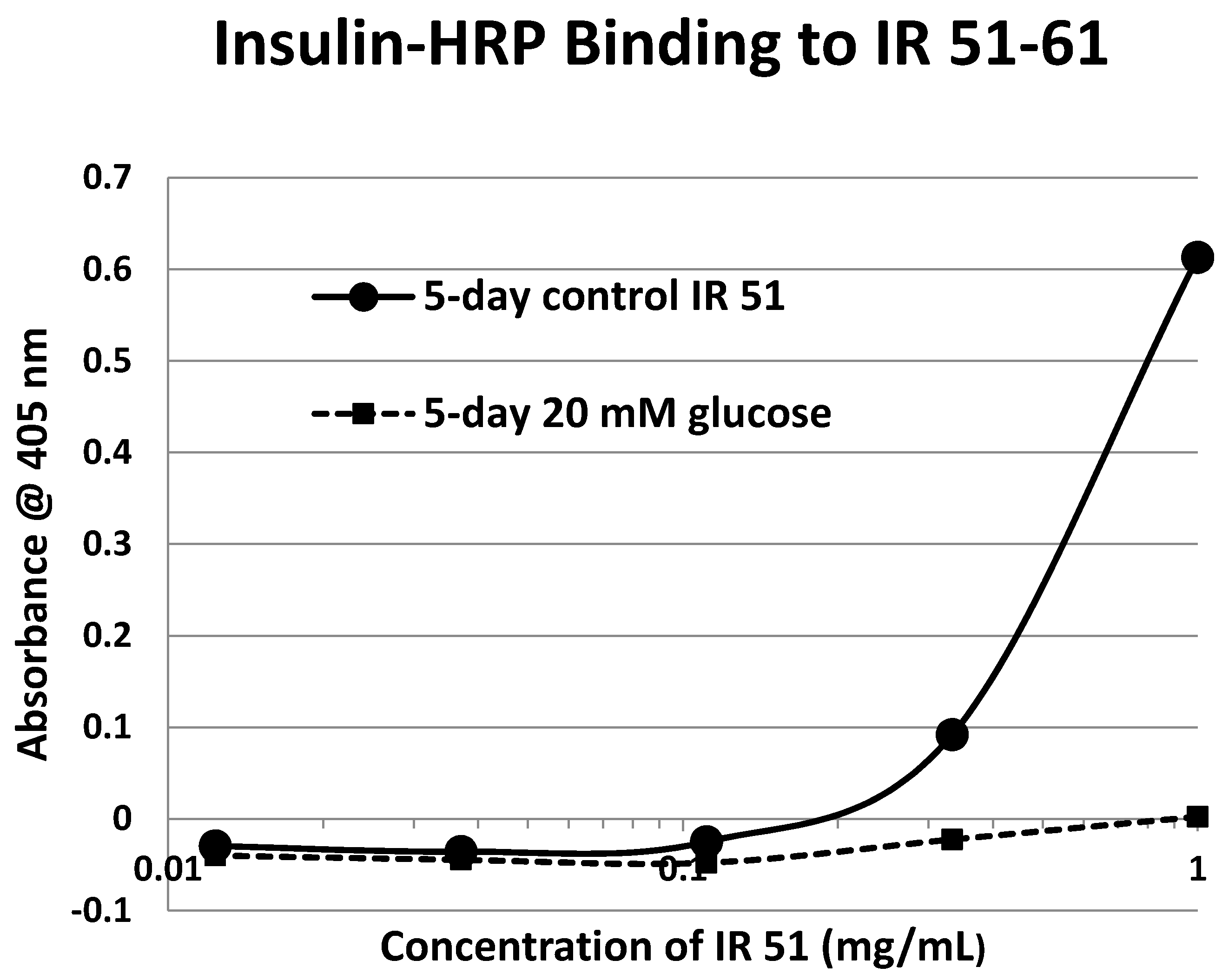
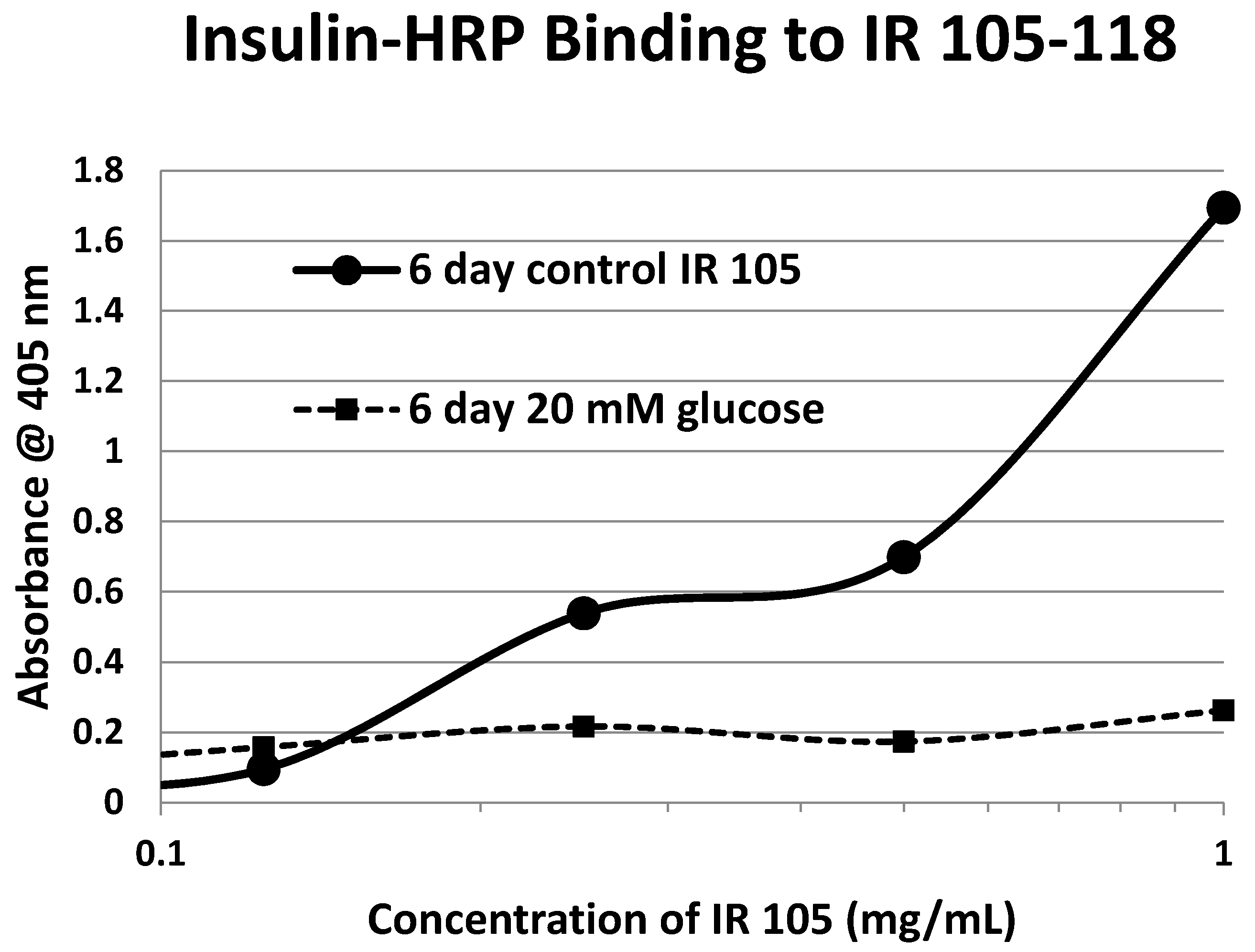
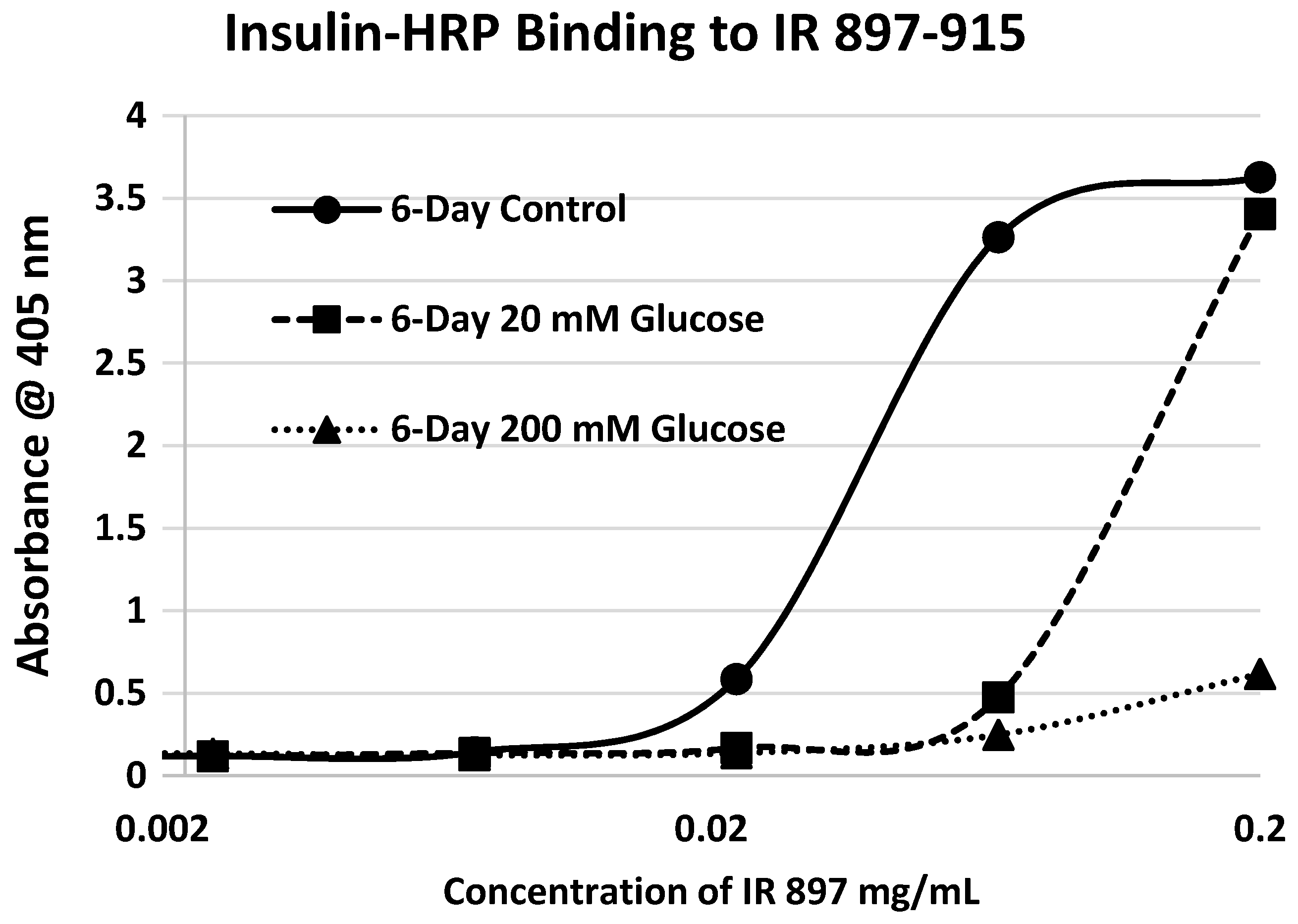
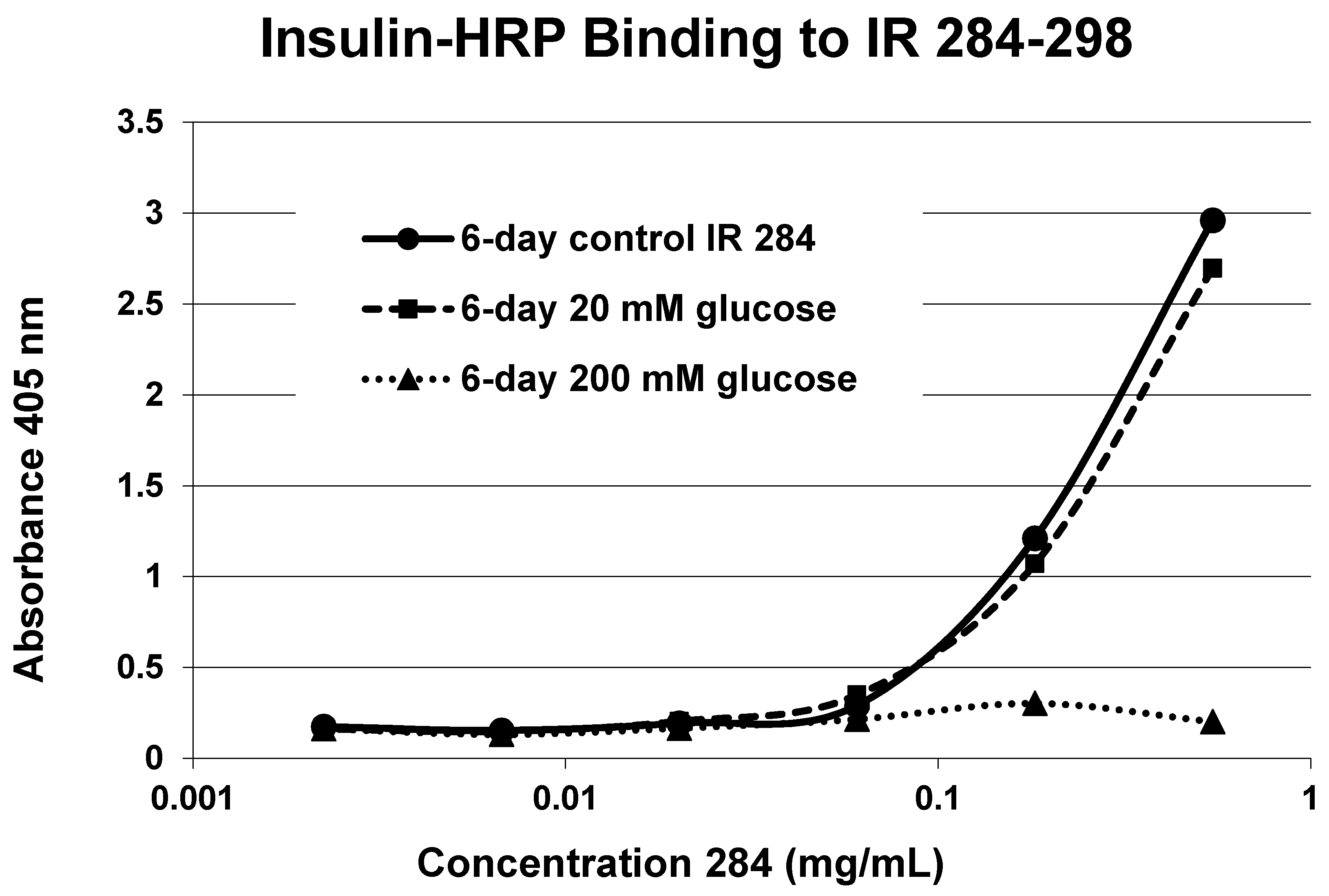
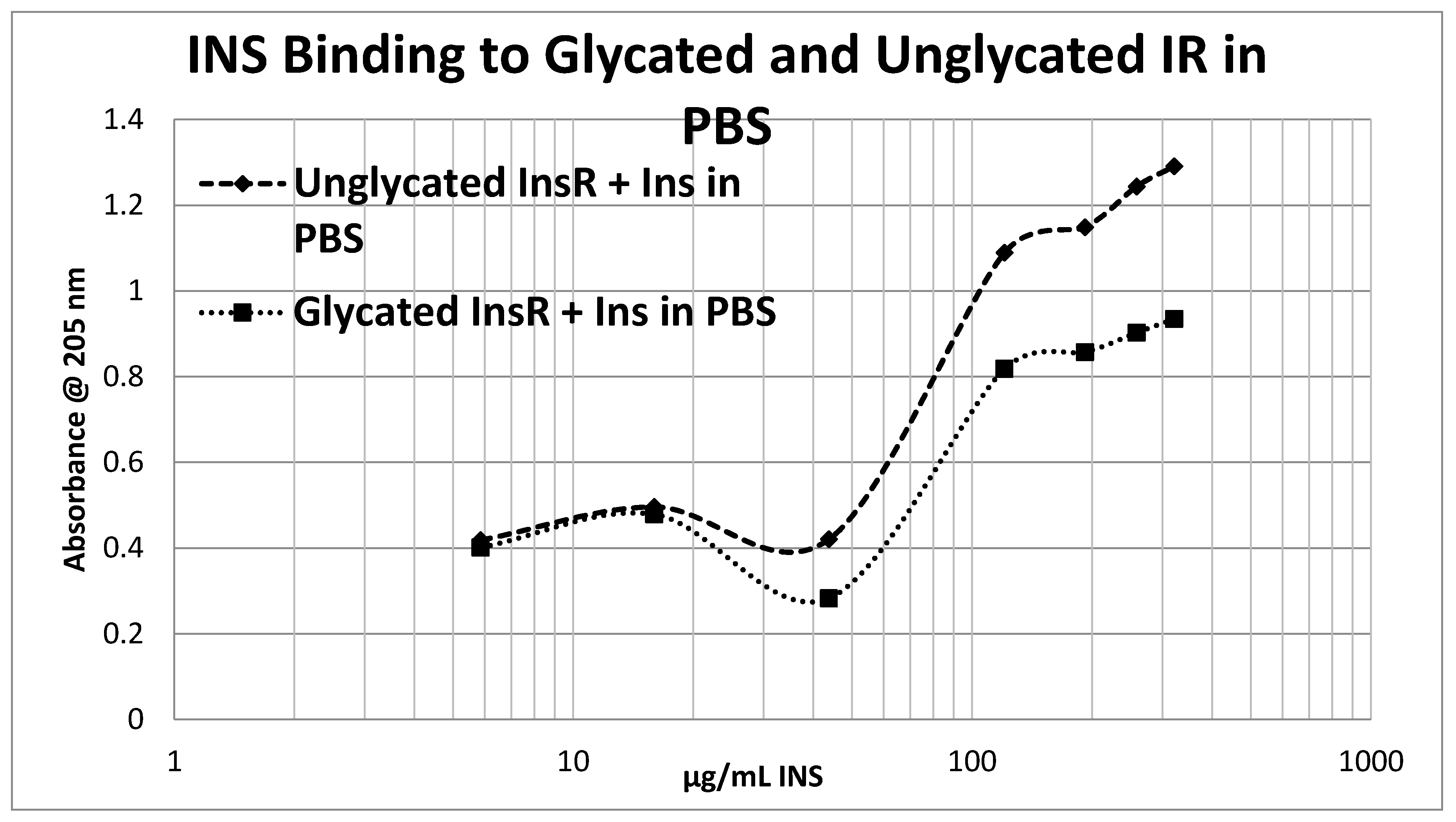
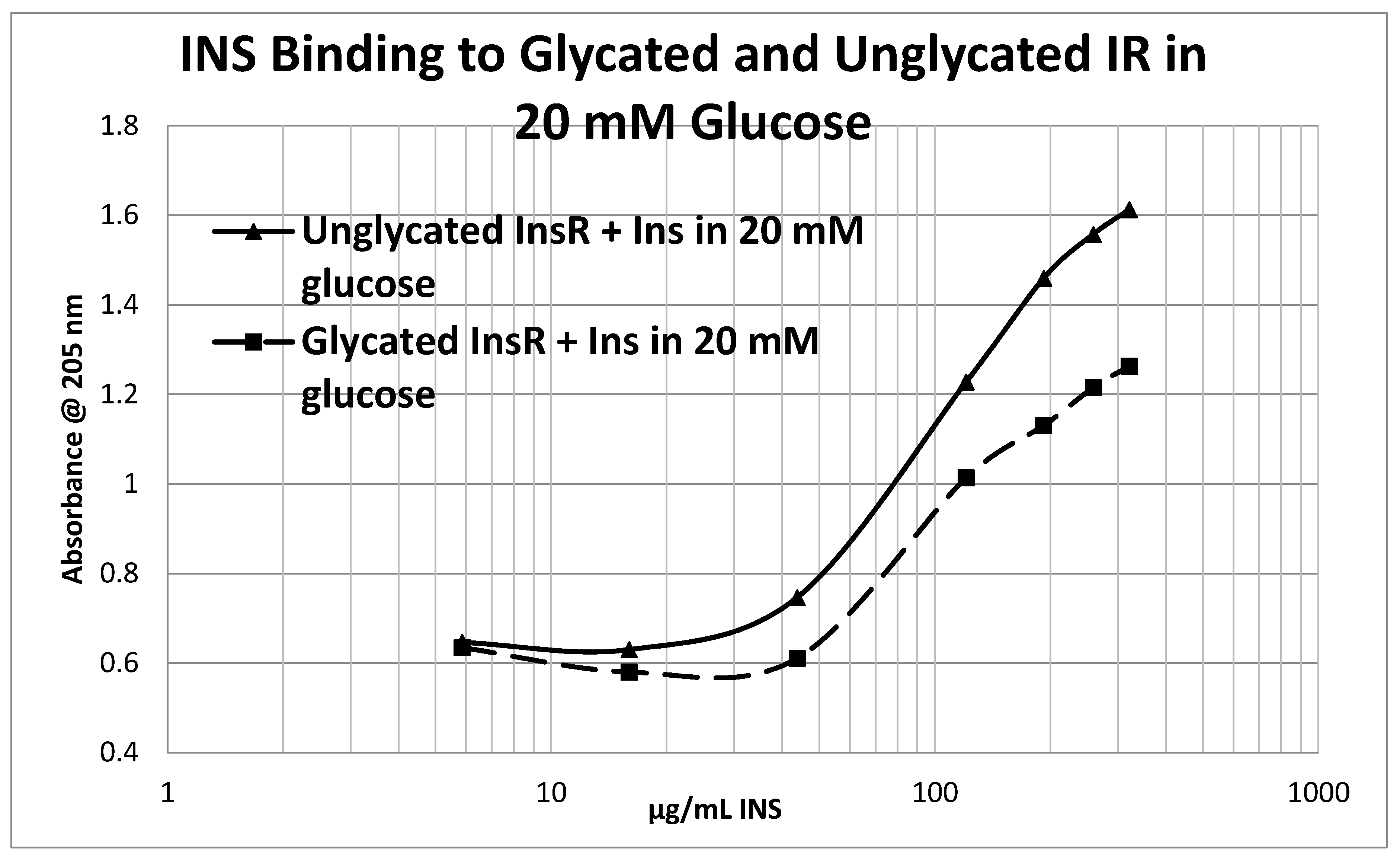
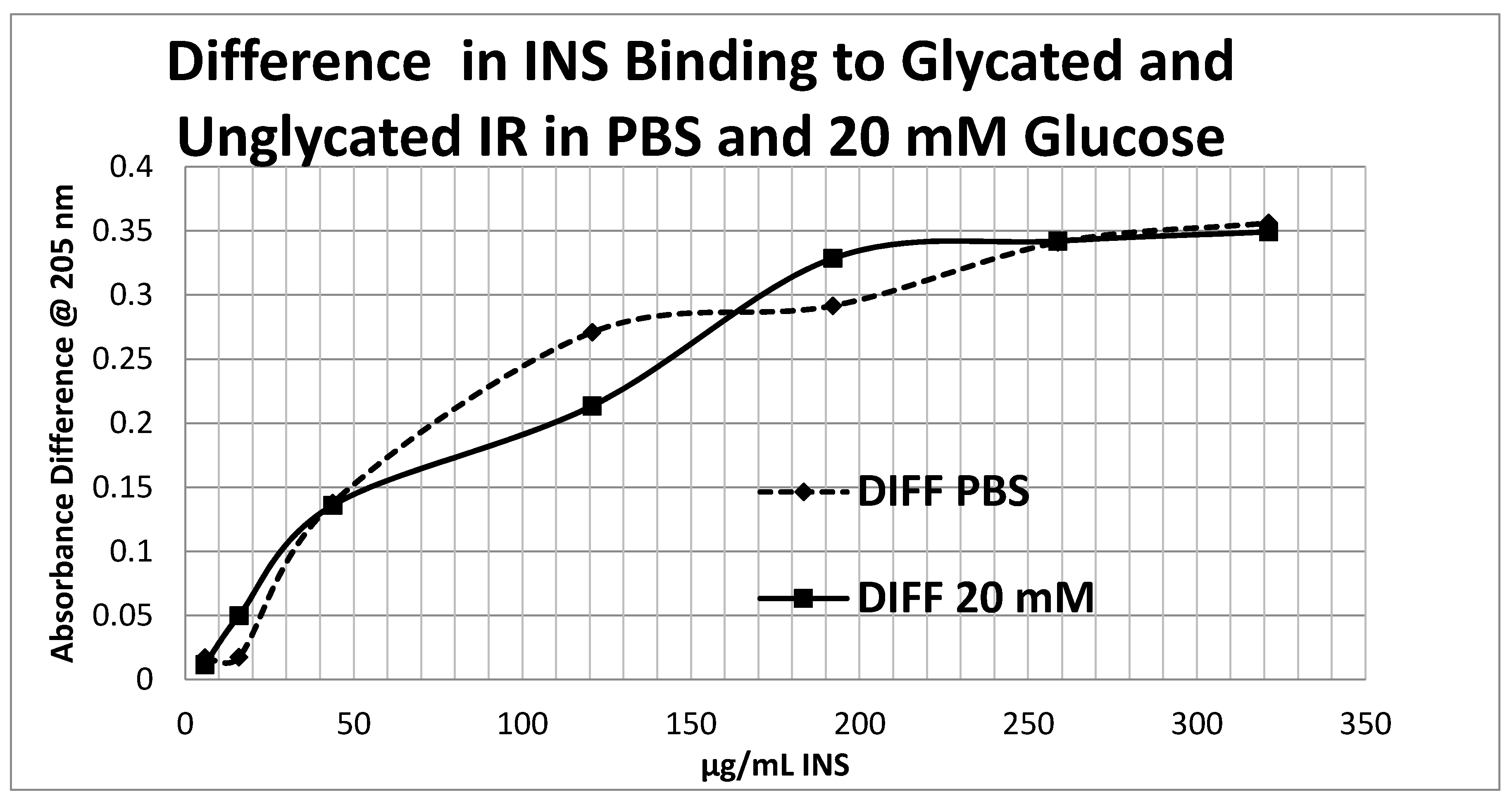
2. Results
3. Discussion
4. Materials and Methods
4.1. Insulin Receptor Peptides
4.2. Mass Spectrometry
4.3. Insulin-HRP Binding to IR Peptides
4.4. Effect of Glycation of Insulin-HRP Binding to Peptides
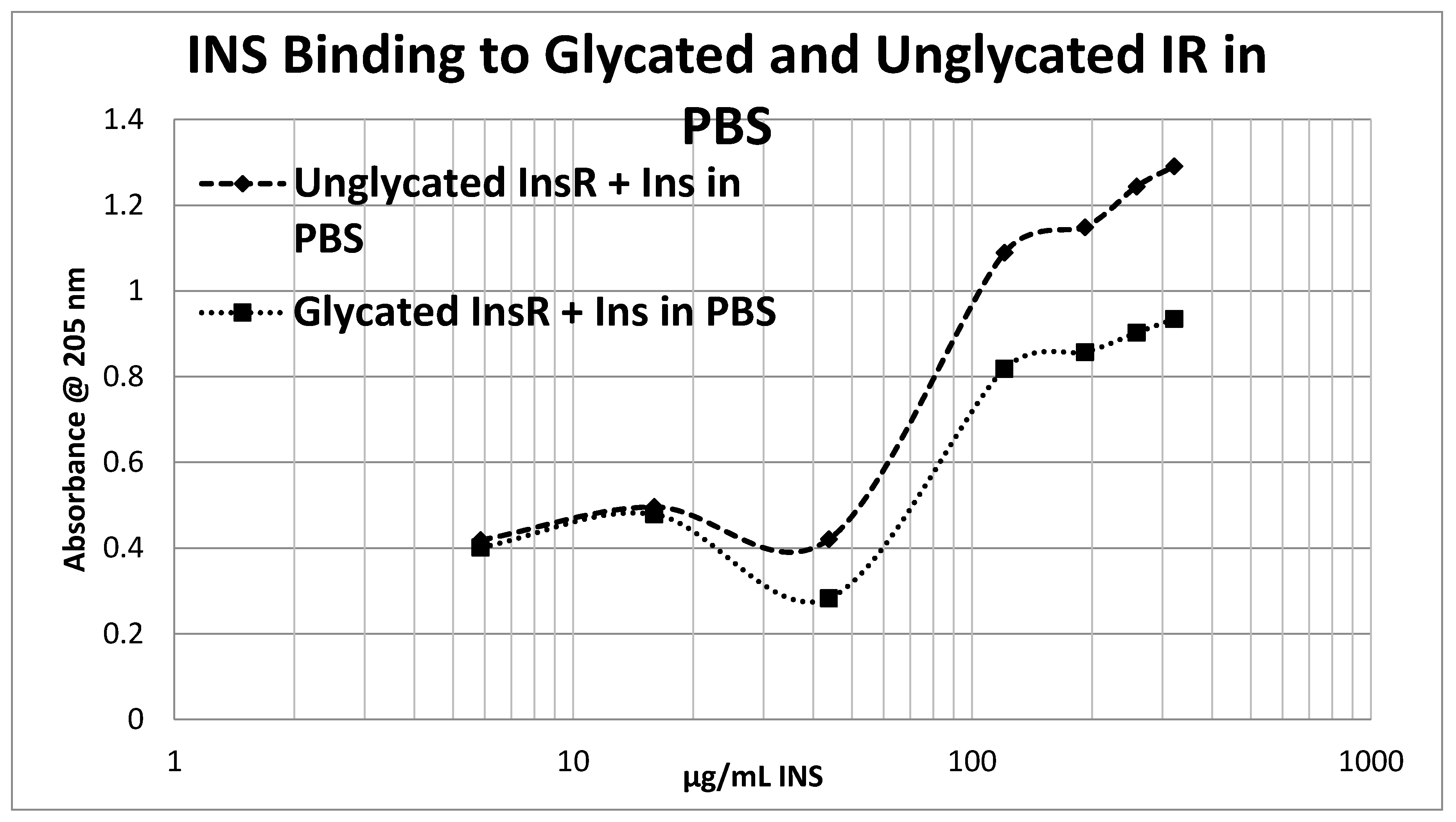
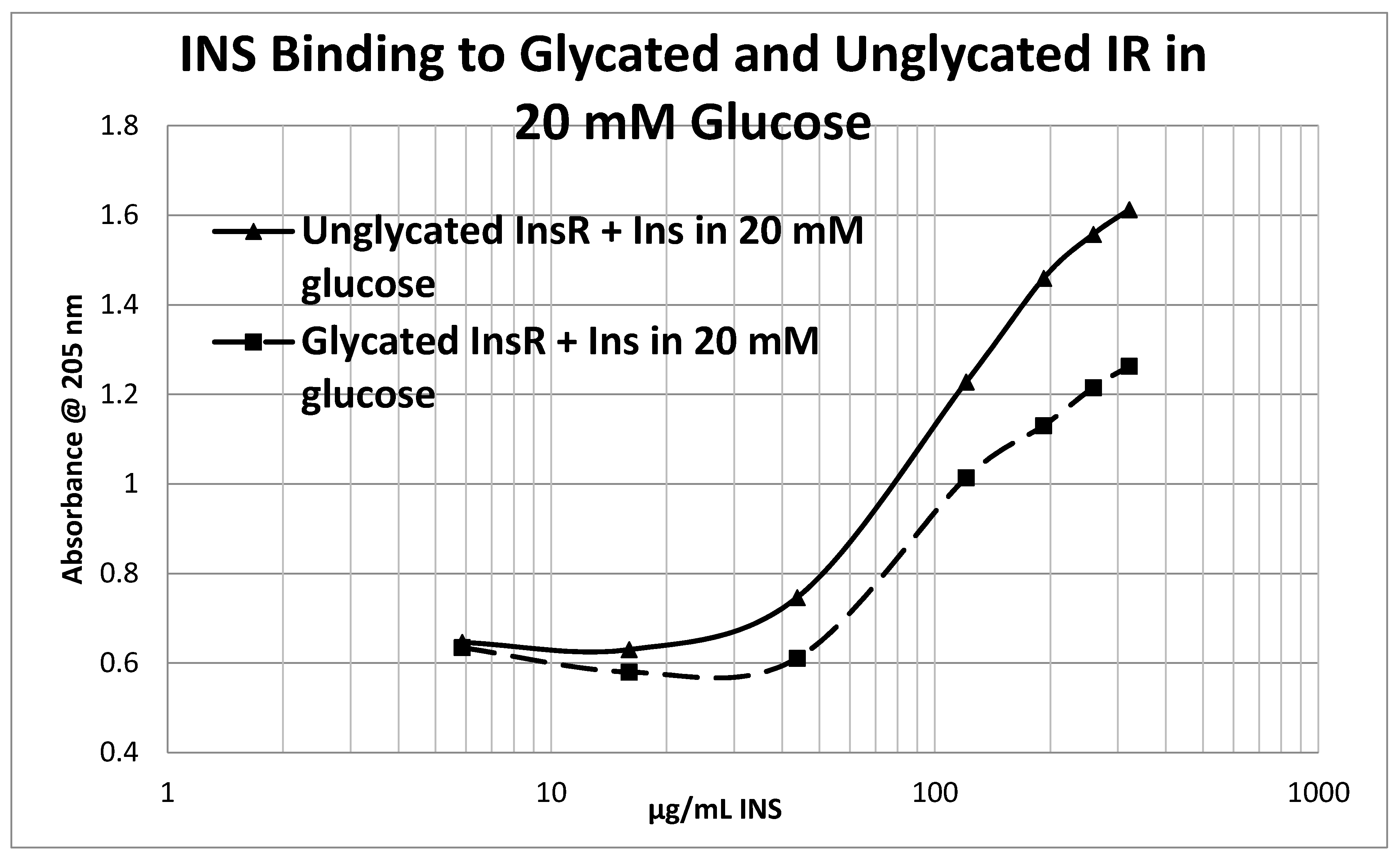
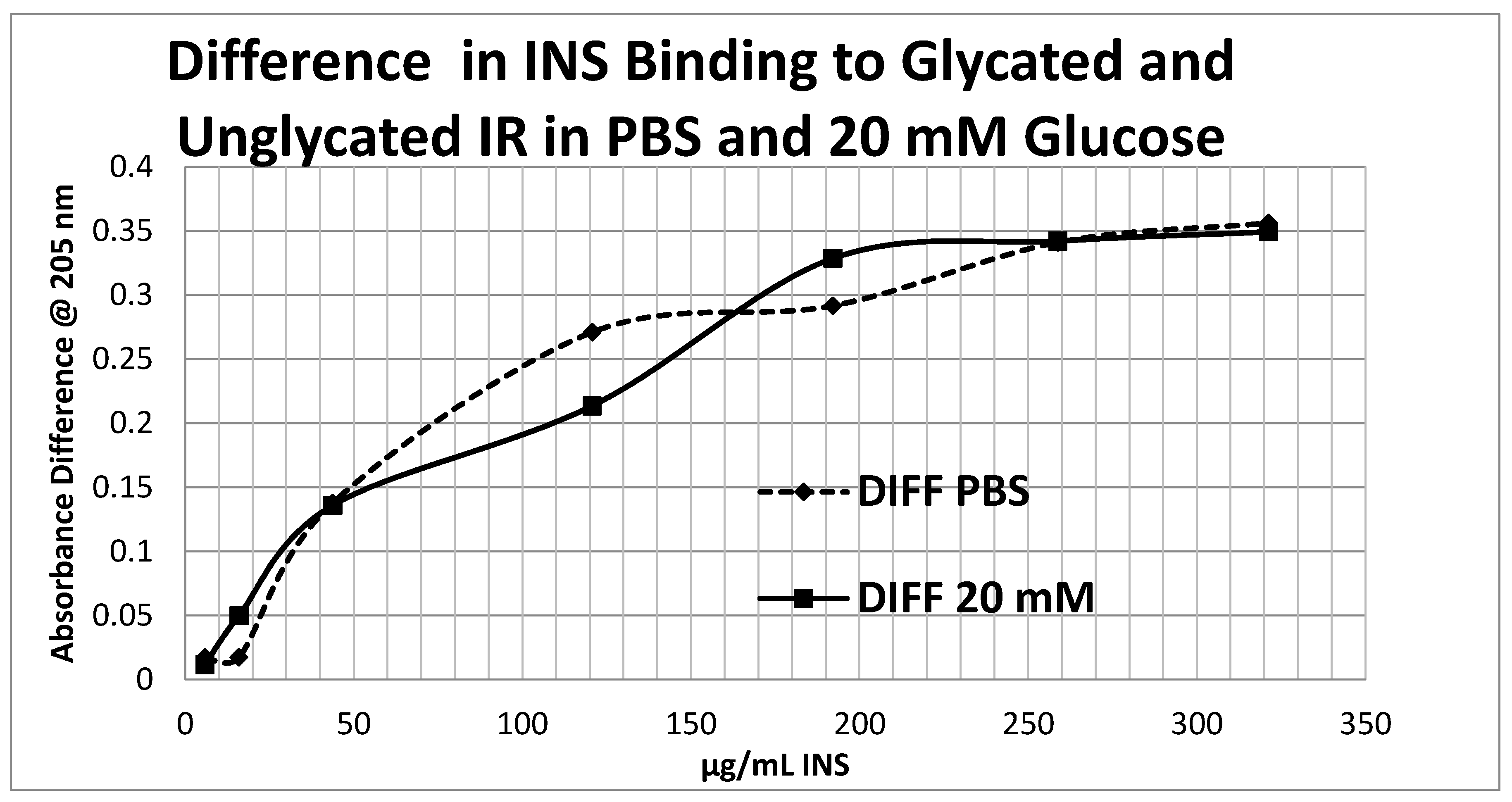
4.5. Effect of Glycation of Insulin Receptor on Insulin Binding
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| INS | insulin; |
| IR | insulin receptor; |
| T1DM | type 1 diabetes mellitus; |
| T2DM | type 2 diabetes mellitus; |
| DMEM | Dulbecco’s modified Eagles growth medium |
References
- Bergman, B.C.; Howard, D.; Schauer, I.E.; Maahs, D.M.; Snell-Bergeon, J.K.; Clement, T.W.; Eckel, R.H.; Perreault, L.; Rewers, M. The importance of palmitoleic acid to adipocyte insulin resistance and whole-body insulin sensitivity in type 1 diabetes. J. Clin. Endocrinol. Metab. 2013, 98, E40–E50. [Google Scholar] [CrossRef] [PubMed]
- Schauer, I.E.; Snell-Bergeon, J.K.; Bergman, B.C.; Maahs, D.M.; Kretowski, A.; Eckel, R.H.; Rewers, M. Insulin resistance, defective insulin-mediated fatty acid suppression, and coronary artery calcification in subjects with and without type 1 diabetes, the CACTI study. Diabetes 2011, 60, 306–314. [Google Scholar] [CrossRef] [PubMed]
- Polsky, S.; Ellis, S.L. Obesity, insulin resistance, and type 1 diabetes mellitus. Curr. Opin. Endocrinol. Diabetes Obes. 2015, 22, 277–282. [Google Scholar] [CrossRef] [PubMed]
- Petersen, K.F.; Shulman, G.I. Etiology of insulin resistance. Am. J. Med. 2006, 119, S10–S16. [Google Scholar] [CrossRef] [PubMed]
- Kershaw, E.E.; Flier, J.S. Adipose tissue as an endocrine organ. J. Clin. Endocrinol. Metab. 2004, 89, 2548–2556. [Google Scholar] [CrossRef] [PubMed]
- Park, H.K.; Ahima, R.S. Physiology of leptin: Energy homeostasis, neuroendocrine function and metabolism. Metabolism 2015, 64, 24–34. [Google Scholar] [CrossRef] [PubMed]
- Díez, J.J.; Iglesias, P. The role of the novel adipocyte-derived hormone adiponectin in human disease. Eur. J. Endocrinol. 2003, 148, 293–300. [Google Scholar] [CrossRef] [PubMed]
- Qatanani, M.; Lazar, M. Mechanisms of obesity-associated insulin resistance, many choices on the menu. Genes Dev. 2007, 21, 1443–1455. [Google Scholar] [CrossRef] [PubMed]
- Hotamisligil, G.S. Inflammatory pathways and insulin action. Int. J. Obes. Relat. Metab. Disord. 2003, 27, S53–S55. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.; Hwang, D.; Bataille, F.; Lefevre, M.; York, D.; Quon, M.J.; Ye, J. Serine phosphorylation of insulin receptor substrate 1 by inhibitor kappa B kinase complex. J. Biol. Chem. 2002, 277, 48115–48121. [Google Scholar] [CrossRef] [PubMed]
- Evans, J.L.; Goldfine, I.D.; Maddux, B.A.; Grodsky, G.M. Are oxidative stress-activated signaling pathways mediators of insulin resistance and β-cell dysfunction? Diabetes 2003, 52, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Montgomery, M.K.; Turner, N. Mitochondrial dysfunction and insulin resistance, an update. Endocr. Connect. 2015, 41, R1–R15. [Google Scholar] [CrossRef] [PubMed]
- Hummasti, S.; Hotamisligil, G.S. Endoplasmic reticulum stress and Inflammation in obesity and diabetes. Circ. Res. 2010, 107, 579–591. [Google Scholar] [CrossRef] [PubMed]
- Mortensen, H.B. Glycated hemoglobin. Reaction and biokinetic studies. Clinical application of hemoglobin A1C in the assessment of metabolic control in children with diabetes mellitus. Dan. Med. Bull. 1985, 32, 309–328. [Google Scholar] [PubMed]
- Kim, M.K.; Yun, K.J.; Kwon, H.S.; Baek, K.H.; Song, K.H. Discordance in the levels of hemoglobin A1C and glycated albumin, calculation of the glycation gap based on glycated albumin level. J. Diabetes Complicat. 2015, 30, 477–481. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Monroe, M.E.; Schepmoes, A.A.; Clauss, T.R.; Gritsenko, M.A.; Meng, D.; Petyuk, V.A.; Smith, R.D.; Metz, T.O. Comprehensive identification of glycated peptides and their glycation motifs in plasma and erythrocytes of control and diabetic subjects. J. Proteome Res. 2011, 10, 3076–3088. [Google Scholar] [CrossRef] [PubMed]
- Priego-Capote, F.; Ramirez-Boo, M.; Hoogland, C.; Scherl, A.; Mueller, M.; Lisacek, F.; Sanchez, J.C. Human hemolysate glycated proteome. Anal. Chem. 2011, 83, 5673–5680. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, H.; Abe, M.; Yoshida, Y.; Suzuki, H.; Maruyama, N.; Okada, K. Glycated albumin versus glycated hemoglobin as a glycemic indicator in diabetic patients on peritoneal dialysis. Int. J. Mol. Sci. 2016, 17, 619. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, P.; Sahoo, R.; Vaidya, A.; Cantel, S.; Kavishwar, A.; Goldfine, A.; Herring, N.; Bry, L.; Chorev, M.; Halperin, J.A. A specific and sensitive assay for blood levels of glycated CD59, a novel biomarker for diabetes. Am. J. Hematol. 2013, 88, 670–676. [Google Scholar] [CrossRef]
- Dev, K.; Sharma, S.B.; Garg, S.; Aggarwal, A.; Madhu, S.V. Glycated apolipoprotein B-A surrogate marker of subclinical atherosclerosis. Diabetes Metab. Syndr. 2015, 10, 78–81. [Google Scholar] [CrossRef] [PubMed]
- Abraham, E.C.; Huaqian, J.; Aziz, A.; Kumarasamy, A.; Datta, P. Role of the specifically targeted lysine residues in the glycation dependent loss of chaperone activity of α A- and α B-crystallins. Mol. Cell. Biochem. 2008, 310, 235–239. [Google Scholar] [CrossRef] [PubMed]
- Nedić, O.; Lagundžin, D.; Masnikosa, R. Posttranslational modifications of the insulin-like growth factor-binding protein 3 in patients with type 2 diabetes mellitus assessed by affinity chromatography. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2012, 904, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Miyata, T.; Inagi, R.; Wada, Y.; Ueda, Y.; Iida, Y.; Takahashi, M.; Taniguchi, N.; Maeda, K. Glycation of human β 2-microglobulin in patients with hemodialysis-associated amyloidosis, identification of the glycated sites. Biochemistry 1994, 33, 12215–12221. [Google Scholar] [CrossRef] [PubMed]
- Hoshi, A.; Takahashi, M.; Fujii, J.; Myint, T.; Kaneto, H.; Suzuki, K.; Yamasaki, Y.; Kamada, T.; Taniguchi, N. Glycation and inactivation of sorbitol dehydrogenase in normal and diabetic rats. Biochem. J. 1996, 318, 119–123. [Google Scholar] [CrossRef] [PubMed]
- Karim, L.; Tang, S.Y.; Sroga, G.E.; Vashishth, D. Differences in non-enzymatic glycation and collagen cross-links between human cortical and cancellous bone. Osteoporos. Int. 2013, 24, 2441–2447. [Google Scholar] [CrossRef] [PubMed]
- Blakytny, R.; Harding, J.J. Glycation non-enzymic glycosylation inactivates glutathione reductase. Biochem. J. 1992, 288, 303–307. [Google Scholar] [CrossRef] [PubMed]
- Sztanke, K.; Pasternak, K. The Maillard reaction and its consequences for a living body. Ann. Univ. Mariae Curie Sklodowska Med. 2003, 58, 159–162. [Google Scholar] [PubMed]
- O’Harte, F.P.; Højrup, P.; Barnett, C.R.; Flatt, P.R. Identification of the site of glycation of human insulin. Peptides 1996, 17, 1323–1330. [Google Scholar] [CrossRef]
- McKillop, A.M.; Meade, A.; Flatt, P.R.; O’Harte, F.P. Evaluation of the sites of glycation in human proinsulin by ion-trap LCQ electrospray ionization mass spectrometry. Regul. Pept. 2003, 113, 1–8. [Google Scholar] [CrossRef]
- Farah, M.A.; Bose, S.; Lee, J.H.; Jung, H.C.; Kim, Y. Analysis of glycated insulin by MALDI-TOF mass spectrometry. Biochim. Biophys. Acta 2005, 1725, 269–282. [Google Scholar] [CrossRef] [PubMed]
- Guedes, S.; Vitorino, R.; Domingues, M.R.; Amado, F.; Domingues, P. Mass spectrometry characterization of the glycation sites of bovine insulin by tandem mass spectrometry. J. Am. Soc. Mass Spectrom. 2009, 20, 1319–1326. [Google Scholar] [CrossRef] [PubMed]
- Anzenbacher, P.; Kalous, V. Binding of D-glucose to insulin. Biochim. Biophys. Acta 1975, 386, 603–607. [Google Scholar] [CrossRef]
- Kalous, V.; Anzenbacher, P. On the mechanism of the insulin-glucose interactions. Acta Diabetol. Lat. 1979, 16, 169–174. [Google Scholar] [CrossRef] [PubMed]
- Zoete, V.; Meuwly, M.; Karplus, M. Investigation of glucose binding sites on insulin. Proteins 2004, 55, 568–581. [Google Scholar] [CrossRef] [PubMed]
- Falconi, M.; Bozzi, M.; Paci, M.; Raudino, A.; Purrello, R.; Cambria, A.; Sette, M.; Cambria, M.T. Spectroscopic and molecular dynamics simulation studies of the interaction of insulin with glucose. Int. J. Biol. Macromol. 2001, 29, 161–168. [Google Scholar] [CrossRef]
- Cheng, R.Z.; Kawakishi, S. Site-specific oxidation of histidine residues in glycated insulin mediated by Cu2+. Eur. J. Biochem. 1994, 223, 759–764. [Google Scholar] [CrossRef] [PubMed]
- Mastrocola, R.; Nigro, D.; Chiazza, F.; Medana, C.; Dal Bello, F.; Boccuzzi, G.; Collino, M.; Aragno, M. Fructose-derived advanced glycation end-products drive lipogenesis and skeletal muscle reprogramming via SREBP-1c dysregulation in mice. Free Radic. Biol. Med. 2016, 91, 224–235. [Google Scholar] [CrossRef] [PubMed]
- Root-Bernstein, R.S. Molecular complementarity III. Molecular complementarity as the basis for peptide hormone evolution: A bioinformatic case study of insulin, glucagon band gastrin. J. Theor. Biol. 2002, 218, 71–84. [Google Scholar] [CrossRef] [PubMed]
- Root-Bernstein, R.S. Peptide self-aggregation and peptide complementarity as bases for the evolution of peptide receptors, a review. J. Mol. Recognit. 2005, 18, 40–49. [Google Scholar] [CrossRef] [PubMed]
- Root-Bernstein, R.S.; Vonck, J. The insulin receptor binds glucose altering the mutual affinity of insulin for its receptor. Cell. Mol. Life Sci. 2009, 66, 2721–2732. [Google Scholar] [CrossRef] [PubMed]
- Root-Bernstein, R.S.; Vonck, J. Modularity in receptor evolution. Insulin- and glucagon-like peptide modules as binding sites for insulin and glucose in the insulin receptor. J. Recept. Ligand Channel Res. 2010, 3, 87–96. [Google Scholar] [CrossRef]
- Alavi, P.; Yousefi, R.; Amirghofran, S.; Karbalaei-Heidari, H.R.; Moosavi-Movahedi, A.A. Structural analysis and aggregation propensity of reduced and nonreduced glycated insulin adducts. Appl. Biochem. Biotechnol. 2013, 170, 623–638. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Wahab, Y.H.; O’Harte, F.P.; Boyd, A.C.; Barnett, C.R.; Flatt, P.R. Glycation of insulin results in reduced biological activity in mice. Acta Diabetol. 1997, 34, 265–270. [Google Scholar] [CrossRef] [PubMed]
- McKillop, A.M.; Abdel-Wahab, Y.H.; Mooney, M.H.; O’Harte, F.P.; Flatt, P.R. Secretion of glycated insulin from pancreatic β-cells in diabetes represents a novel aspect of β-cell dysfunction and glucose toxicity. Diabetes Metab. 2002, 28, S61–S69. [Google Scholar]
- Yu, B.; Caspar, D.L. Structure of cubic insulin crystals in glucose solutions. Biophys. J. 1998, 74, 616–622. [Google Scholar] [CrossRef]
- Hunter, S.J.; Boyd, A.C.; O’Harte, F.P.; McKillop, A.M.; Wiggam, M.I.; Mooney, M.H.; McCluskey, J.T.; Lindsay, J.R.; Ennis, C.N.; Gamble, R.; et al. Demonstration of glycated insulin in human diabetic plasma and decreased biological activity assessed by euglycemic-hyperinsulinemic clamp technique in humans. Diabetes 2003, 52, 492–498. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, S.; Cuatrecasas, P. Insulin receptors. Ann. Rev. Pharmacol. Toxicol. 1983, 23, 461–479. [Google Scholar] [CrossRef] [PubMed]
- Robertson, R.P.; Olson, L.K.; Zhang, H.J. Differentiating glucose toxicity from glucose desensitization, a new message from the insulin gene. Diabetes 1994, 43, 1085–1089. [Google Scholar] [CrossRef] [PubMed]
- Root-Bernstein, R.S.; Busik, J.; Henry, D. Are diabetic neuropathy, retinopathy, and nephropathy caused by hyperglycemic exclusion of ascorbate uptake by glucose transporters? J. Theor. Biol. 2002, 216, 345–359. [Google Scholar] [CrossRef] [PubMed]
- Root-Bernstein, R. An insulin-like modular basis for the evolution of glucose transporters GLUT with implications for diabetes. Evol. Bioinform. Online 2007, 3, 317–331. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Wahab, Y.H.; O’Harte, F.P.; Mooney, M.H.; Barnett, C.R.; Flatt, P.R. Vitamin C supplementation decreases insulin glycation and improves glucose homeostasis in obese hyperglycemic ob/ob mice. Metabolism 2002, 51, 514–517. [Google Scholar] [CrossRef] [PubMed]
- Ajiboye, R.; Harding, J.J. The non-enzymic glycosylation of bovine lens proteins by glucosamine and its inhibition by aspirin, ibuprofen and glutathione. Exp. Eye Res. 1989, 49, 31–41. [Google Scholar] [CrossRef]
- Mirmiranpour, H.; Khaghani, S.; Bathaie, S.Z.; Nakhjavani, M.; Kebriaeezadeh, A.; Ebadi, M.; Gerayesh-Nejad, S.; Zangooei, M. The preventive effect of l-lysine on lysozyme glycation in type 2 diabetes. Acta Med. Iran. 2016, 54, 24–31. [Google Scholar] [PubMed]
- Chen, L.; Jia, R.-H.; Qiu, C.-J.; Ding, G. Hyperglycemia inhibits the uptake of dehydroascorbate in tubular epithelial cell. Am. J. Nephrol. 2005, 25, 459–465. [Google Scholar] [CrossRef] [PubMed]
- Song, F.; Schmidt, A.M. Glycation and insulin resistance, novel mechanisms and unique targets? Arterioscler. Thromb. Vasc. Biol. 2012, 32, 1760–1765. [Google Scholar] [CrossRef] [PubMed]


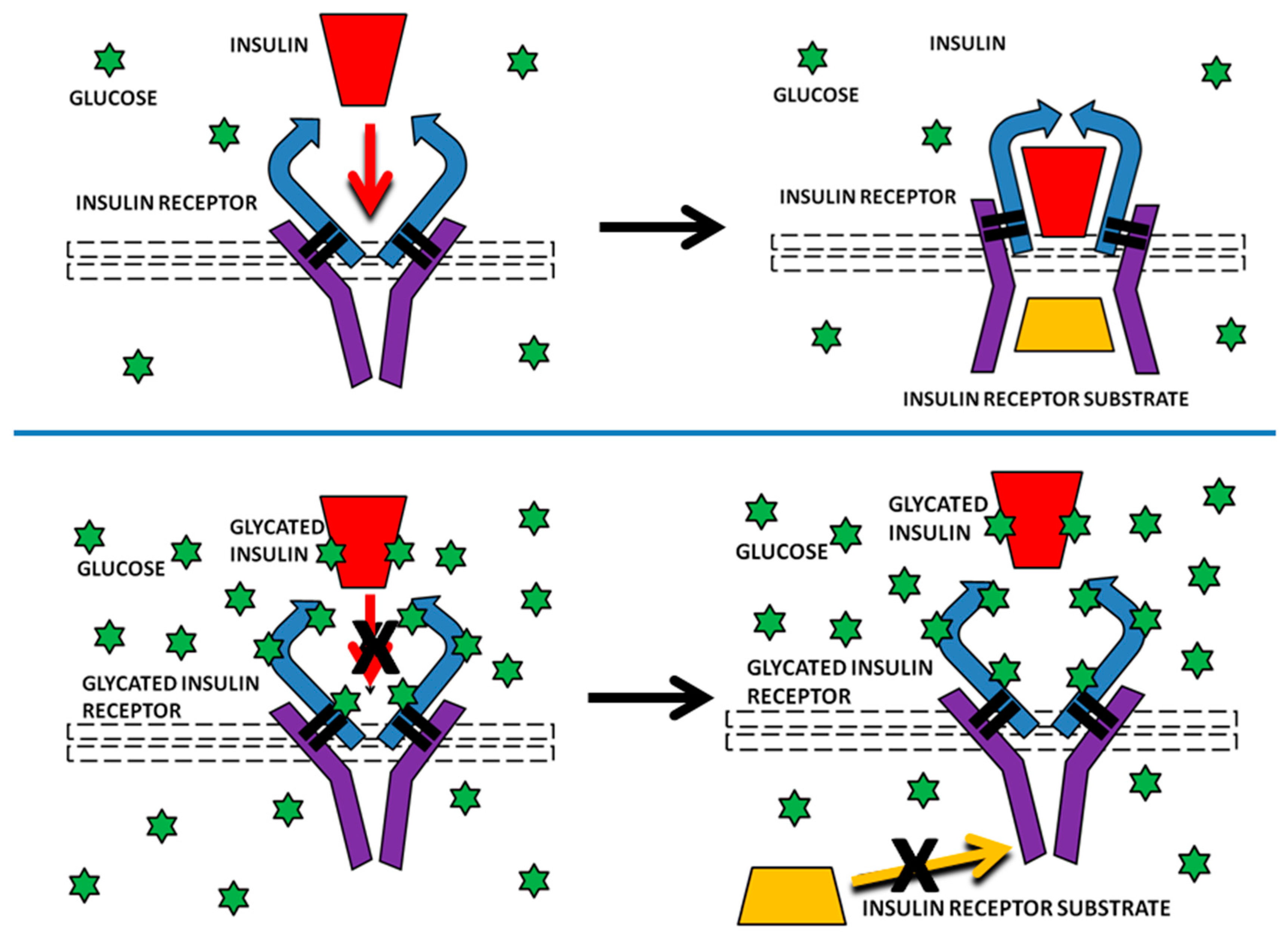

{kind=link}
{kind=link}
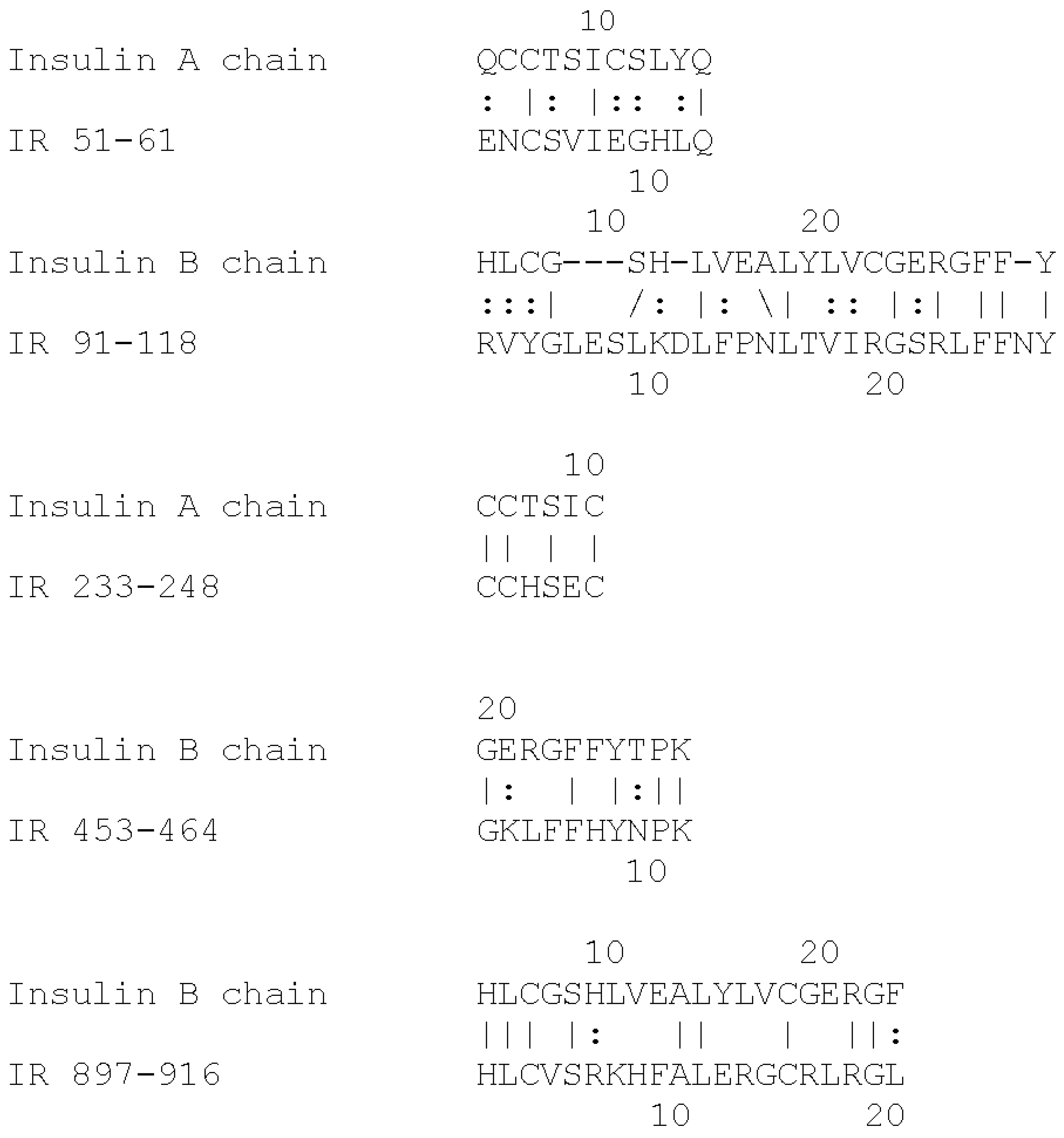
{kind=link}
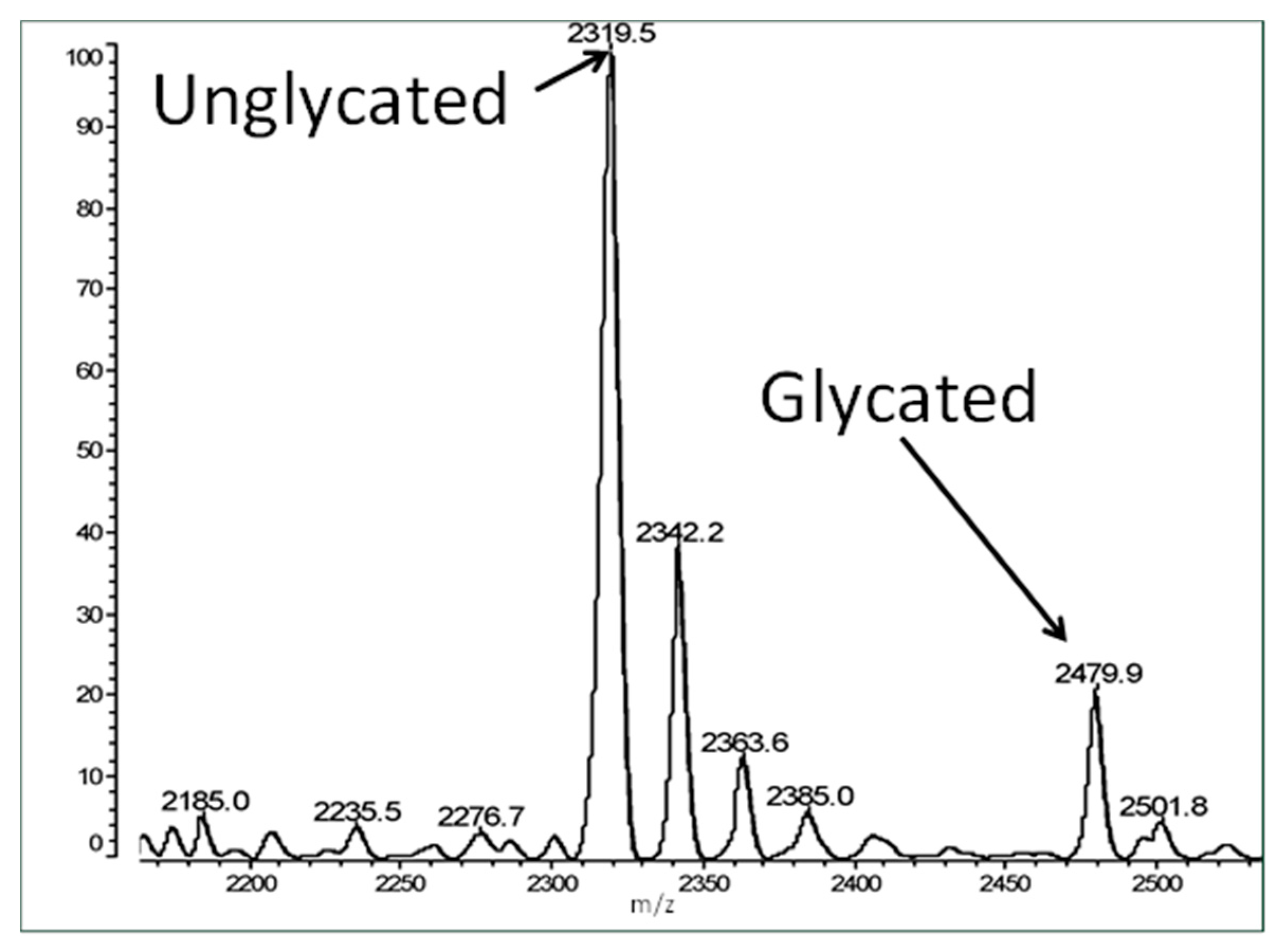
{kind=link}
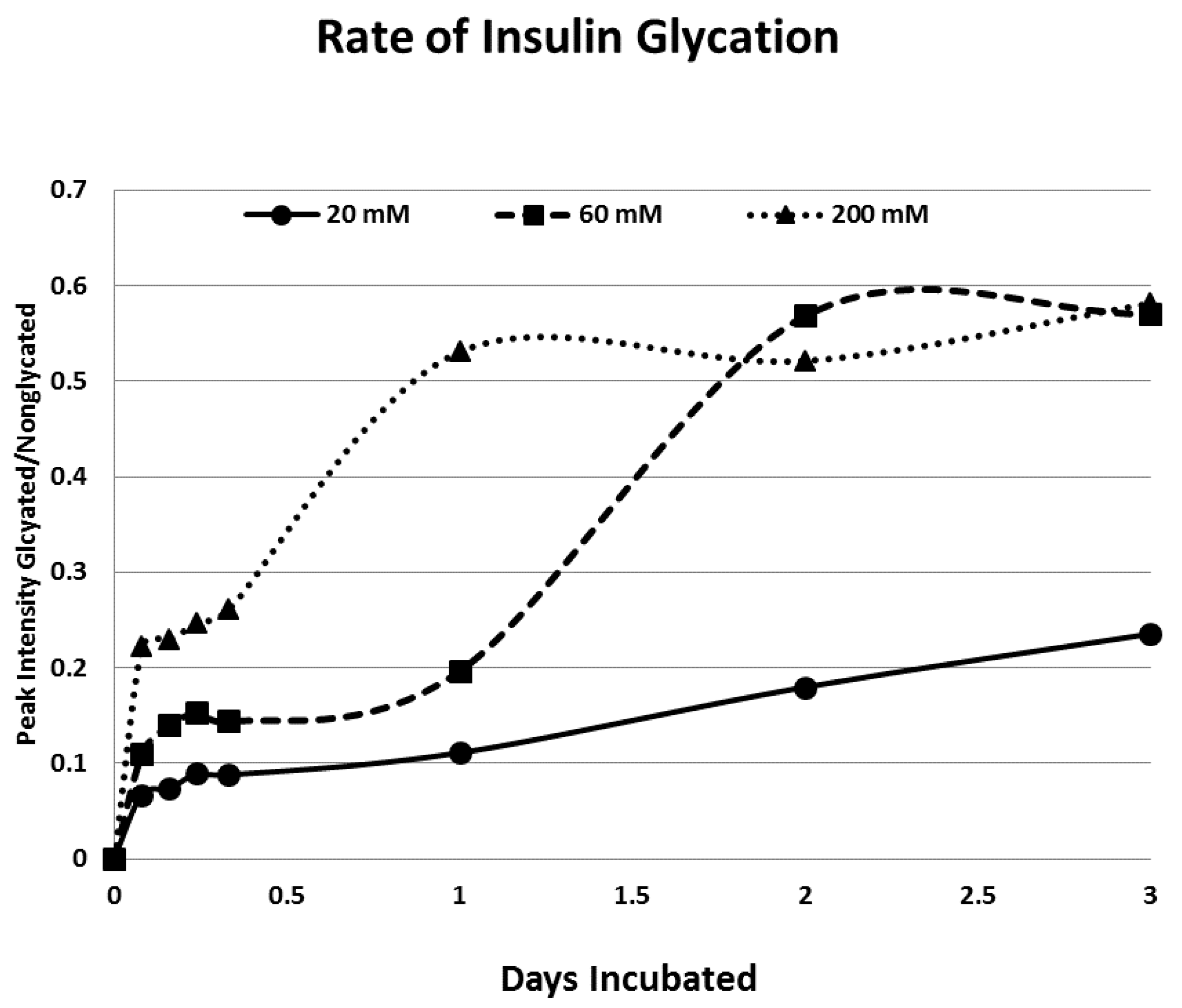
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
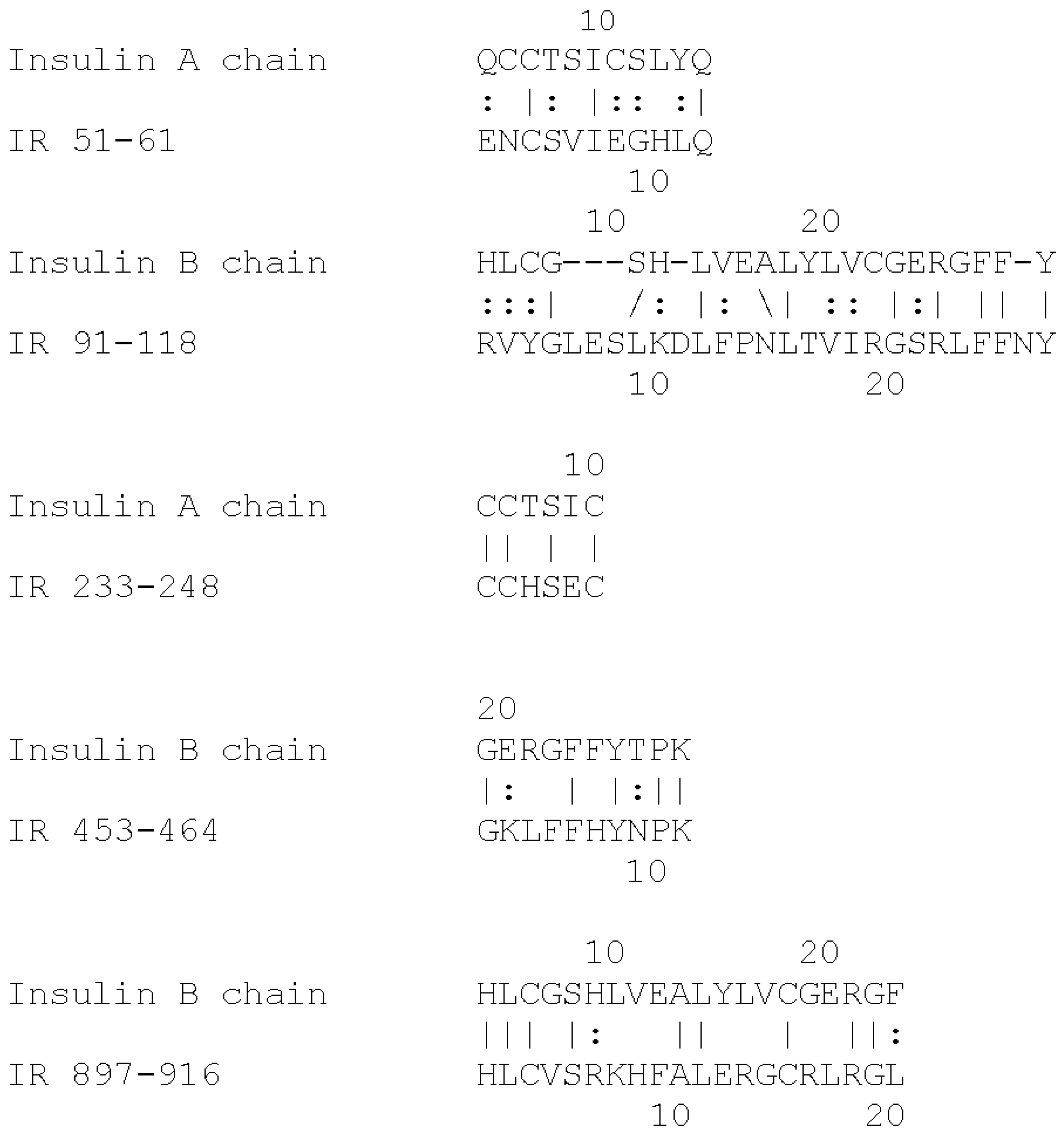
| Insulin Receptor Peptides | Peptide Molecular Weight | Glucose Binding Kd (mM) | % Glycation 6 Day 20 mM | Ins-HRP Kd (µM) | 10 nM Ins-HRP Binding (Native) | Ins-HRP Binding Inhibition (Glycated) |
|---|---|---|---|---|---|---|
| Insulin | 5808 | 0.25 & 30.0 | 24/6 | Not Done | Not Done | Not Done |
| 28-43 HLYFGEVCPGMDIRNN | 1906 | 50 | 0/0 | ND | no | no |
| 37-51 GMDIRNNLTRLHELE | 1677 | 40 | 11/0 | ND | no | no |
| 51-61 (IL) ENCSVIEGHLQ | 1316 | 52 | 6/0 | ND | YES | YES |
| 91-103 (IL) FRVYGLESLKDLF | 1570 | 37 | 13/4 | 2.5 | YES | YES |
| 105-118 (IL) NLTVIRGSRLFFNY | 1568 | 35 | 2.4/0 | 6.2 | YES | YES |
| 157-166 (GL) TIDWSRILDS | 1202 | 57 | 4/2 | >100 | no | no |
| 233-248 (IL) CCHSECLGNCSQPDD | 2376 | 46 | 14/0 | >100 | no | no |
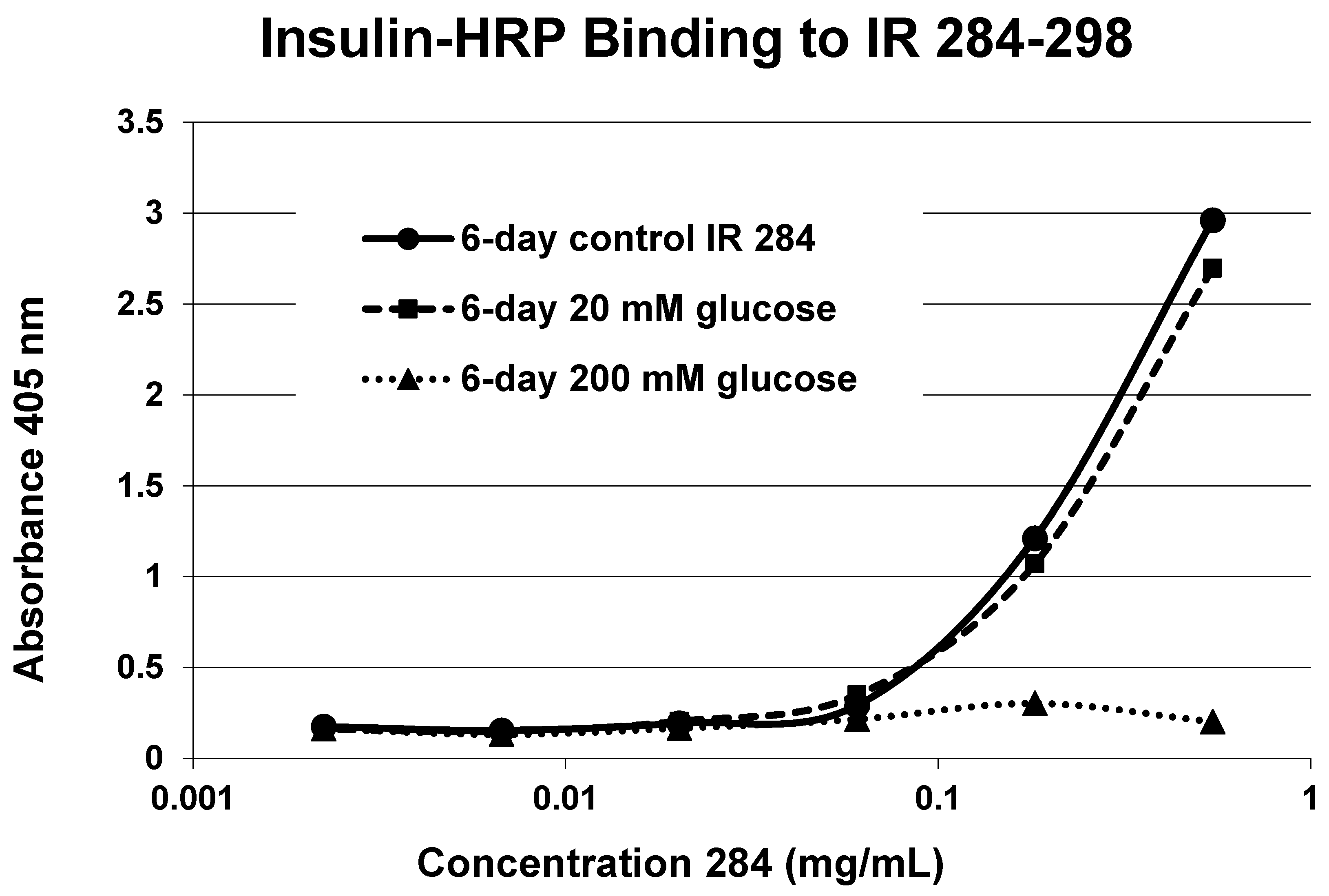
| 284–300 (GL) SFCQDLHHKCKNSRRQG | 1835 | >500 | 0.1/0 | 3.0 | YES | INSIGNIF. |
| 390-405 (GL) EISGYLKIRRSYALVS | 1510 | 30 | 13/2 | 55.0 | no | no |
| 425-444 (GL) YSFYALDNQNLRQLWDWSKH | 2316 | >500 | 0/0 | >100 | no | no |
| 453-464 (IL) TQGKLFFHYNPK | 1467 | 57 | 9/0 | >100 | no | no |
| 660-679 ERQAEDSELFELDYCLKGLK | 2474 | 70 | 0/0 | >100 | no | no |
| 689-705 ESEDSQKHNQSEYEDS | 1912 | 63 | 0/0 | ND | no | no |
| 730-742 KTFEDYLHNVVFV | 1261 | 60 | 11/0 | ND | no | no |
| 897-916 (IL) HLCVSRKHFALERGCRLRGL | 2314 | 14 | 4.5/0 | 0.3 | YES | YES |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rhinesmith, T.; Turkette, T.; Root-Bernstein, R. Rapid Non-Enzymatic Glycation of the Insulin Receptor under Hyperglycemic Conditions Inhibits Insulin Binding In Vitro: Implications for Insulin Resistance. Int. J. Mol. Sci. 2017, 18, 2602. https://doi.org/10.3390/ijms18122602
Rhinesmith T, Turkette T, Root-Bernstein R. Rapid Non-Enzymatic Glycation of the Insulin Receptor under Hyperglycemic Conditions Inhibits Insulin Binding In Vitro: Implications for Insulin Resistance. International Journal of Molecular Sciences. 2017; 18(12):2602. https://doi.org/10.3390/ijms18122602
Chicago/Turabian StyleRhinesmith, Tyler, Thomas Turkette, and Robert Root-Bernstein. 2017. "Rapid Non-Enzymatic Glycation of the Insulin Receptor under Hyperglycemic Conditions Inhibits Insulin Binding In Vitro: Implications for Insulin Resistance" International Journal of Molecular Sciences 18, no. 12: 2602. https://doi.org/10.3390/ijms18122602
APA StyleRhinesmith, T., Turkette, T., & Root-Bernstein, R. (2017). Rapid Non-Enzymatic Glycation of the Insulin Receptor under Hyperglycemic Conditions Inhibits Insulin Binding In Vitro: Implications for Insulin Resistance. International Journal of Molecular Sciences, 18(12), 2602. https://doi.org/10.3390/ijms18122602