
An In Silico Identification of Common Putative Vaccine Candidates against Treponema pallidum: A Reverse Vaccinology and Subtractive Genomics Based Approach


 and
and
Abstract
:1. Introduction
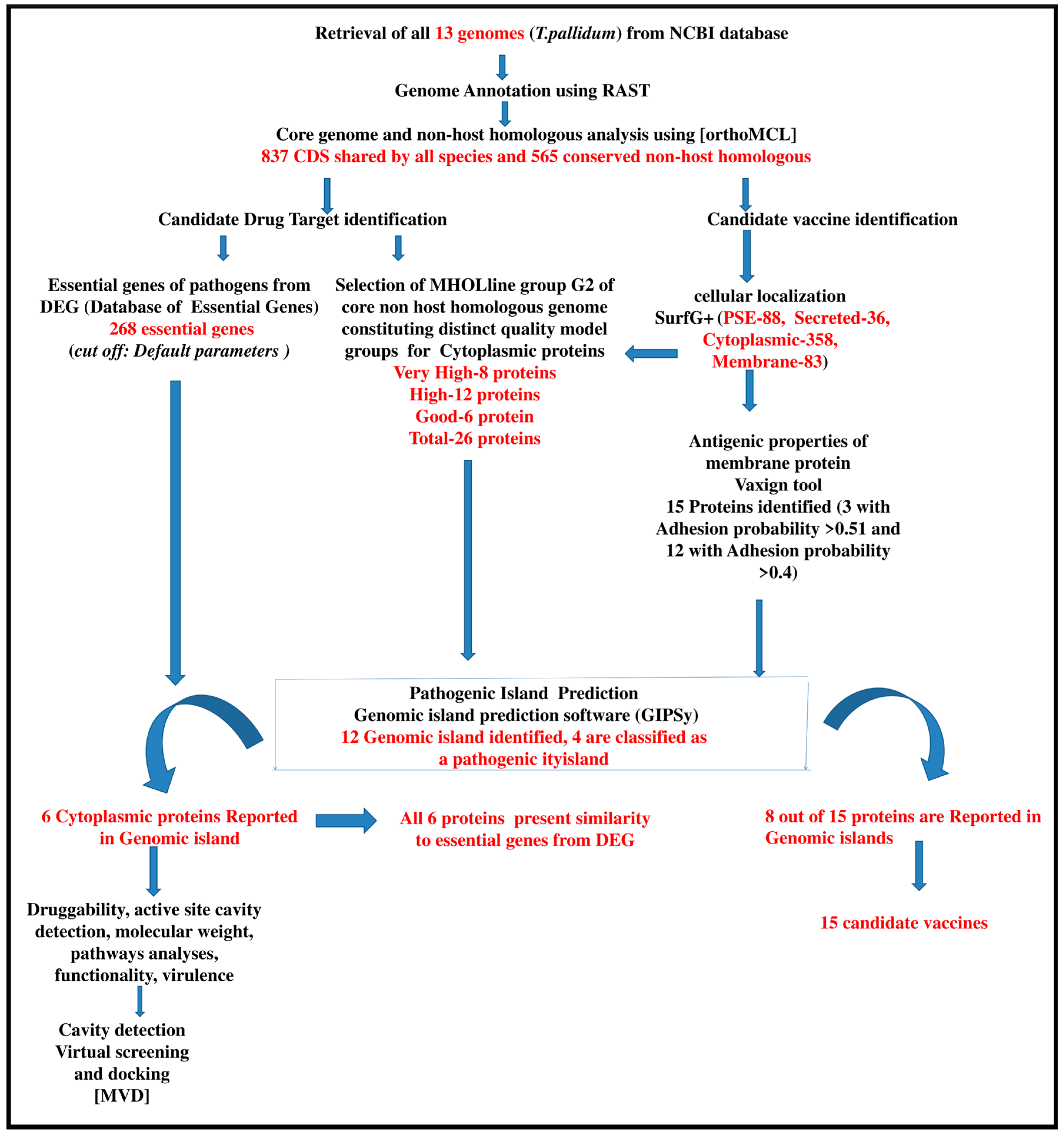
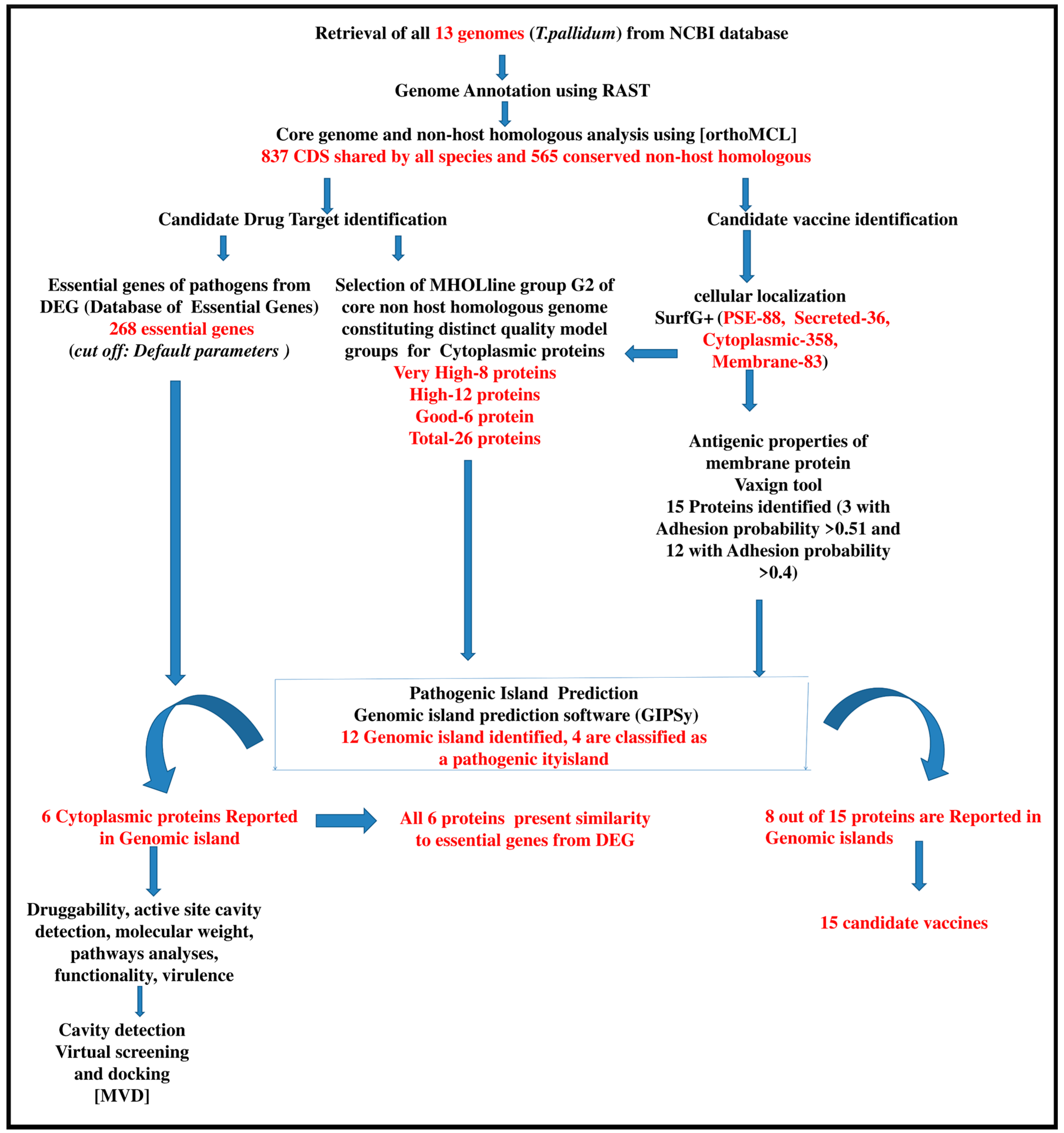
2. Result and Discussion
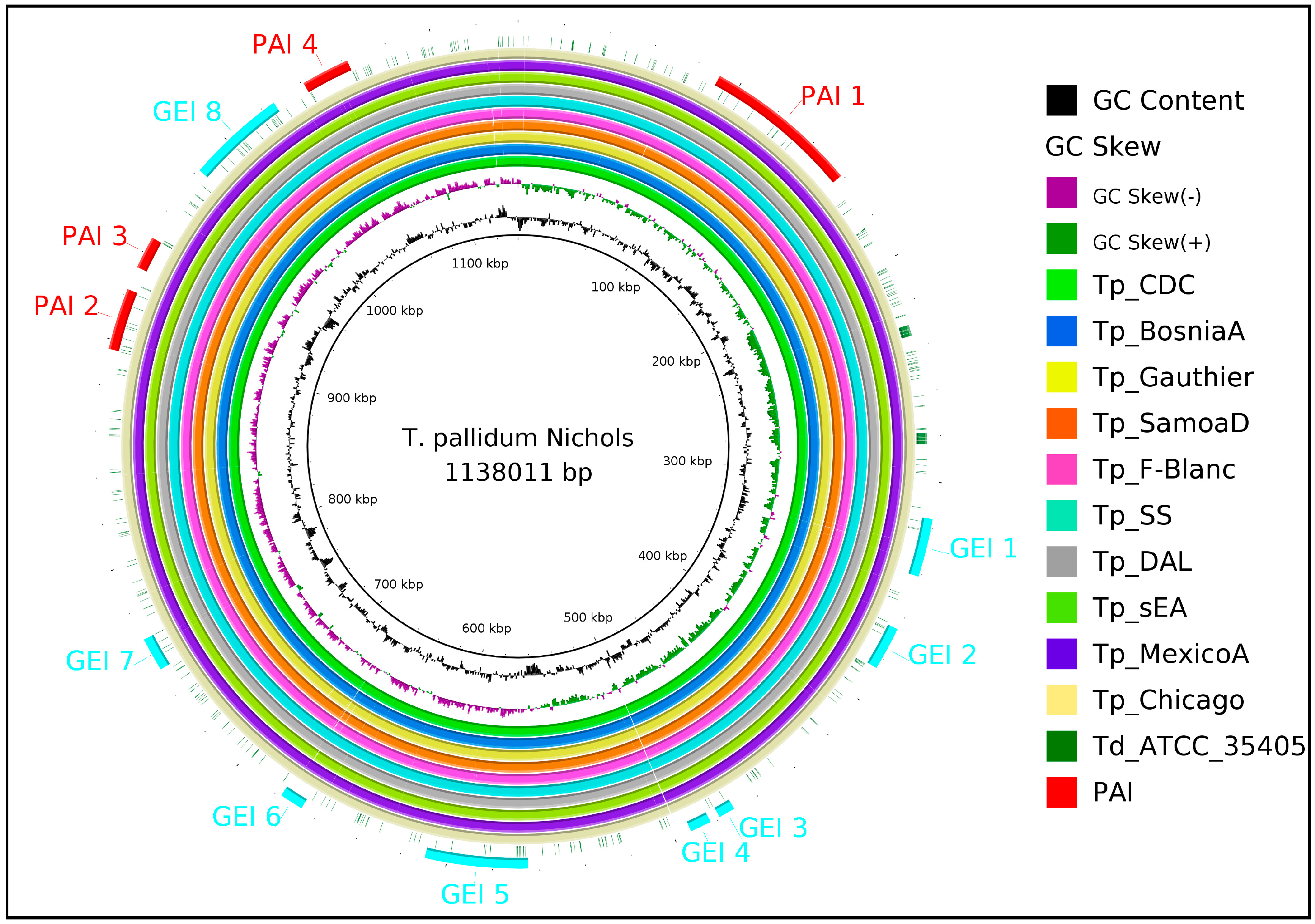
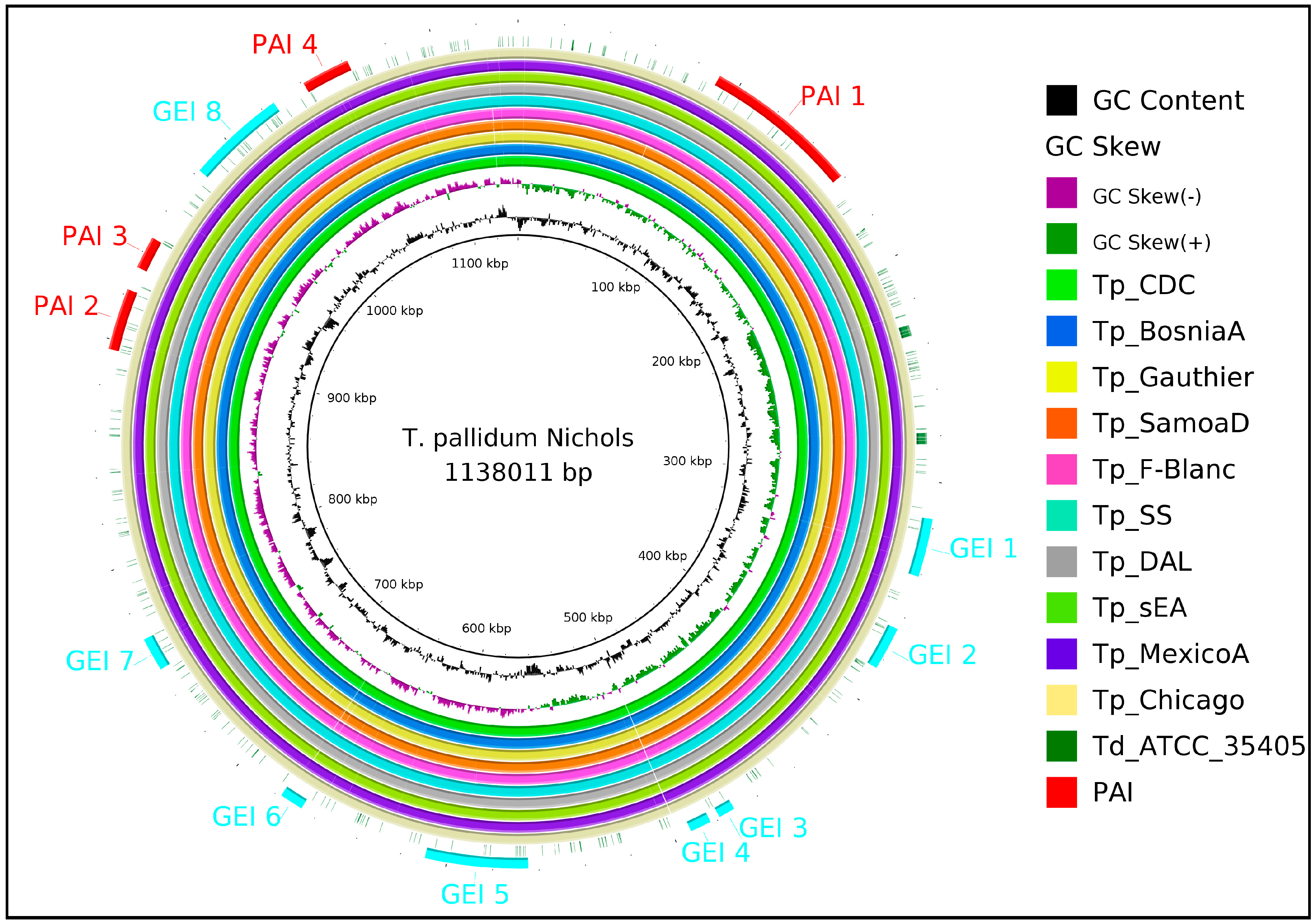
2.1. Identification of Intra-Species Conserved Non-Host Homologous Proteinsand Pathogenicity Islands
2.2. Assessment of Essential Genes
2.3. Prediction of Candidate Vaccine Target for T. pallidum
2.4. High Throughput Structural Modeling
2.5. Analyses of Non-Host Homologous Targets and Molecular Docking
3. Materials and Methods
3.1. Selection of Data
3.2. Identification of Intra-Species Conserved Non-Host Homologous Proteins
3.3. Identification of Pathogenicity Islands
3.4. Assessment of Essential Genes
3.5. Reverse Vaccinology Approach for Prediction of Putative T. pallidum Vaccine Targets
3.6. High Throughput Structural Modeling
3.7. Ligand Libraries and Docking Analyses
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Wagenlehner, F.M.; Brockmeyer, N.H.; Discher, T.; Friese, K.; Wichelhaus, T.A. The presentation, diagnosis, and treatment of sexually transmitted infections. Dtsch. Arztebl. Int. 2016, 113, 11–22. [Google Scholar] [PubMed]
- Nyatsanza, F.; Tipple, C. Syphilis: Presentations in general medicine. Clin. Med. 2016, 16, 184–188. [Google Scholar] [CrossRef] [PubMed]
- Newman, L.; Rowley, J.; Vander Hoorn, S.; Wijesooriya, N.S.; Unemo, M.; Low, N.; Stevens, G.; Gottlieb, S.; Kiarie, J.; Temmerman, M. Global estimates of the prevalence and incidence of four curable sexually transmitted infections in 2012 based on systematic review and global reporting. PLoS ONE 2015, 10, e0143304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lafeta, K.R.; Martelli Junior, H.; Silveira, M.F.; Paranaiba, L.M. Maternal and congenital syphilis, underreported and difficult to control. Rev. Bras. Epidemiol. 2016, 19, 63–74. [Google Scholar] [PubMed]
- Deperthes, B.D.; Meheus, A.; O’Reilly, K.; Broutet, N. Maternal and congenital syphilis programmes: Case studies in Bolivia, Kenya and South Africa. Bull World Health Organ 2004, 82, 410–416. [Google Scholar] [PubMed]
- Cameron, C.E.; Lukehart, S.A. Current status of syphilis vaccine development: Need, challenges, prospects. Vaccine 2014, 32, 1602–1609. [Google Scholar] [CrossRef] [PubMed]
- Radolf, J.D. Treponema. In Medical Microbiology, 4th ed.; Baron, S., Ed.; University of Texas Medical Branch at Galveston: Galveston, TX, USA, 1996. [Google Scholar]
- Jakopanec, I.; Grjibovski, A.M.; Nilsen, O.; Aavitsland, P. Syphilis epidemiology in Norway, 1992–2008: Resurgence among men who have sex with men. BMC Infect. Dis. 2010, 10. [Google Scholar] [CrossRef] [PubMed]
- Tucker, J.D.; Cohen, M.S. China’s syphilis epidemic: Epidemiology, proximate determinants of spread, and control responses. Curr. Opin. Infect. Dis. 2011, 24, 50–55. [Google Scholar] [CrossRef] [PubMed]
- Abara, W.E.; Hess, K.L.; Neblett Fanfair, R.; Bernstein, K.T.; Paz-Bailey, G. Syphilis trends among men who have sex with men in the United States and Western Europe: A systematic review of trend studies published between 2004 and 2015. PLoS ONE 2016, 11, e0159309. [Google Scholar] [CrossRef] [PubMed]
- Cabie, A.; Rollin, B.; Pierre-Francois, S.; Abel, S.; Desbois, N.; Richard, P.; Hochedez, P.; Theodose, R.; Quist, D.; Helenon, R.; et al. Reemergence of syphilis in Martinique, 2001–2008. Emerg. Infect. Dis. 2010, 16, 106–109. [Google Scholar] [CrossRef] [PubMed]
- Plotkin, S.A. Why certain vaccines have been delayed or not developed at all. Health Aff. 2005, 24, 631–634. [Google Scholar] [CrossRef] [PubMed]
- Barh, D.; Tiwari, S.; Jain, N.; Ali, A.; Santos, A.R.; Misra, A.N.; Azevedo, V.; Kumar, A. In silico subtractive genomics for target identification in human bacterial pathogens. Drug Dev. Res. 2011, 72, 162–177. [Google Scholar] [CrossRef]
- Barh, D.; Gupta, K.; Jain, N.; Khatri, G.; Leon-Sicairos, N.; Canizalez-Roman, A.; Tiwari, S.; Verma, A.; Rahangdale, S.; Shah Hassan, S.; et al. Conserved host-pathogen PPIs. Globally conserved inter-species bacterial PPIs based conserved host-pathogen interactome derived novel target in C. pseudotuberculosis, C. diphtheriae, M. tuberculosis, C. ulcerans, Y. pestis, and E. coli targeted by piper betel compounds. Integr. Biol. 2013, 5, 495–509. [Google Scholar]
- Perumal, D.; Lim, C.S.; Sakharkar, K.R.; Sakharkar, M.K. Differential genome analyses of metabolic enzymes in Pseudomonas aeruginosa for drug target identification. In Silico Biol. 2007, 7, 453–465. [Google Scholar] [PubMed]
- Pizza, M.; Scarlato, V.; Masignani, V.; Giuliani, M.M.; Arico, B.; Comanducci, M.; Jennings, G.T.; Baldi, L.; Bartolini, E.; Capecchi, B.; et al. Identification of vaccine candidates against serogroup B meningococcus by whole-genome sequencing. Science 2000, 287, 1816–1820. [Google Scholar] [CrossRef] [PubMed]
- Asif, S.M.; Asad, A.; Faizan, A.; Anjali, M.S.; Arvind, A.; Neelesh, K.; Hirdesh, K.; Sanjay, K. Dataset of potential targets for Mycobacterium tuberculosis H37Rv through comparative genome analysis. Bioinformation 2009, 4, 245–248. [Google Scholar] [CrossRef] [PubMed]
- Dutta, A.; Singh, S.K.; Ghosh, P.; Mukherjee, R.; Mitter, S.; Bandyopadhyay, D. In silico identification of potential therapeutic targets in the human pathogen Helicobacter pylori. In Silico Biol 2006, 6, 43–47. [Google Scholar] [PubMed]
- Chong, C.E.; Lim, B.S.; Nathan, S.; Mohamed, R. In silico analysis of Burkholderia pseudomallei genome sequence for potential drug targets. In Silico Biol 2006, 6, 341–346. [Google Scholar] [PubMed]
- Sakharkar, K.R.; Sakharkar, M.K.; Chow, V.T. A novel genomics approach for the identification of drug targets in pathogens, with special reference to Pseudomonas aeruginosa. In Silico Biol 2004, 4, 355–360. [Google Scholar] [PubMed]
- Rathi, B.; Sarangi, A.N.; Trivedi, N. Genome subtraction for novel target definition in Salmonella typhi. Bioinformation 2009, 4, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Barh, D.; Kumar, A. In silico identification of candidate drug and vaccine targets from various pathways in Neisseria gonorrhoeae. In Silico Biol 2009, 9, 225–231. [Google Scholar] [PubMed]
- Barh, D.; Jain, N.; Tiwari, S.; Parida, B.P.; D’Afonseca, V.; Li, L.; Ali, A.; Santos, A.R.; Guimaraes, L.C.; de Castro Soares, S.; et al. A novel comparative genomics analysis for common drug and vaccine targets in Corynebacterium pseudotuberculosis and other CMN group of human pathogens. Chem. Biol. Drug Des. 2011, 78, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Aronov, A.M.; Verlinde, C.L.; Hol, W.G.; Gelb, M.H. Selective tight binding inhibitors of trypanosomal glyceraldehyde-3-phosphate dehydrogenase via structure-based drug design. J. Med. Chem. 1998, 41, 4790–4799. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Malik, B.K.; Sharma, D.K. Molecular modeling and docking analysis of entamoeba histolytica glyceraldehyde-3 phosphate dehydrogenase, a potential target enzyme for anti-protozoal drug development. Chem. Biol. Drug Des. 2008, 71, 554–562. [Google Scholar] [CrossRef] [PubMed]
- Li, L. Orthomcl: Identification of ortholog groups for eukaryotic genomes. Genome Res. 2003, 13, 2178–2189. [Google Scholar] [CrossRef] [PubMed]
- Soares, S.C.; Geyik, H.; Ramos, R.T.; de Sa, P.H.; Barbosa, E.G.; Baumbach, J.; Figueiredo, H.C.; Miyoshi, A.; Tauch, A.; Silva, A.; et al. Gipsy: Genomic island prediction software. J. Biotechnol. 2016, 232, 2–11. [Google Scholar] [CrossRef] [PubMed]
- Naz, A.; Awan, F.M.; Obaid, A.; Muhammad, S.A.; Paracha, R.Z.; Ahmad, J.; Ali, A. Identification of putative vaccine candidates against Helicobacter pylori exploiting exoproteome and secretome: A reverse vaccinology based approach. Infect. Genet. Evol. 2015, 32, 280–291. [Google Scholar] [CrossRef] [PubMed]
- Barinov, A.; Loux, V.; Hammani, A.; Nicolas, P.; Langella, P.; Ehrlich, D.; Maguin, E.; van de Guchte, M. Prediction of surface exposed proteins in streptococcus pyogenes, with a potential application to other gram-positive bacteria. Proteomics 2009, 9, 61–73. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Xiang, Z.; Mobley, H.L.T. Vaxign: The first web-based vaccine design program for reverse vaccinology and applications for vaccine development. J. Biomed. Biotechnol. 2010, 2010, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Capriles, P.V.S.Z.; Guimarães, A.C.R.; Otto, T.D.; Miranda, A.B.; Dardenne, L.E.; Degrave, W.M. Structural modelling and comparative analysis of homologous, analogous and specific proteins from Trypanosoma cruzi versus Homo sapiens: Putative drug targets for chagas’ disease treatment. BMC Genomics 2010, 11, 610. [Google Scholar] [CrossRef] [PubMed]
- Mondal, S.I.; Ferdous, S.; Jewel, N.A.; Akter, A.; Mahmud, Z.; Islam, M.M.; Afrin, T.; Karim, N. Identification of potential drug targets by subtractive genome analysis of Escherichia coli O157:H7: An in silico approach. Adv. Appl. Bioinform. Chem. 2015, 8, 49–63. [Google Scholar] [CrossRef] [PubMed]
- Duffield, M.; Cooper, I.; McAlister, E.; Bayliss, M.; Ford, D.; Oyston, P. Predicting conserved essential genes in bacteria: In silico identification of putative drug targets. Mol. Biosyst. 2010, 6, 2482–2489. [Google Scholar] [CrossRef] [PubMed]
- Delcour, A.H. Outer membrane permeability and antibiotic resistance. Biochim. Biophys. Acta 2009, 1794, 808–816. [Google Scholar] [CrossRef] [PubMed]
- Vaara, M. Agents that increase the permeability of the outer membrane. Microbiol. Rev. 1992, 56, 395–411. [Google Scholar] [PubMed]
- Gasteiger, E.; Hoogland, C.; Gattiker, A.; Duvaud, S.E.; Wilkins, M.R.; Appel, R.D.; Bairoch, A. Protein identification and analysis tools on the expasy server. In The Proteomics Protocols Handbook; Walker, J.M., Ed.; Humana Press: New York, NY, USA, 2005; pp. 571–607. [Google Scholar]
- Thomsen, R.; Christensen, M.H. Moldock: A new technique for high-accuracy molecular docking. J. Med. Chem. 2006, 49, 3315–3321. [Google Scholar] [CrossRef] [PubMed]
- Hwang, B.; Lee, J.; Liu, Q.H.; Woo, E.R.; Lee, D.G. Antifungal effect of (+)-pinoresinol isolated from Sambucus williamsii. Molecules 2010, 15, 3507–3516. [Google Scholar] [CrossRef] [PubMed]
- Cespedes, C.L.; Avila, J.G.; Garcia, A.M.; Becerra, J.; Flores, C.; Aqueveque, P.; Bittner, M.; Hoeneisen, M.; Martinez, M.; Silva, M. Antifungal and antibacterial activities of Araucaria araucana (Mol.) K. Koch heartwood lignans. Z. Naturforsch. C 2006, 61, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Coordinators, N.R. Database resources of the national center for biotechnology information. Nucleic Acids Res. 2016, 44, 7–19. [Google Scholar]
- Brettin, T.; Davis, J.J.; Disz, T.; Edwards, R.A.; Gerdes, S.; Olsen, G.J.; Olson, R.; Overbeek, R.; Parrello, B.; Pusch, G.D.; et al. Rasttk: A modular and extensible implementation of the RAST algorithm for building custom annotation pipelines and annotating batches of genomes. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Ou, H.Y.; Zhang, C.T. Deg: A database of essential genes. Nucleic Acids Res. 2004, 32, D271–D272. [Google Scholar] [CrossRef] [PubMed]
- Petersen, T.N.; Brunak, S.; von Heijne, G.; Nielsen, H. Signalp 4.0: Discriminating signal peptides from transmembrane regions. Nat. Methods 2011, 8, 785–786. [Google Scholar] [CrossRef] [PubMed]
- Sonnhammer, E.L.; von Heijne, G.; Krogh, A. A hidden markov model for predicting transmembrane helices in protein sequences. Proc. Int. Conf. Intell. Syst. Mol. Biol. 1998, 6, 175–182. [Google Scholar] [PubMed]
- Mitchell, A.; Chang, H.Y.; Daugherty, L.; Fraser, M.; Hunter, S.; Lopez, R.; McAnulla, C.; McMenamin, C.; Nuka, G.; Pesseat, S.; et al. The interpro protein families database: The classification resource after 15 years. Nucleic Acids Res. 2015, 43, D213–D221. [Google Scholar] [CrossRef] [PubMed]
- Hassan, S.S.; Tiwari, S.; Guimaraes, L.C.; Jamal, S.B.; Folador, E.; Sharma, N.B.; de Castro Soares, S.; Almeida, S.; Ali, A.; Islam, A.; et al. Proteome scale comparative modeling for conserved drug and vaccine targets identification in Corynebacterium pseudotuberculosis. BMC Genom. 2014, 15. [Google Scholar] [CrossRef]
- Gutmanas, A.; Alhroub, Y.; Battle, G.M.; Berrisford, J.M.; Bochet, E.; Conroy, M.J.; Dana, J.M.; Fernandez Montecelo, M.A.; van Ginkel, G.; Gore, S.P.; et al. PDBE: Protein data bank in Europe. Nucleic Acids Res. 2014, 42, D285–D291. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, S.; da Costa, M.P.; Almeida, S.; Hassan, S.S.; Jamal, S.B.; Oliveira, A.; Folador, E.L.; Rocha, F.; de Abreu, V.A.; Dorella, F.; et al. C. pseudotuberculosis PhoP confers virulence and may be targeted by natural compounds. Integr. Biol. 2014, 6, 1088–1099. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. Ucsf chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S. Kegg: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
| Strain | Size (Mb) | GC% | Gene | Protein |
|---|---|---|---|---|
| Tp_Nichols | 1.13 | 52.80 | 1044 | 970 |
| Tp_Sea81-4 | 1.13 | 52.80 | 1032 | 931 |
| Tp_SS14 | 1.13 | 52.80 | 1042 | 971 |
| Tp_Chicago | 1.13 | 52.80 | 1030 | 969 |
| Tp_SamoaD | 1.13 | 52.80 | 1027 | 971 |
| Tp_CDC2 | 1.13 | 52.80 | 1030 | 973 |
| Tp_Gautheir | 1.13 | 52.80 | 1029 | 971 |
| Tp_DAL1 | 1.13 | 52.80 | 1030 | 969 |
| Tp_MexicoA | 1.14 | 52.80 | 1029 | 968 |
| Tp_Fribourg-Blanc | 1.14 | 52.80 | 1030 | 970 |
| Tp_SS14 (14.8.2015) | 1.13 | 52.80 | 1029 | 970 |
| Tp_BosniaA | 1.13 | 52.80 | 1027 | 970 |
| Tp_pallidum | 1.13 | 52.70 | 1033 | 964 |
| Localization | Number of Proteins |
|---|---|
| Cytoplasmic Protein | 358 |
| Membrane Protein | 83 |
| PSE a | 88 |
| Secreted Protein | 36 |
| Tp_Nichols | Protein ID | Gene Name | Subcellular Localization | SignalP Result (Cleavage Site) | TMHMM Result | InterProScan (Domain) | Gene Product | Adhesion Probability |
|---|---|---|---|---|---|---|---|---|
| Tp_Nichols797 | WP_010882178.1 | - | SEC | Yes (between 25 and 26) | TMH = 0 | Outer membrane protein/outer membrane enzyme PagP, beta-barrel—IPR011250 (65–219) | Hypothetical protein | 0.552 |
| Tp_Nichols141 | WP_014342713.1 | - | PSE | No | TMH = 1 | Outer membrane protein/outer membrane enzyme PagP, beta-barrel—IPR011250 (100–225) | Hypothetical protein | 0.525 |
| Tp_Nichols466 | WP_010881878.1 | ntpK | MEM | No | TMH = 4 | V-ATPase proteolipid subunit C-like domain—IPR002379 (76–138) | Two-sector ATPase, V(0) subunit K | 0.590 |
| Tp_Nichols930 | WP_010882306.1 | slyD | PSE | No | TMH = 1 | Peptidyl-prolyl cis-trans isomerase, FKBP-type, N-terminal—IPR000774 (66–143) | FKBP-type peptidyl-prolyl cis-trans isomerase SlyD | 0.488 |
| Tp_Nichols471 | WP_010881883.1 | nlpE | SEC | Yes (between 23 and 24) | TMH = 0 | No | Copper resistance lipoprotein NlpE | 0.475 |
| Tp_Nichols650 | WP_010882040.1 | - | PSE | No | TMH = 2 | Domain of unknown function DUF2147—IPR019223 (71–193) | Hypothetical Protein | 0.474 |
| Tp_Nichols1046 | WP_010882416.1 | ftr1 | MEM | No | TMH = 6 | No | Conserved hypothetical integral membrane protein | 0.44 |
| Tp_Nichols52 | WP_010881498.1 | TPANIC_0600 | PSE | No | TMH = 1 | Duplicated hybrid motif—Ipr011055 (196–355) | Zinc metalloprotease | 0.428 |
| Tp_Nichols610 | WP_010882004.1 | - | SEC | No | TMH = 1 | Zinc finger, CHCC-type—IPR019401 (8–34) | Hypothetical Protein | 0.425 |
| Tp_Nichols323 | WP_010881746.1 | - | SEC | No | TMH = 1 | Sporulation-related domain—IPR007730 (172–252) | Hypothetical Protein | 0.41 |
| Tp_Nichols852 | WP_010882234.1 | TP_0453 | SEC | Yes (between 23 and 24) | TMH = 0 | No | Outer membrane protein TP0453 | 0.408 |
| Tp_Nichols350 | WP_014342788.1 | tp92 | SEC | Yes (between 37 and 38) | TMH = 1 | Bacterial surface antigen (D15)—IPR000184 (478–849) | Putative outer membrane protein assembly factor TP_0326 | 0.405 |
| Tp_Nichols98 | WP_010881537.1 | - | PSE | No | TMH = 0 | No | Hypothetical Protein | 0.401 |
| Tp_Nichols347 | WP_010881771.1 | TP_0323 | MEM | No | No | Ribose/galactose ABC transporter, permease protein (RbsC-2) | 0.401 | |
| Tp_Nichols362 | WP_010881783.1 | TPANIC_0335 | MEM | No | TMH = 2 | No | Putative membrane protein | 0.401 |
| Locus Tag, Gene, and Protein ID | Official Full Name | Mol. Wt (KDa) a | Functions b | Cellular Component c | Pathways d | Virulence e | DEG Analyses |
|---|---|---|---|---|---|---|---|
| Tp_Nichols130, uvrB, WP_010881565.1 | UvrABC system protein B | 76.19 | MF: ATP (Adenosine triphosphate) binding, DNA binding, excinuclease ABC activity, helicase activity. BP: nucleotide-excision repair, SOS response. | Cytoplasm | Unknown | Yes | Essential gene |
| Tp_Nichols593, Pfp, WP_010881989.1 | Pyrophosphate-fructose 6-phosphate 1-phosphotransferase | 62.43 | -- | Cytoplasm | Glycolysis | Yes | Essential gene |
| Tp_Nichols609, asnA, WP_010882003.1 | Aspartate-ammonia ligase | 36.86 | MF: Aminoacyl-tRNA ligase activity, aspartate-ammonia ligase activity, ATP binding.BP: l-asparagine biosynthetic process, tRNA aminoacylation for protein translation. | Cytoplasm | l-asparaginebiosynthesis | Yes | Essential gene |
| Tp_Nichols754, recA, WP_010882137.1 | Protein RecA | 45.33 | MF: ATP binding, damaged DNA binding, DNA-dependent ATPase activity, single stranded DNA binding.BP: DNA recombination, DNA repair, SOS response. | Cytoplasm | Unknown | Yes | Essential gene |
| Tp_Nichols990, Ndh, WP_010882364.1 | NADH (Nicotinamide adenine dinucleotide) dehydrogenase | 48.64 | MF: flavin adenine dinucleotide binding, NADH dehydrogenase activity.BP: cell redox homeostasis. | Cytoplasmic | Unknown | Yes | Essential gene |
| Tp_Nichols1011, Dxs, WP_010882382.1 | 1-deoxy-d-xylulose-5-phosphate synthase | 129.82 | MF: 1-deoxy-d-xylulose-5-phosphate synthase activity, magnesium ion binding, thiamine pyrophosphate binding.BP: 1-deoxy-d-xylulose-5-phosphate biosynthetic process, terpenoid biosynthesis process, Thiamine biosynthesis process. | Cytoplasmic | 1-deoxy-d-xylulose 5-phosphate biosynthesis | Yes | Essential gene |
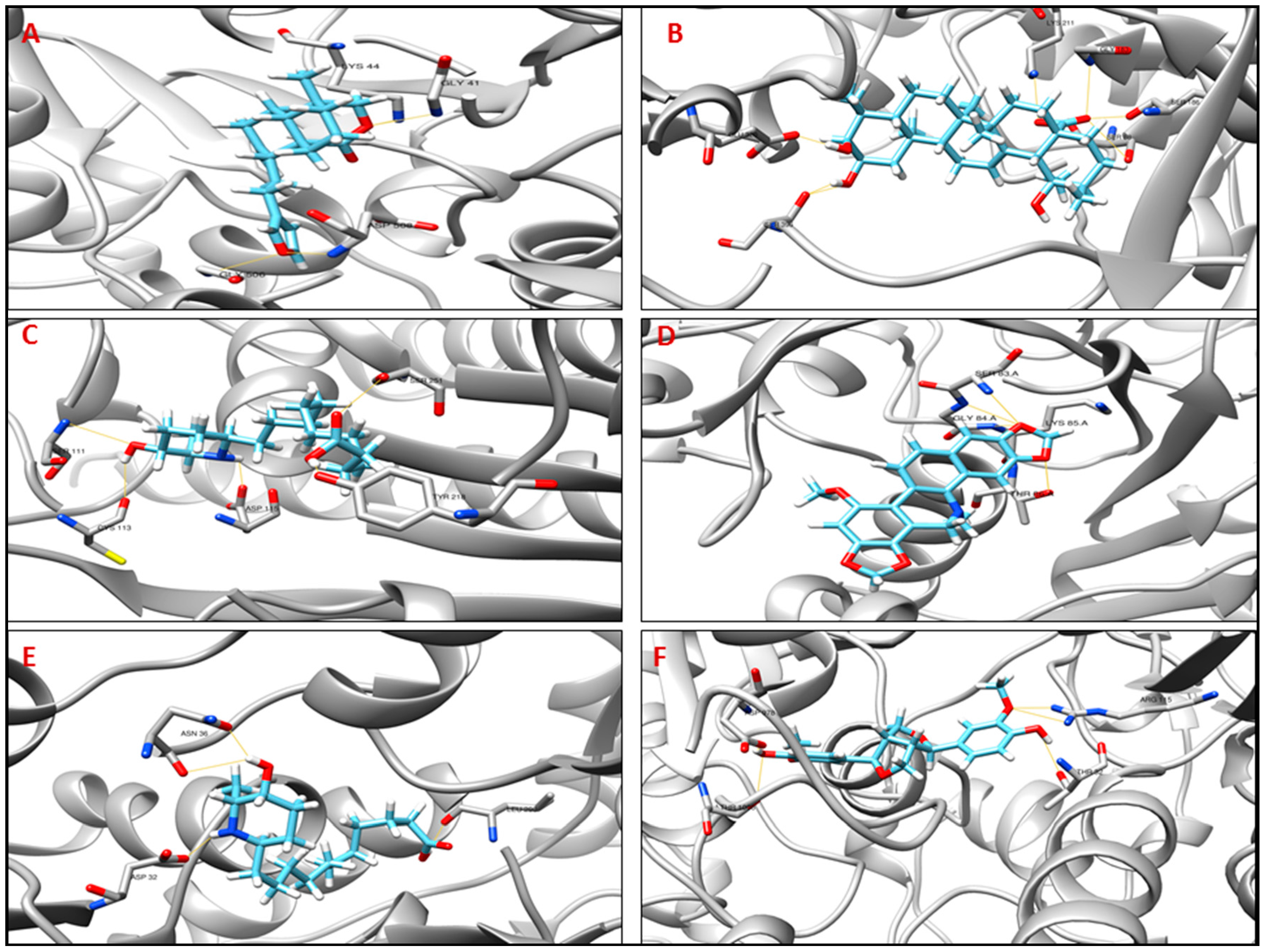
| Compounds Name | MolDock Score | Number of H-Bond | Residues Interacting |
|---|---|---|---|
| Tp_Nichols130 (UvrB, Uvr ABC System Protein B) | |||
| Diospyrin (CID 308140) MW: ~374.3 g/mol | −119.83 | 4 | Gly506, Asp508 |
| Pinoresinol (CID 234817) MW: ~358.4 g/mol | −114.82 | 2 | His64, Asp508 |
| Potamogetonin (CID 5742898) MW: ~314.4 g/mol | −97.81 | 4 | Gly41, Lys44, Gly506, Asp508 |
| Tp_Nichols593 (pfp, Pyrophosphate-fructose 6-phosphate 1-phosphotransferase) | |||
| Pinoresinol (CID 234817) MW: ~358.4 g/mol | −112.67 | 5 | Ser88, Lys211, Gly260, Glu320 |
| Jacarandic acid (CID 73645) MW: ~488.7 g/mol | −62.15 | 7 | Ser88, Ser186, Gly183, Lys211, Glu320, Ser396 |
| Texalin (CID 473253) MW: ~266.3 g/mol | −91.57 | 4 | Gly90, Thr212, Ser186, Ile213 |
| Tp_Nichols609 (asnA, Aspartate-ammonia ligase) | |||
| Leptophyllin B (CID 10447482) MW: ~299.4 g/mol | −141.21 | 5 | Ser111, Cys113, Asp115, Tyr218, Ser251 |
| Pinoresinol (CID 234817) MW: ~358.4 g/mol | −132.814 | 5 | Ser49, Lys77, Ser251, Arg255 |
| Liriodenine (CID 10144) MW: ~275.1 g/mol | −95.65 | 2 | Lys77, Arg255 |
| Tp_Nichols754 (recA, Protein RecA) | |||
| Dihydrochelirubine (CID 440589) MW: ~363.4 g/mol | −138.94 | 4 | Gly84, Lys85, Ser83, Thr86 |
| Piperine (CID 638024) MW: ~285.3 g/mol | −17.14 | 5 | Ser83, Gly84, Lys84, Gln207, Gly279 |
| Rhein (CID 10168) MW: ~284.2 g/mol | −96.11 | 7 | Ser83, Gly84, Thr86, Tyr116, Asn254, Gly279 |
| Tp_Nichols990 (ndh, NADH dehydrogenase) | |||
| Leptophyllin B (CID 10447482) MW: ~299.4 g/mol | −122.62 | 4 | Leu298, Asp32, Asn36 |
| Dicentrinone (CID 177744) MW: ~335.3 g/mol | −111.09 | 4 | Arg33, Ala11 |
| Isosakuranetin (CID 160481) MW: ~286.3 g/mol | −109.35 | 3 | Arg33, Ala11, Cyc42 |
| Tp_Nichols1011 (dxs, 1-deoxy-d-xylulose-5-phosphate synthase) | |||
| Pinoresinol (CID 234817) MW: ~358.4 g/mol | −146.18 | 5 | Asp978, Thr1006, Thr32, Arg115 |
| Piperine (CID 638024) MW: ~285.3 g/mol | −131.40 | 3 | Thr32, Arg115, Trp980 |
| Berberine (CID 2353) MW: ~336.4 g/mol | −115.94 | 3 | Thr32, Gly979, Asn1011 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kumar Jaiswal, A.; Tiwari, S.; Jamal, S.B.; Barh, D.; Azevedo, V.; Soares, S.C. An In Silico Identification of Common Putative Vaccine Candidates against Treponema pallidum: A Reverse Vaccinology and Subtractive Genomics Based Approach. Int. J. Mol. Sci. 2017, 18, 402. https://doi.org/10.3390/ijms18020402
Kumar Jaiswal A, Tiwari S, Jamal SB, Barh D, Azevedo V, Soares SC. An In Silico Identification of Common Putative Vaccine Candidates against Treponema pallidum: A Reverse Vaccinology and Subtractive Genomics Based Approach. International Journal of Molecular Sciences. 2017; 18(2):402. https://doi.org/10.3390/ijms18020402
Chicago/Turabian StyleKumar Jaiswal, Arun, Sandeep Tiwari, Syed Babar Jamal, Debmalya Barh, Vasco Azevedo, and Siomar C. Soares. 2017. "An In Silico Identification of Common Putative Vaccine Candidates against Treponema pallidum: A Reverse Vaccinology and Subtractive Genomics Based Approach" International Journal of Molecular Sciences 18, no. 2: 402. https://doi.org/10.3390/ijms18020402