
Inhibition of CCAR1, a Coactivator of β-Catenin, Suppresses the Proliferation and Migration of Gastric Cancer Cells

Abstract
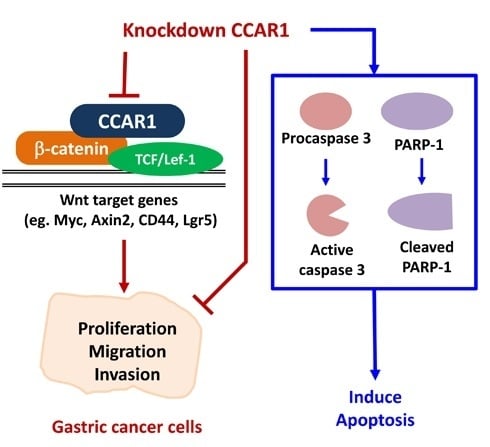
:
1. Introduction
2. Results
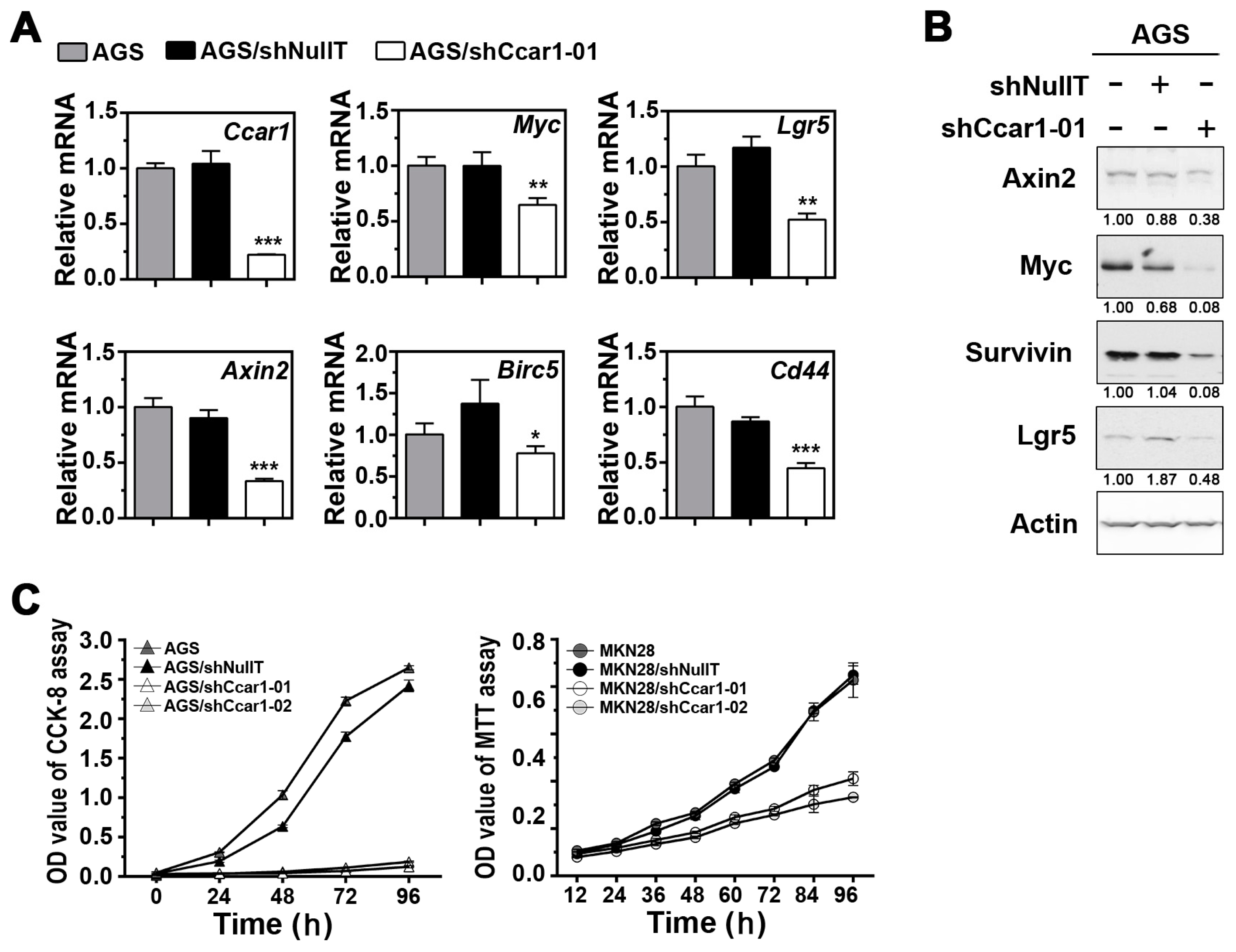
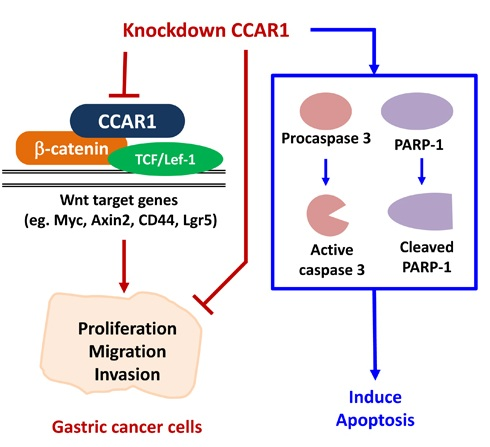
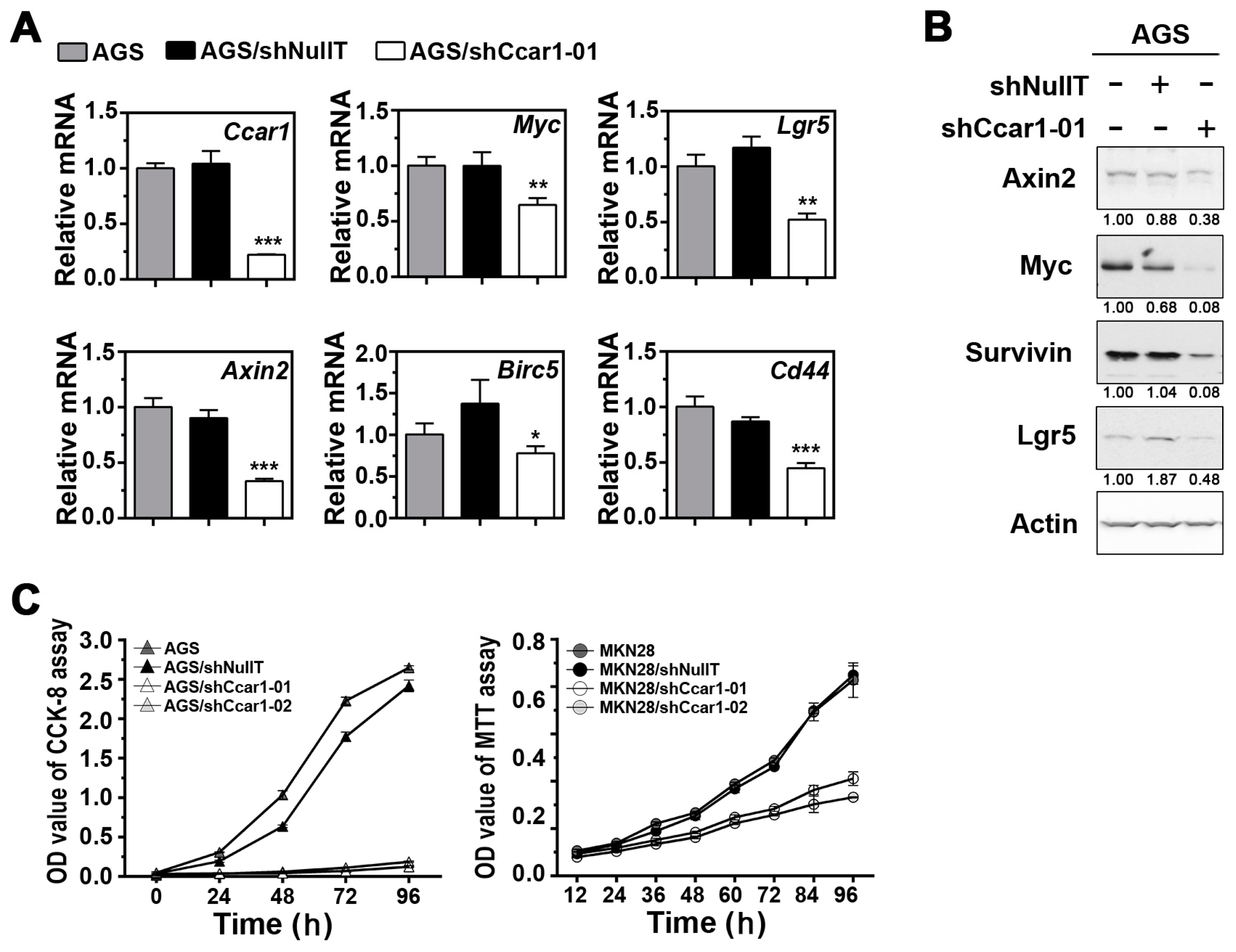
2.1. Suppression of CCAR1 Reduces the Expression of Wnt Signaling Target Genes and Inhibits Gastric Cancer Cell Growth
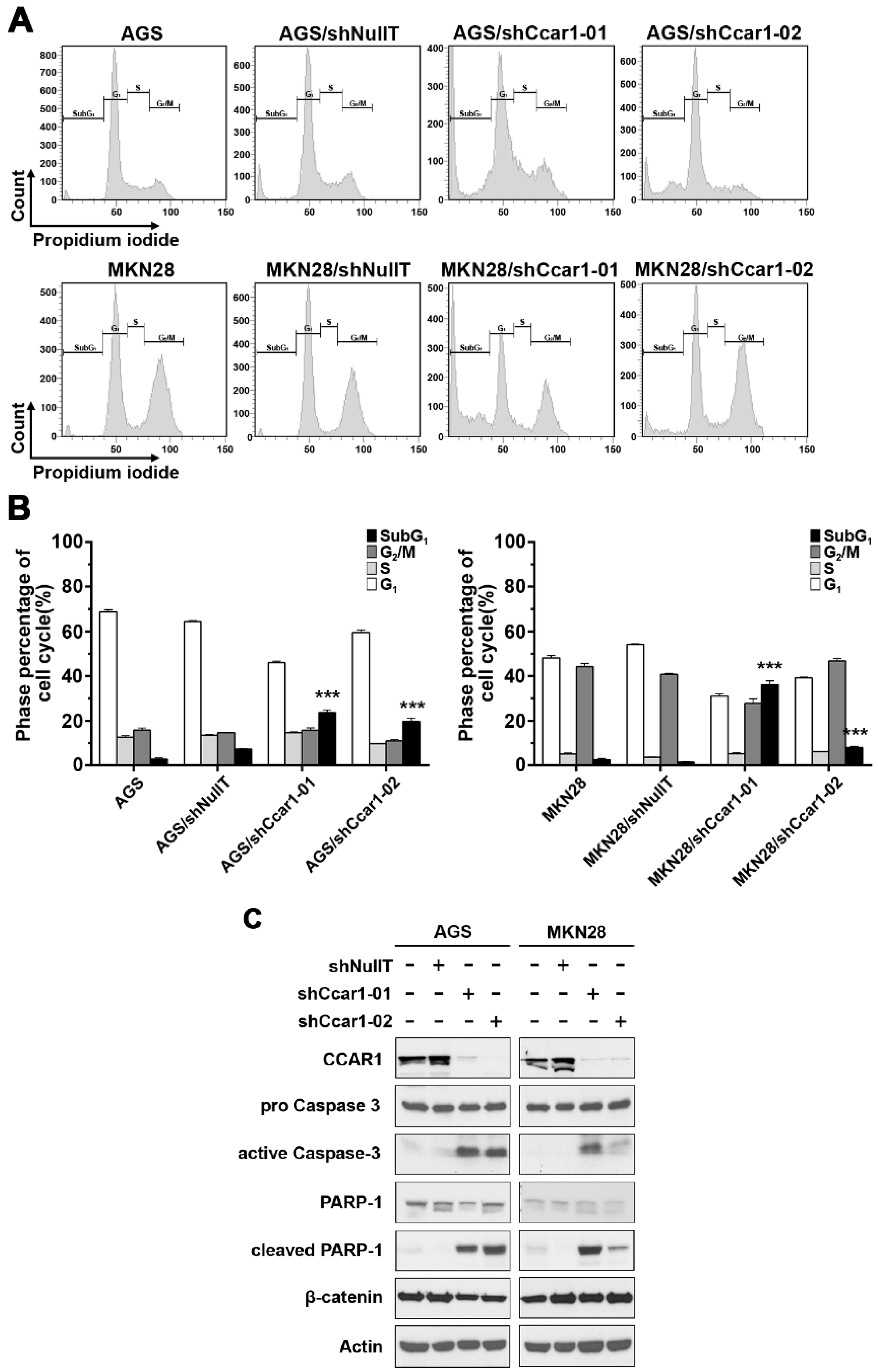
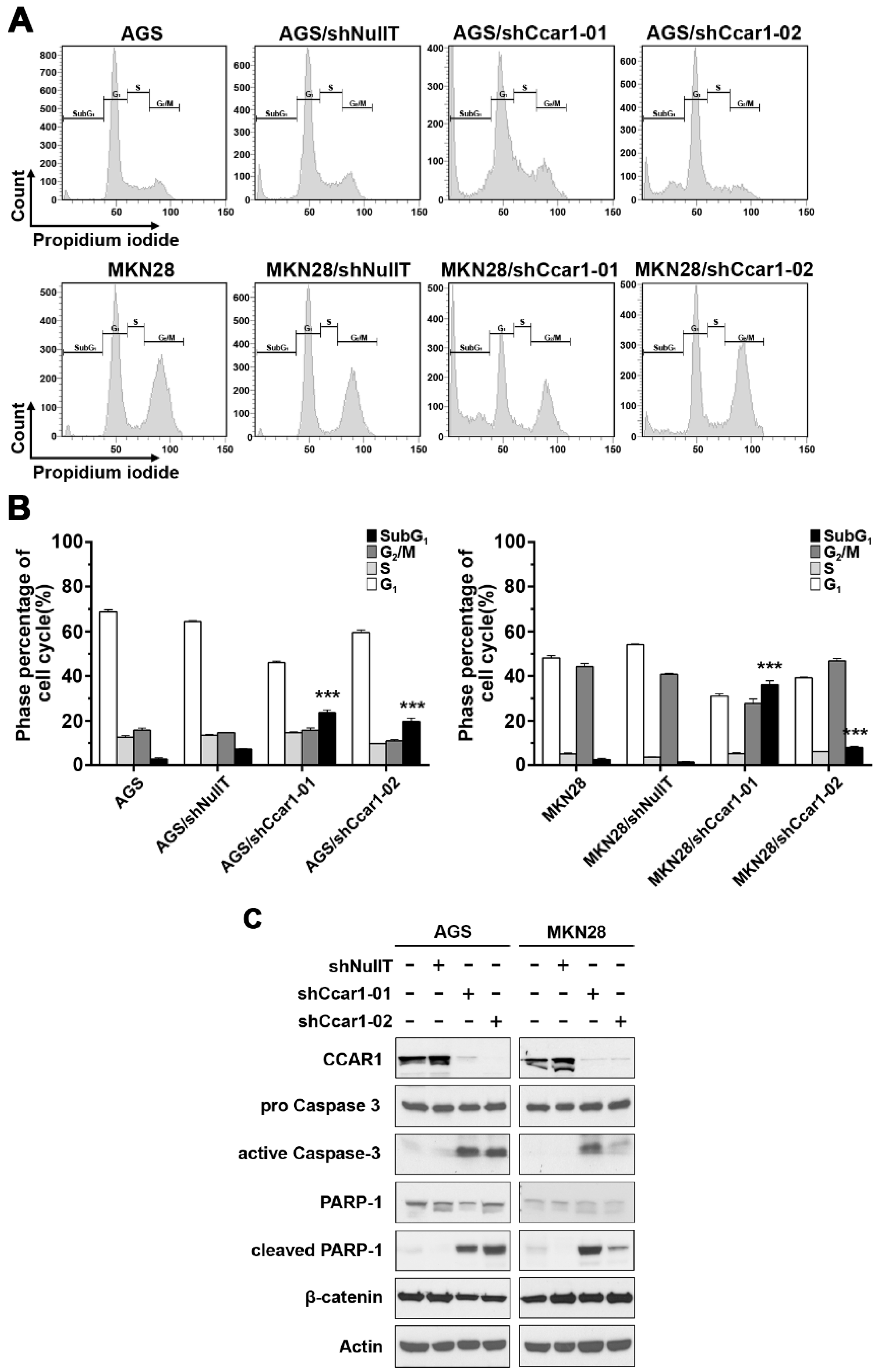
2.2. Suppression of CCAR1 Induces Apoptotic Cell Death in Gastric Cancer Cells
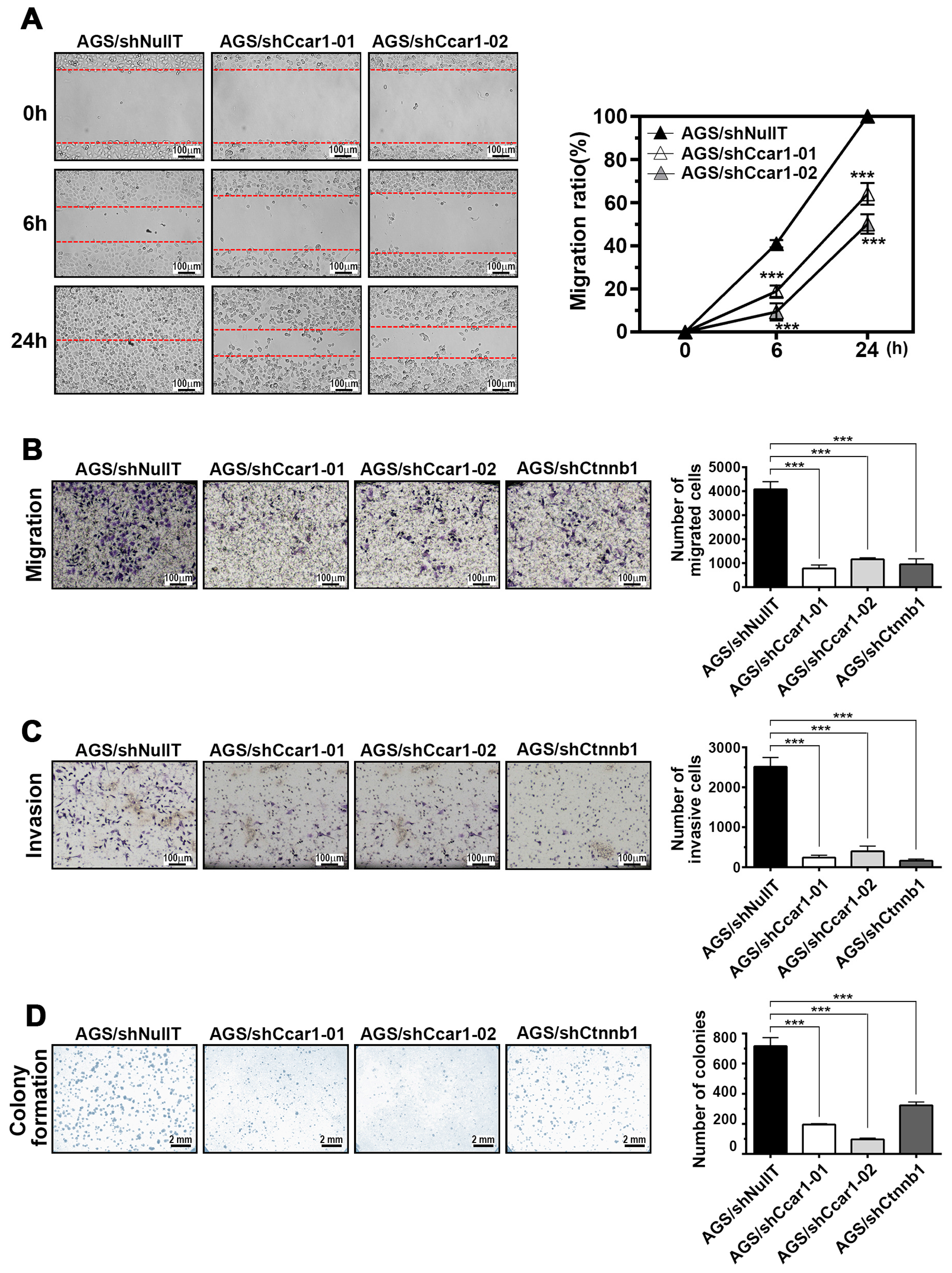
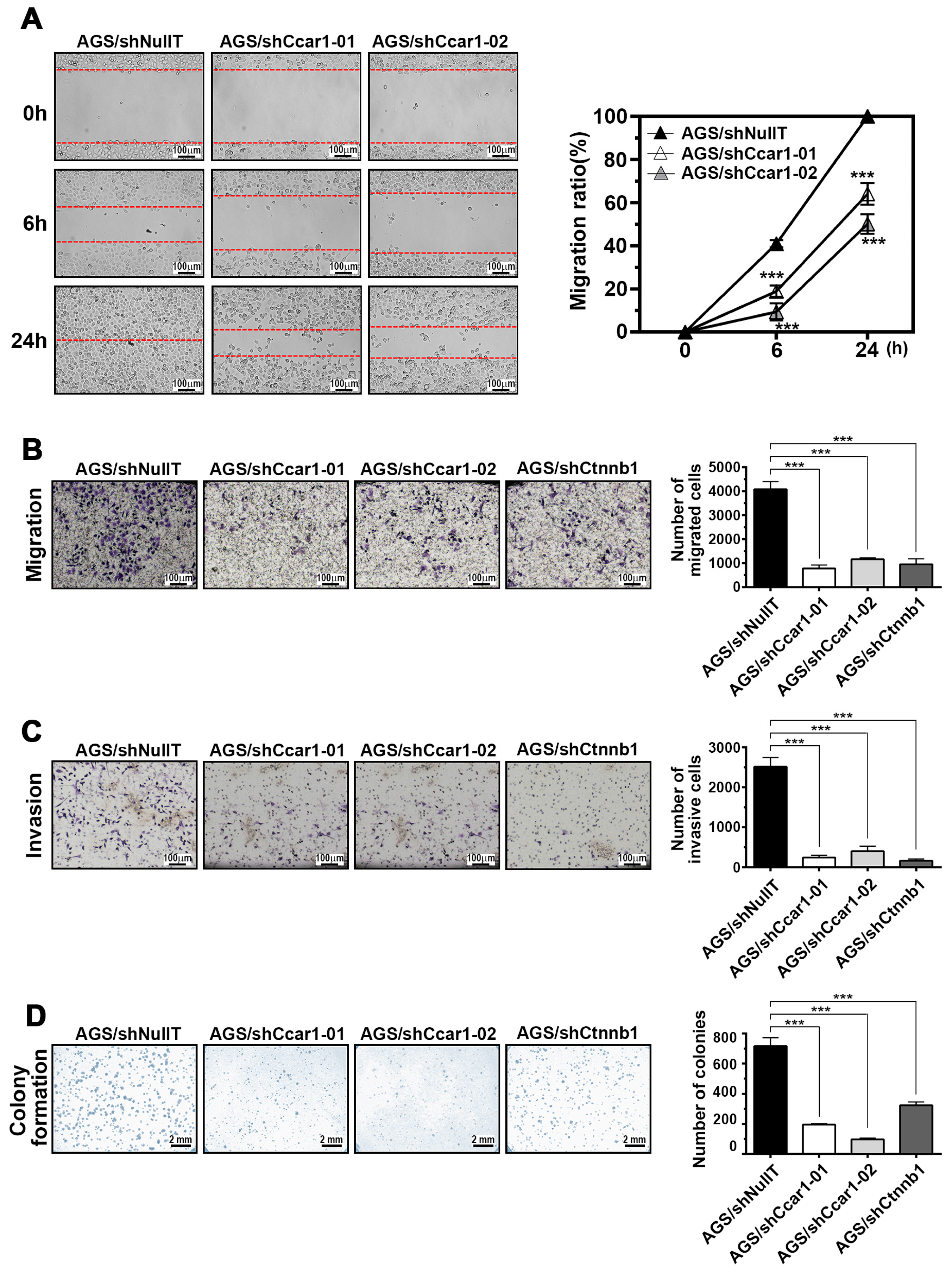
2.3. CCAR1 Mediates the Invasive Characters of Gastric Cancer Cells
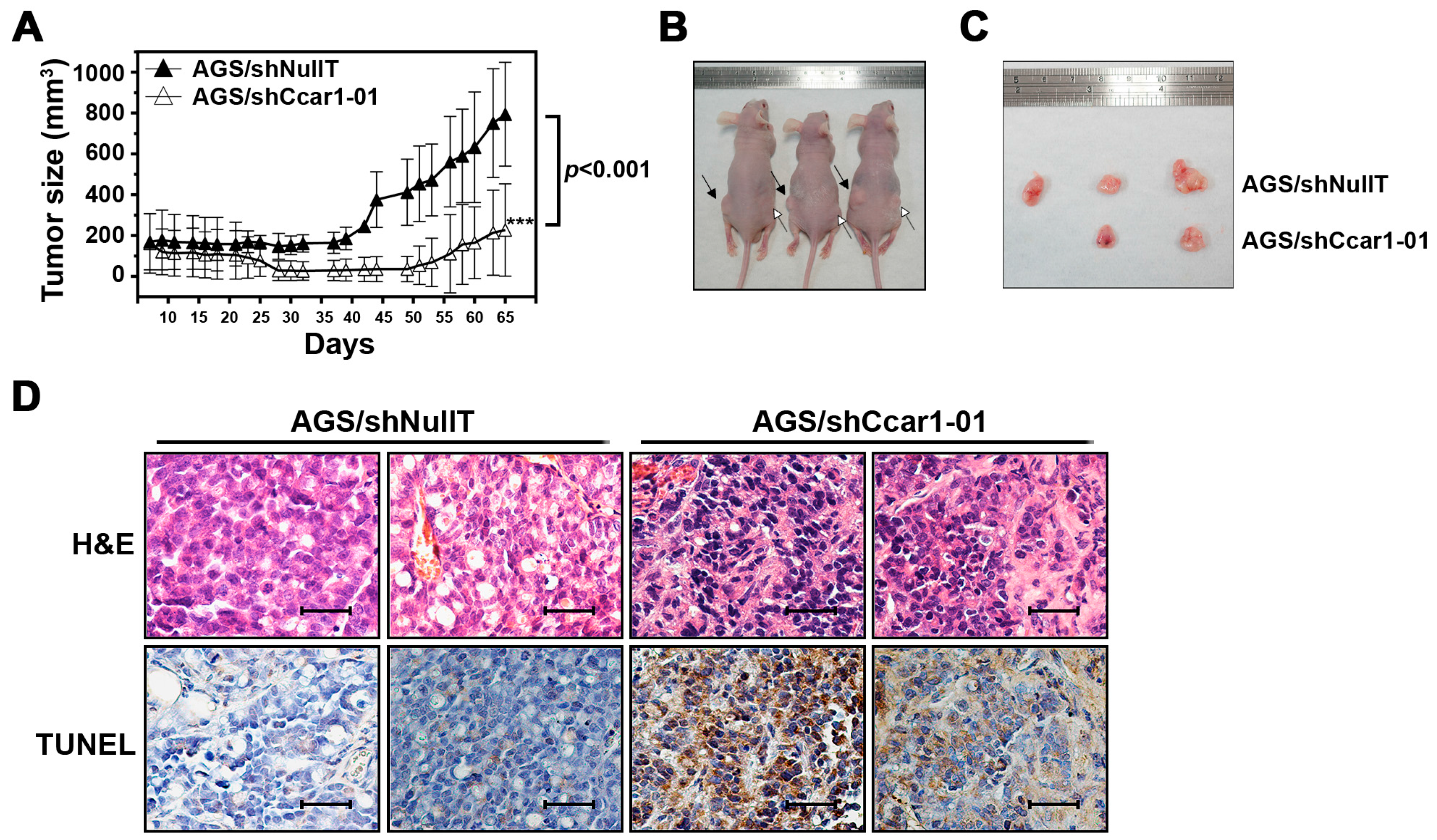
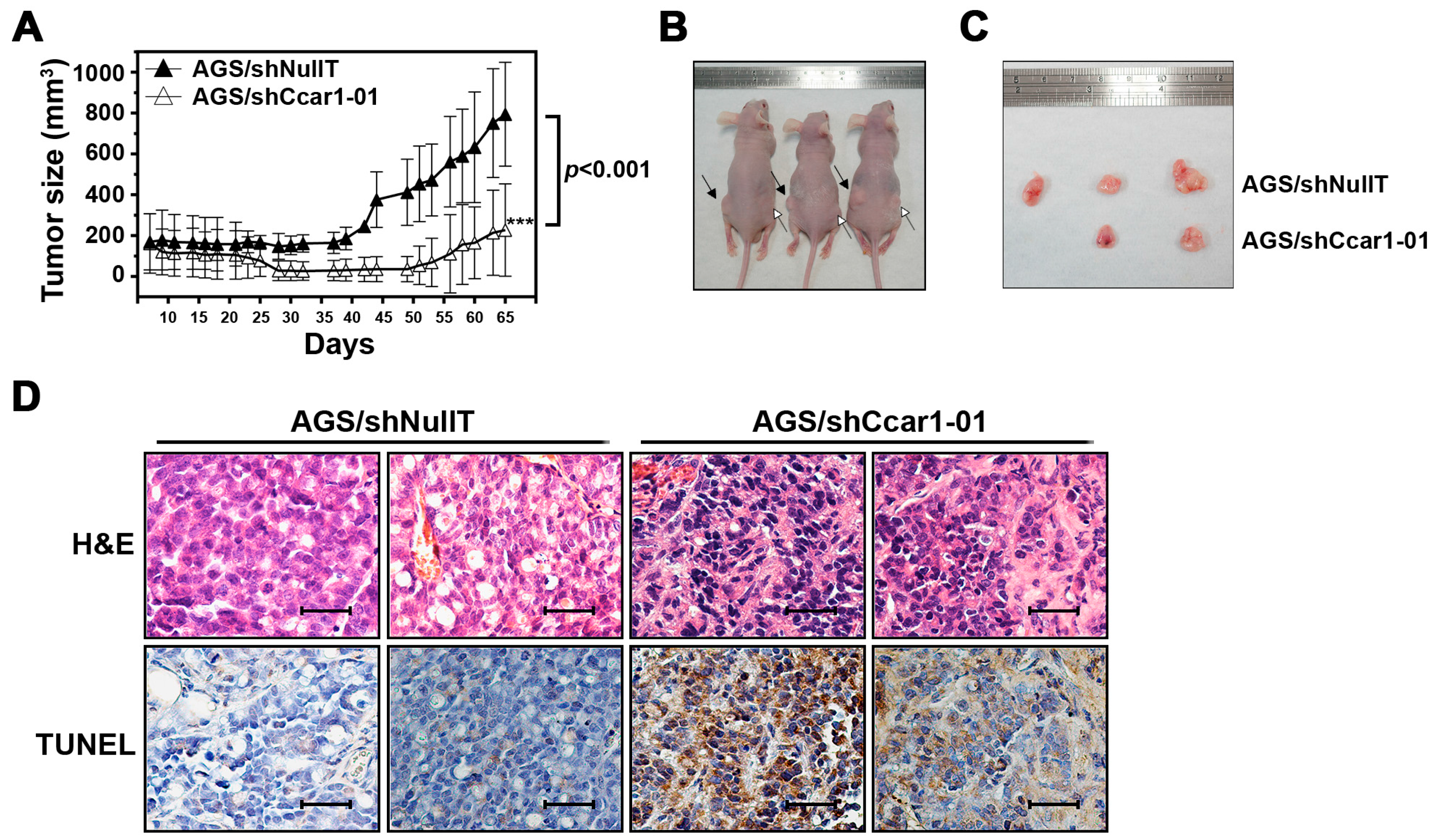
2.4. CCAR1 Is Required for the Expansion of the Gastric Cancer Xenograft in the Animal Model
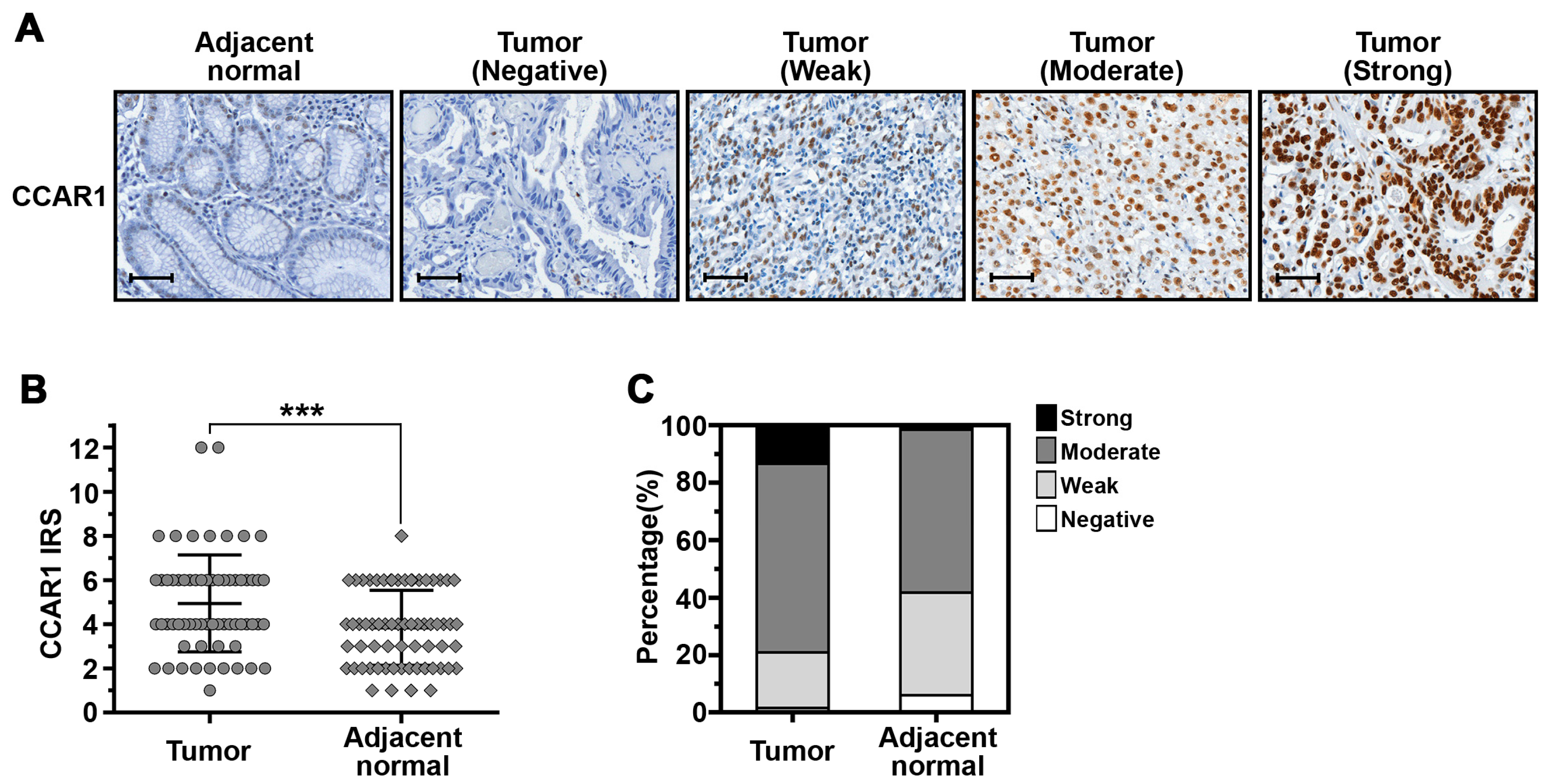
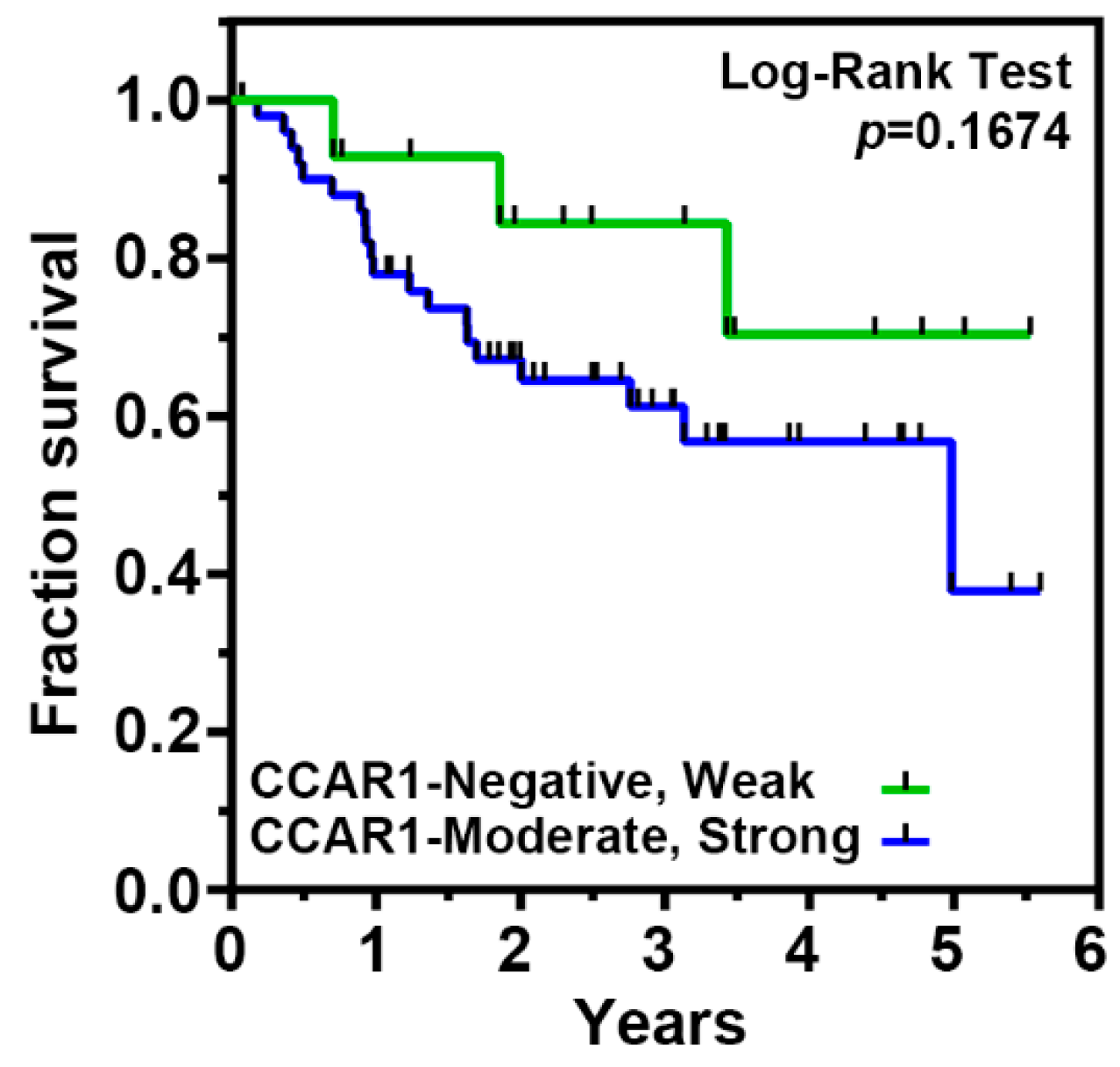
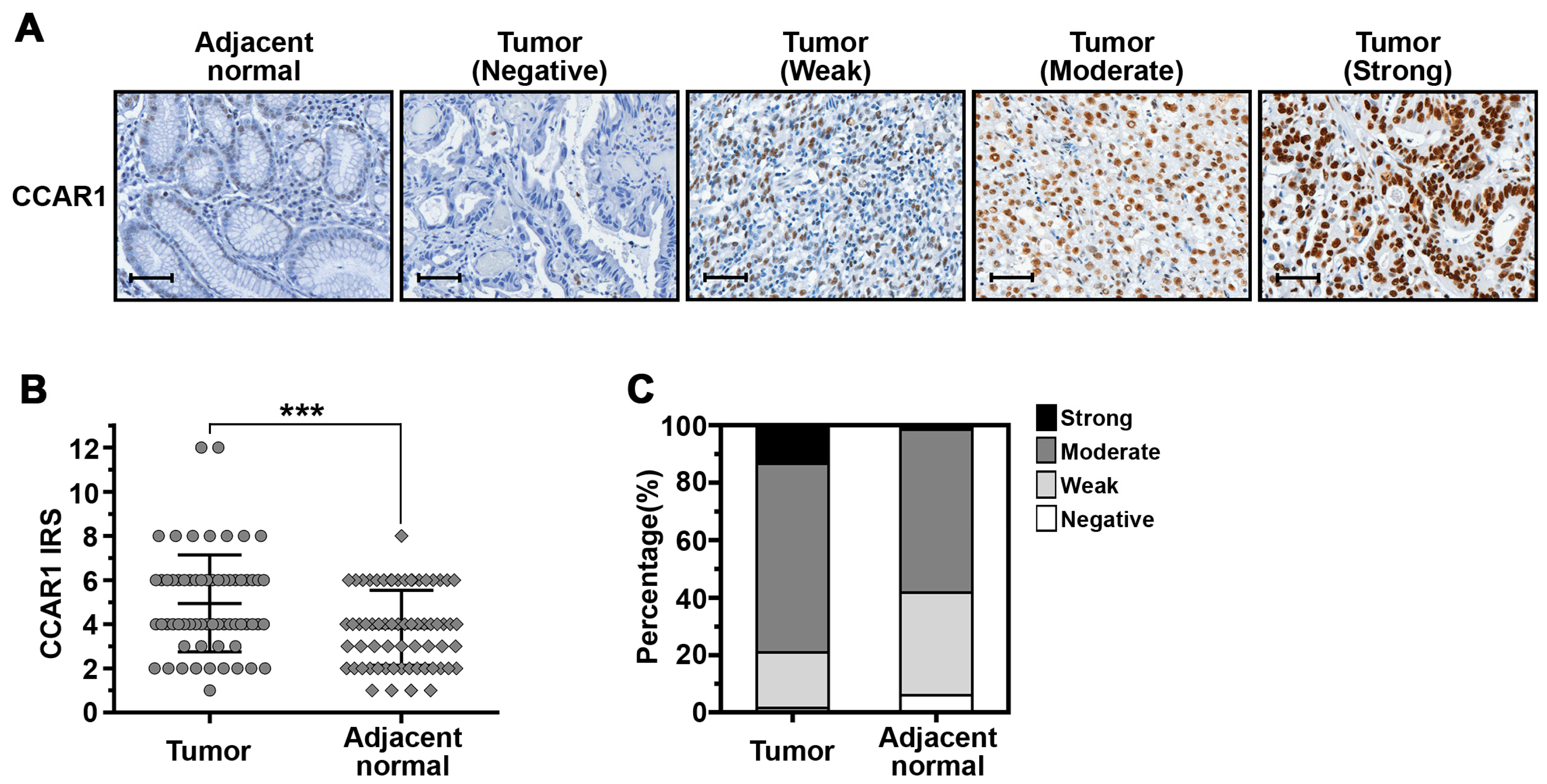
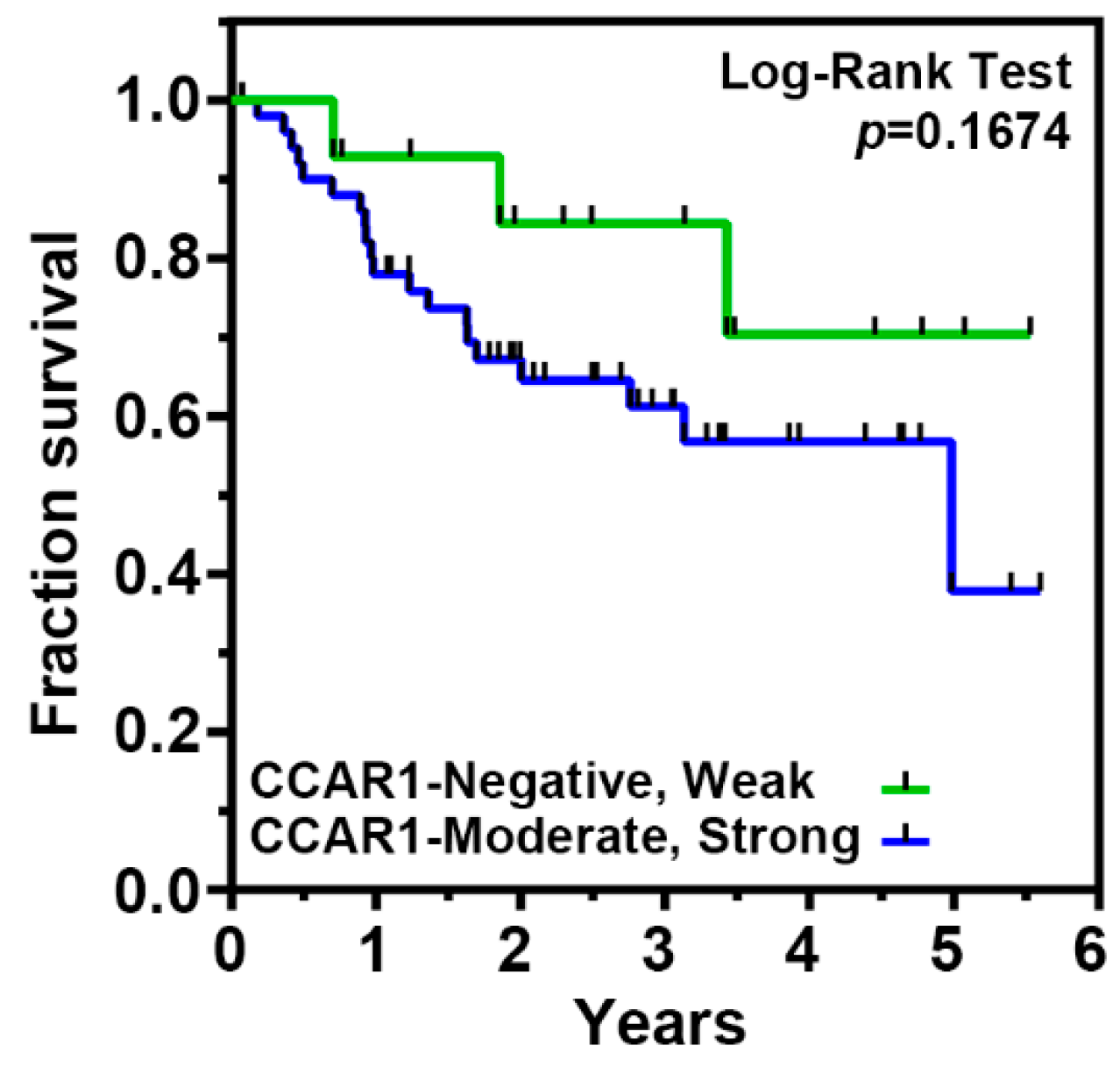
2.5. The Expression of CCAR1 Is Significantly Stronger in Human Gastric Cancer Tissues Than Their Adjacent Normal Parts
3. Discussion
4. Materials and Methods
4.1. Cell Lines, Reagents, and Patients
4.2. Tissue Microarray
4.3. Immunohistochemical Staining
4.4. Lentiviral Production and Infection
4.5. Colony Formation Assay
4.6. Wound Healing and Transwell Migration Assay
4.7. Invasion Assay
4.8. RNA Isolation and RT-Quantitative PCR
- Ccar1 forward 5′-CTGATGGCTAGCCCTAGTATGGA-3′,
- Ccar1 reverse 5′-TGCCTTTCATGCCCACTAAAA-3′,
- Axin2 forward 5′-GGAATCATTCGGCCACTGTT-3′,
- Axin2 reverse 5′-TGGACACCTGCCAGTTTCTTT-3′,
- Cd44 forward 5′-ACCTTTCCCCCACCAGCTA-3′,
- Cd44 reverse 5′-GCTATGGAAGGGCAAAATGG-3′,
- Lgr5 forward 5′-CACTTGCTTTCCAAATGGGTTT-3′,
- Lgr5 reverse 5′-ATCACAGCCTCTACCTAGCAATGTAG-3,
- Myc forward 5′-GGCCCCCAAGGTAGTTATCC-3′,
- Myc reverse 5′-GTTTCCGCAACAAGTCCTCTTC-3′,
- Birc5 forward 5′-GACCCACTTATTTCTGCCACATC-3′,
- Birc5 reverse 5′-GAGTACAGAGGCTGGAGTGCATT-3′,
- Actb forward 5′-TGGCATTGCCGACAGGAT-3′,
- Actb reverse 5′-CGCTCAGGAGGAGCAATGAT-3′.
4.9. Tumorigenicity in Nude Mice
4.10. Cell Proliferation Assay
4.11. Statistical Analysis
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Parkin, D.M.; Bray, F.; Ferlay, J.; Pisani, P. Global Cancer Statistics, 2002. CA Cancer J. Clin. 2005, 55, 74–108. [Google Scholar] [CrossRef] [PubMed]
- Gatta, G.; Ciccolallo, L.; Kunkler, I.; Capocaccia, R.; Berrino, F.; Coleman, M.P.; De Angelis, R.; Faivre, J.; Lutz, J.M.; Martinez, C.; et al. Survival from rare cancer in adults: A population-based study. Lancet Oncol. 2006, 7, 132–140. [Google Scholar] [CrossRef]
- Health Promotion Administration, Ministry of Health and Welfare, Taiwan. Cancer Registry Annual Report, 2013. Available online: http://www.hpa.gov.tw/Pages/Detail.aspx?nodeid=269&pid=5191 (accessed on 25 February 2015).
- Milne, A.N.; Carneiro, F.; O’Morain, C.; Offerhaus, G.J. Nature meets nurture: Molecular genetics of gastric cancer. Hum. Genet. 2009, 126, 615–628. [Google Scholar] [CrossRef] [PubMed]
- Barker, N.; Ridgway, R.A.; van Es, J.H.; van de Wetering, M.; Begthel, H.; van den Born, M.; Danenberg, E.; Clarke, A.R.; Sansom, O.J.; Clevers, H. Crypt stem cells as the cells-of-origin of intestinal cancer. Nature 2009, 457, 608–611. [Google Scholar] [CrossRef] [PubMed]
- Barker, N.; van Es, J.H.; Kuipers, J.; Kujala, P.; van den Born, M.; Cozijnsen, M.; Haegebarth, A.; Korving, J.; Begthel, H.; Peters, P.J.; et al. Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature 2007, 449, 1003–1007. [Google Scholar] [CrossRef] [PubMed]
- Zon, L.I. Intrinsic and extrinsic control of haematopoietic stem-cell self-renewal. Nature 2008, 453, 306–313. [Google Scholar] [CrossRef] [PubMed]
- Bilic, J.; Huang, Y.L.; Davidson, G.; Zimmermann, T.; Cruciat, C.M.; Bienz, M.; Niehrs, C. Wnt induces LRP6 signalosomes and promotes dishevelled-dependent LRP6 phosphorylation. Science 2007, 316, 1619–1622. [Google Scholar] [CrossRef] [PubMed]
- Schwarz-Romond, T.; Fiedler, M.; Shibata, N.; Butler, P.J.; Kikuchi, A.; Higuchi, Y.; Bienz, M. The DIX domain of Dishevelled confers Wnt signaling by dynamic polymerization. Nat. Struct. Mol. Biol. 2007, 14, 484–492. [Google Scholar] [CrossRef] [PubMed]
- Behrens, J.; von Kries, J.P.; Kuhl, M.; Bruhn, L.; Wedlich, D.; Grosschedl, R.; Birchmeier, W. Functional interaction of β-catenin with the transcription factor LEF-1. Nature 1996, 382, 638–642. [Google Scholar] [CrossRef] [PubMed]
- Molenaar, M.; van de Wetering, M.; Oosterwegel, M.; Peterson-Maduro, J.; Godsave, S.; Korinek, V.; Roose, J.; Destrée, O.; Clevers, H. XTcf-3 transcription factor mediates β-catenin-induced axis formation in xenopus embryos. Cell 1996, 86, 391–399. [Google Scholar] [CrossRef]
- Grigoryan, T.; Wend, P.; Klaus, A.; Birchmeier, W. Deciphering the function of canonical Wnt signals in development and disease: Conditional loss- and gain-of-function mutations of β-catenin in mice. Genes Dev. 2008, 22, 2308–2341. [Google Scholar] [CrossRef] [PubMed]
- Polakis, P. Wnt signaling and cancer. Genes Dev. 2000, 14, 1837–1851. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.R.; Hocking, A.M.; Brown, J.D.; Moon, R.T. Mechanism and function of signal transduction by the Wnt/β-catenin and Wnt/Ca2+ pathways. Oncogene 1999, 18, 7860–7872. [Google Scholar] [CrossRef] [PubMed]
- Kinzler, K.W.; Vogelstein, B. Lessons from hereditary colorectal cancer. Cell 1996, 87, 159–170. [Google Scholar] [CrossRef]
- Rishi, A.K.; Zhang, L.; Boyanapalli, M.; Wali, A.; Mohammad, R.M.; Yu, Y.; Fontana, J.A.; Hatfield, J.S.; Dawson, M.I.; Majumdar, A.P.; et al. Identification and characterization of a cell cycle and apoptosis regulatory protein-1 as a novel mediator of apoptosis signaling by retinoid CD437. J. Biol. Chem. 2003, 278, 33422–33435. [Google Scholar] [CrossRef] [PubMed]
- Rishi, A.K.; Zhang, L.; Yu, Y.; Jiang, Y.; Nautiyal, J.; Wali, A.; Fontana, J.A.; Levi, E.; Majumdar, A.P. Cell cycle- and apoptosis-regulatory protein-1 is involved in apoptosis signaling by epidermal growth factor receptor. J. Biol. Chem. 2006, 281, 13188–13198. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Yang, C.K.; Heo, K.; Roeder, R.G.; An, W.; Stallcup, M.R. CCAR1, a key regulator of mediator complex recruitment to nuclear receptor transcription complexes. Mol. Cell 2008, 31, 510–519. [Google Scholar] [CrossRef] [PubMed]
- Ou, C.Y.; Kim, J.H.; Yang, C.K.; Stallcup, M.R. Requirement of cell cycle and apoptosis regulator 1 for target gene activation by Wnt and β-catenin and for anchorage-independent growth of human colon carcinoma cells. J. Biol. Chem. 2009, 284, 20629–20637. [Google Scholar] [CrossRef] [PubMed]
- Fedchenko, N.; Reifenrath, J. Different approaches for interpretation and reporting of immunohistochemistry analysis results in the bone tissue—A review. Diagn. Pathol. 2014, 9, 221. [Google Scholar] [CrossRef] [PubMed]
- Chiurillo, M.A. Role of the Wnt/β-catenin pathway in gastric cancer: An in-depth literature review. World J. Exp. Med. 2015, 5, 84–102. [Google Scholar] [CrossRef] [PubMed]
- Qian, C.; Liu, F.; Ye, B.; Zhang, X.; Liang, Y.; Yao, J. Notch4 promotes gastric cancer growth through activation of Wnt1/β-catenin signaling. Mol. Cell. Biochem. 2015, 401, 165–174. [Google Scholar] [CrossRef] [PubMed]
- Takagi, H.; Sasaki, S.; Suzuki, H.; Toyota, M.; Maruyama, R.; Nojima, M.; Yamamoto, H.; Omata, M.; Tokino, T.; Imai, K.; et al. Frequent epigenetic inactivation of SFRP genes in hepatocellular carcinoma. J. Gastroenterol. 2008, 43, 378–389. [Google Scholar] [CrossRef] [PubMed]
- Pan, K.F.; Liu, W.G.; Zhang, L.; You, W.C.; Lu, Y.Y. Mutations in components of the Wnt signaling pathway in gastric cancer. World J. Gastroenterol. 2008, 14, 1570–1574. [Google Scholar] [CrossRef] [PubMed]
- Clements, W.M.; Lowy, A.M.; Groden, J. Adenomatous polyposis coli/β-catenin interaction and downstream targets: Altered gene expression in gastrointestinal tumors. Clin. Colorectal Cancer 2003, 3, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Nakatsuru, S.; Yanagisawa, A.; Ichii, S.; Tahara, E.; Kato, Y.; Nakamura, Y.; Horii, A. Somatic mutation of the APC gene in gastric cancer: Frequent mutations in very well differentiated adenocarcinoma and signet-ring cell carcinoma. Hum. Mol. Genet. 1992, 1, 559–563. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.J.; Kim, J.H.; Yoon, J.; Cho, S.J.; Ko, Y.S.; Park, J.W.; Lee, H.S.; Lee, H.E.; Kim, W.H.; Lee, B.L. Constitutive activation of glycogen synthase kinase-3beta correlates with better prognosis and cyclin-dependent kinase inhibitors in human gastric cancer. BMC Gastroenterol. 2010, 10, 91. [Google Scholar] [CrossRef] [PubMed]
- Clements, W.M.; Wang, J.; Sarnaik, A.; Kim, O.J.; MacDonald, J.; Fenoglio-Preiser, C.; Groden, J.; Lowy, A.M. β-Catenin mutation is a frequent cause of Wnt pathway activation in gastric cancer. Cancer Res. 2002, 62, 3503–3506. [Google Scholar] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | CCAR1 Expression | p-Value | |||
|---|---|---|---|---|---|
| Negative (%) | Weak (%) | Moderate (%) | Strong (%) | ||
| Adjacent normal | 4 (6.0%) | 24 (35.8%) | 38 (56.7%) | 1 (1.5%) | 0.005 |
| Tumor | 1 (1.5%) | 13 (19.4%) | 44 (65.7%) | 9 (13.4%) | |
| Features | CCAR1 Expression of Tumor Part | p-Value | |
|---|---|---|---|
| Negative, Weak (%) | Moderate, Strong (%) | ||
| Gender | |||
| Male | 11 (23.4%) | 36 (76.6%) | 0.528 |
| Female | 3 (15.0%) | 17 (85.0%) | |
| Age | |||
| <65 years | 5 (23.8%) | 16 (76.2%) | 0.692 |
| ≥65 years | 9 (19.6%) | 37 (80.4%) | |
| Pathological Stage | |||
| I + II | 7 (24.1%) | 22 (75.9%) | 0.569 |
| III + IV | 7 (18.4%) | 31 (81.6%) | |
| Depth of invasion (pT) | |||
| T1 + T2 | 5 (23.8%) | 16 (76.2%) | 0.751 |
| T3 + T4 | 9 (19.6%) | 37 (80.4%) | |
| Lymph nodes metastasis | |||
| Negative | 6 (26.1%) | 17 (73.9%) | 0.450 |
| Positive | 8 (18.2%) | 36 (81.8%) | |
| Distant metastasis | |||
| Negative | 11 (19.3%) | 46 (80.7%) | 0.425 |
| Positive | 3 (30.0%) | 7 (70.0%) | |
| H. pylori | |||
| Negative | 3 (13.0%) | 20 (87.0%) | 0.349 |
| Positive | 11 (25.0%) | 33 (75.0%) | |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chang, T.-S.; Wei, K.-L.; Lu, C.-K.; Chen, Y.-H.; Cheng, Y.-T.; Tung, S.-Y.; Wu, C.-S.; Chiang, M.-K. Inhibition of CCAR1, a Coactivator of β-Catenin, Suppresses the Proliferation and Migration of Gastric Cancer Cells. Int. J. Mol. Sci. 2017, 18, 460. https://doi.org/10.3390/ijms18020460
Chang T-S, Wei K-L, Lu C-K, Chen Y-H, Cheng Y-T, Tung S-Y, Wu C-S, Chiang M-K. Inhibition of CCAR1, a Coactivator of β-Catenin, Suppresses the Proliferation and Migration of Gastric Cancer Cells. International Journal of Molecular Sciences. 2017; 18(2):460. https://doi.org/10.3390/ijms18020460
Chicago/Turabian StyleChang, Te-Sheng, Kuo-Liang Wei, Chung-Kuang Lu, Yi-Hsing Chen, Ying-Tung Cheng, Shui-Yi Tung, Cheng-Shyong Wu, and Ming-Ko Chiang. 2017. "Inhibition of CCAR1, a Coactivator of β-Catenin, Suppresses the Proliferation and Migration of Gastric Cancer Cells" International Journal of Molecular Sciences 18, no. 2: 460. https://doi.org/10.3390/ijms18020460