
Hotspot Selective Preference of the Chimeric Sequences Formed in Multiple Displacement Amplification


Abstract
:1. Introduction
2. Materials and Methods
2.1. Data Source, Sample Information and Experimental Procedure
2.2. Recognition of Chimeras
2.3. Chimeric Hotspots Screening in Hg19
2.4. Analysis of Chimeras and Chimeric Hotspots
2.5. Td Estimation
3. Results
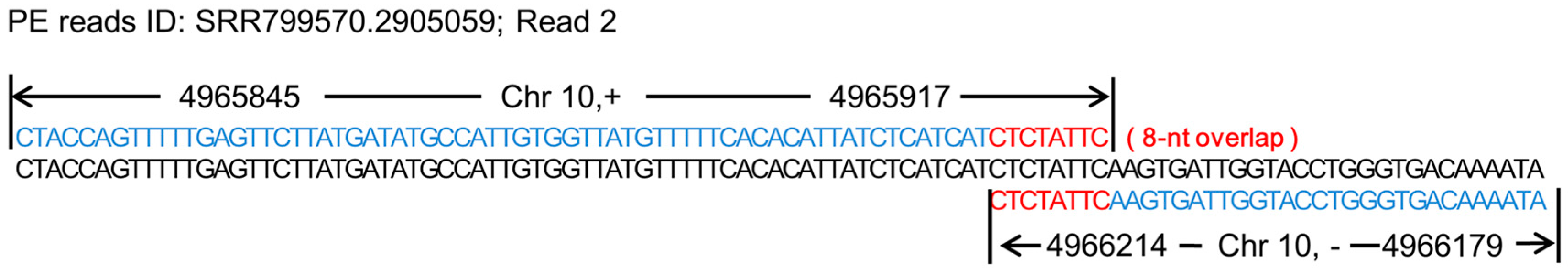
3.1. Chimeras and the Formation of Chimeras
3.2. Screening Results of Chimeras and Chimeric Hotspots
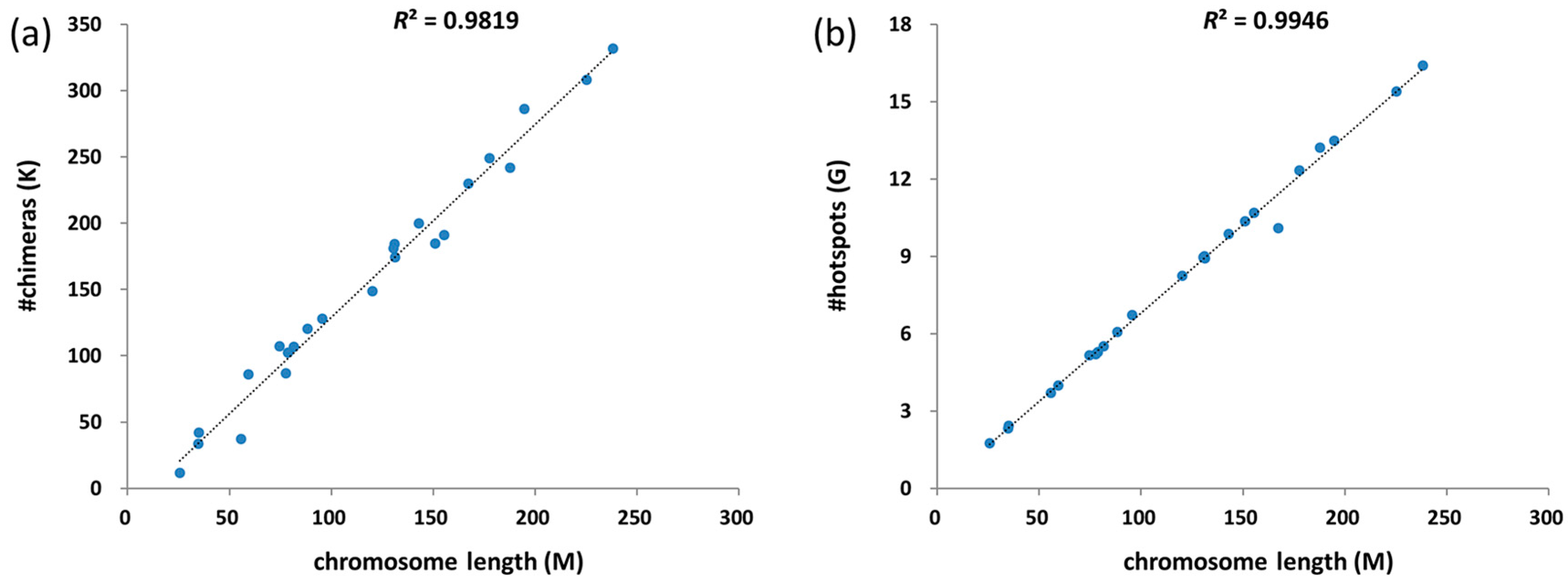
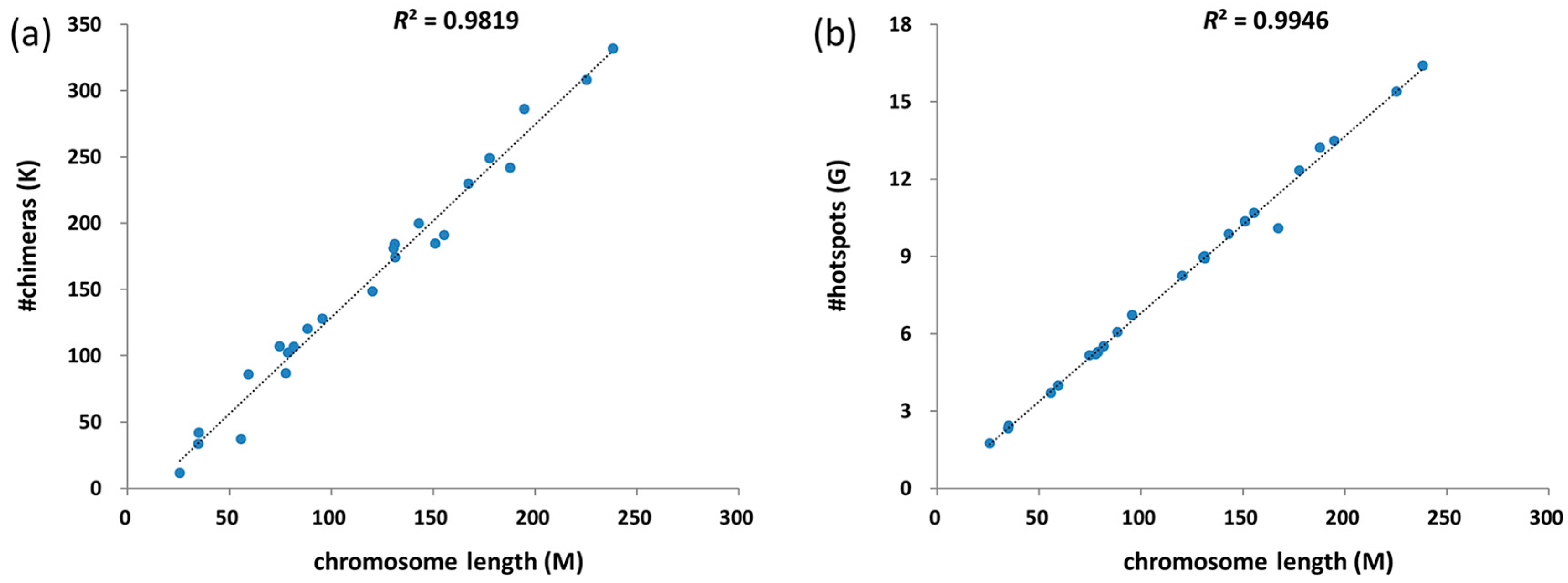
3.3. Chimeras and Hotspots Distribution in Chromosomes
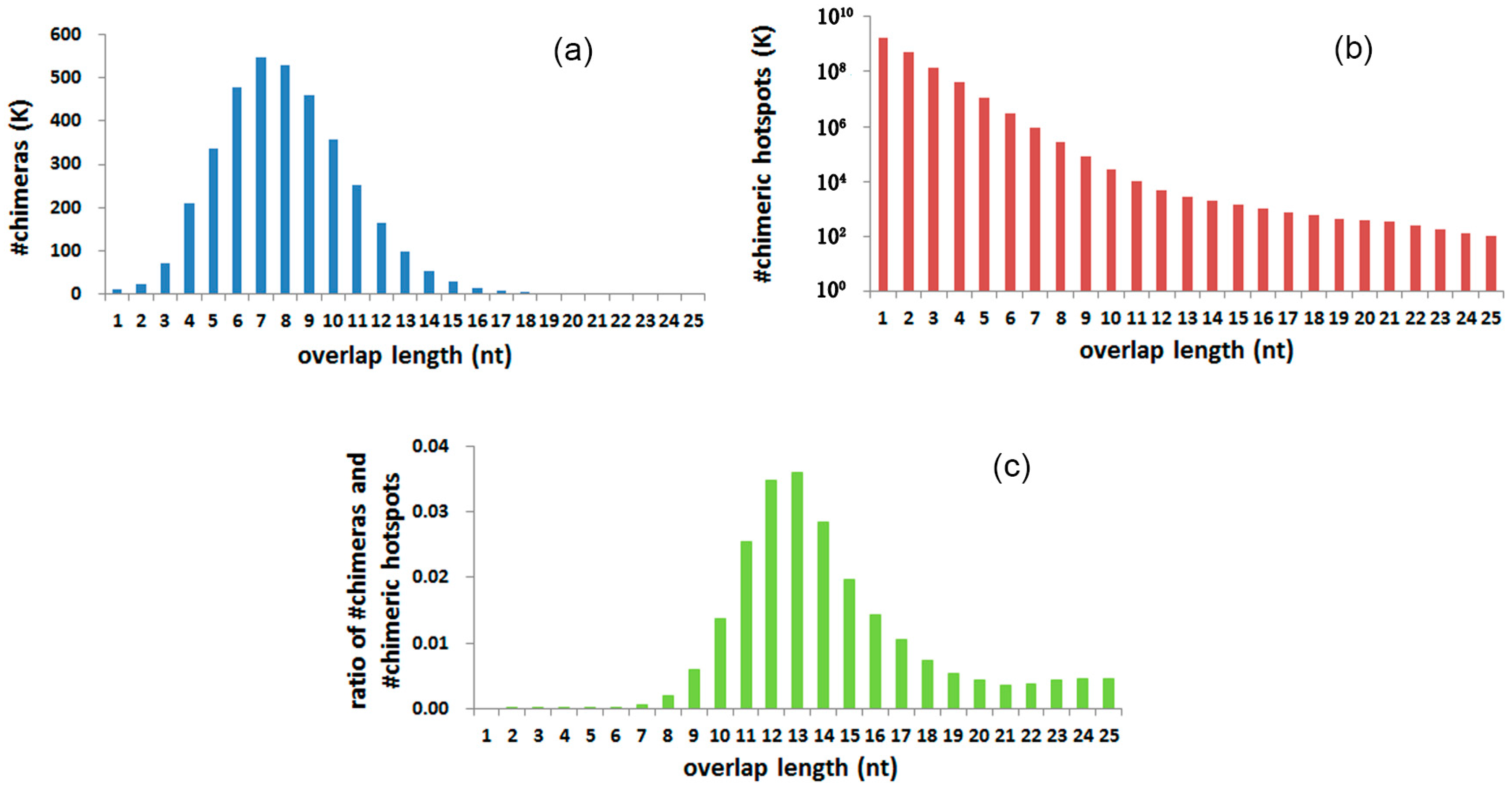
3.4. Chimeras and Hotspots Distribution in Overlap Length
3.5. GC Content and Td of Overlaps
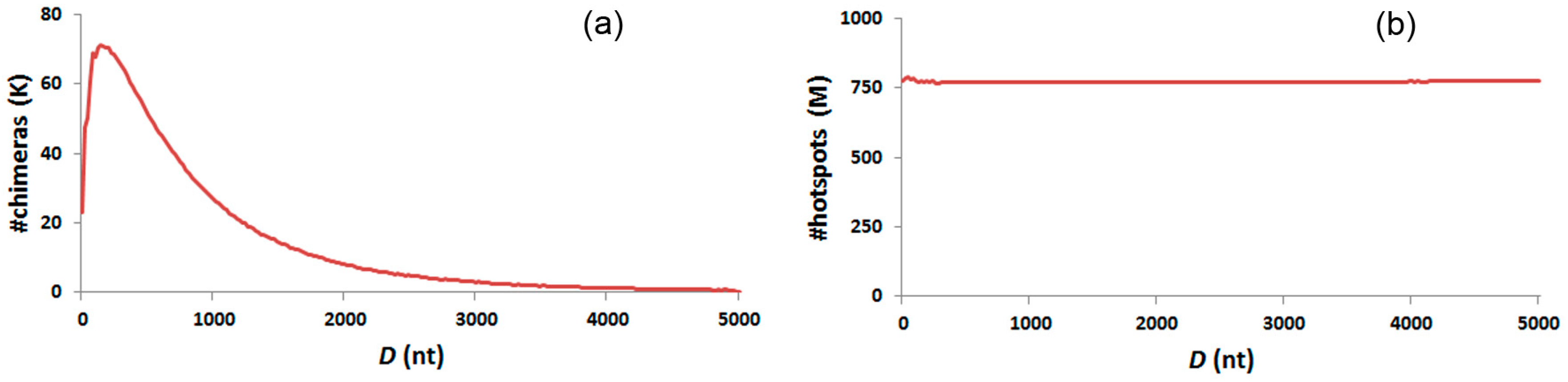
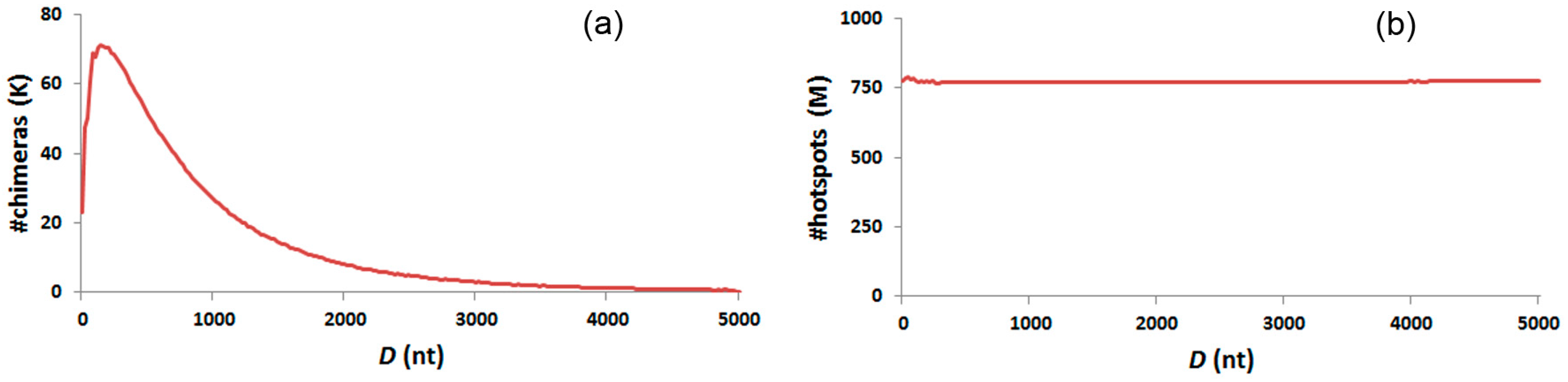
3.6. Distance Distribution
4. Discussion
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Dean, F.B.; Hosono, S.; Fang, L.; Wu, X.; Faruqi, A.F.; Bray-Ward, P.; Sun, Z.; Zong, Q.; Du, Y.; Du, J.; et al. Comprehensive human genome amplification using multiple displacement amplification. Proc. Natl. Acad. Sci. USA 2002, 99, 5261–5266. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Zong, C.; Fan, W.; Yang, M.; Li, J.; Chapman, A.R.; Zhu, P.; Hu, X.; Xu, L.; Yan, L.; et al. Probing meiotic recombination and aneuploidy of single sperm cells by whole-genome sequencing. Science 2012, 338, 1627–1630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirkness, E.F.; Grindberg, R.V.; Yee-Greenbaum, J.; Marshall, C.R.; Scherer, S.W.; Lasken, R.S.; Venter, J.C. Sequencing of isolated sperm cells for direct haplotyping of a human genome. Genome Res. 2013, 23, 826–832. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Fan, H.C.; Behr, B.; Quake, S.R. Genome-wide single-cell analysis of recombination activity and de novo mutation rates in human sperm. Cell 2012, 150, 402–412. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Fan, W.; Yan, L.; Li, R.; Lian, Y.; Huang, J.; Li, J.; Xu, L.; Tang, F.; Xie, X.S.; et al. Genome analyses of single human oocytes. Cell 2013, 155, 1492–1506. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Xiao, Y.; Huang, H.; Wang, Q.; Rao, W.; Feng, Y.; Zhang, K.; Song, Q. Direct determination of molecular haplotypes by chromosome microdissection. Nat. Methods 2010, 7, 299–301. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Chen, X.; Wong, W.H. Completely phased genome sequencing through chromosome sorting. Proc. Natl. Acad. Sci. USA 2011, 108, 12–17. [Google Scholar] [CrossRef] [PubMed]
- Peters, B.A.; Kermani, B.G.; Sparks, A.B.; Alferov, O.; Hong, P.; Alexeev, A.; Jiang, Y.; Dahl, F.; Tang, Y.T.; Haas, J.; et al. Accurate whole-genome sequencing and haplotyping from 10 to 20 human cells. Nature 2012, 487, 190–195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaper, F.; Swamy, S.; Klotzle, B.; Munchel, S.; Cottrell, J.; Bibikova, M.; Chuang, H.Y.; Kruglyak, S.; Ronaghi, M.; Eberle, M.A.; et al. Whole-genome haplotyping by dilution, amplification, and sequencing. Proc. Natl. Acad. Sci. USA 2013, 110, 5552–5557. [Google Scholar] [CrossRef] [PubMed]
- Paez, J.G.; Lin, M.; Beroukhim, R.; Lee, J.C.; Zhao, X.; Richter, D.J.; Gabriel, S.; Herman, P.; Sasaki, H.; Altshuler, D.; et al. Genome coverage and sequence fidelity of φ29 polymerase-based multiple strand displacement whole genome amplification. Nucleic Acids Res. 2004, 32, e71. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Martiny, A.C.; Reppas, N.B.; Barry, K.W.; Malek, J.; Chisholm, S.W.; Church, G.M. Sequencing genomes from single cells by polymerase cloning. Nat. Biotechnol. 2006, 24, 680–686. [Google Scholar] [CrossRef] [PubMed]
- Lasken, R.S.; Stockwell, T.B. Mechanism of chimera formation during the Multiple Displacement Amplification reaction. BMC Biotechnol. 2007, 7, 19. [Google Scholar] [CrossRef] [PubMed]
- Tu, J.; Guo, J.; Li, J.; Gao, S.; Yao, B.; Lu, Z. Systematic Characteristic Exploration of the Chimeras Generated in Multiple Displacement Amplification through Next Generation Sequencing Data Reanalysis. PLoS ONE 2015, 10, e0139857. [Google Scholar] [CrossRef] [PubMed]
- Evrony, G.D.; Lee, E.; Mehta, B.K.; Benjamini, Y.; Johnson, R.M.; Cai, X.; Yang, L.; Haseley, P.; Lehmann, H.S.; Park, P.J.; et al. Cell lineage analysis in human brain using endogenous retroelements. Neuron 2015, 85, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Jeffreys, A.J.; Kauppi, L.; Neumann, R. Intensely punctate meiotic recombination in the class II region of the major histocompatibility complex. Nat. Genet. 2001, 29, 217–222. [Google Scholar] [CrossRef] [PubMed]
- Winkelmann, A.; You, X.; Grunewald, N.; Haussler, U.; Krestel, H.; Haas, C.A.; Schwarz, G.; Chen, W.; Meier, J.C. Identification of a new genomic hot spot of evolutionary diversification of protein function. PLoS ONE 2015, 10, e0125413. [Google Scholar] [CrossRef] [PubMed]
- Meyer, L.R.; Zweig, A.S.; Hinrichs, A.S.; Karolchik, D.; Kuhn, R.M.; Wong, M.; Sloan, C.A.; Rosenbloom, K.R.; Roe, G.; Rhead, B.; et al. The UCSC Genome Browser database: Extensions and updates 2013. Nucleic Acids Res. 2013, 41, D64–D69. [Google Scholar] [CrossRef] [PubMed]
- Wallace, R.B.; Shaffer, J.; Murphy, R.F.; Bonner, J.; Hirose, T.; Itakura, K. Hybridization of synthetic oligodeoxyribonucleotides to φ χ 174 DNA: The effect of single base pair mismatch. Nucleic Acids Res. 1979, 6, 3543–3557. [Google Scholar] [CrossRef] [PubMed]
- Suggs, T.H.S.V.; Miyake, T.; Kawashima, E.H.; Johnson, M.J.; Itakura, K.; Wallace, R.B. Use of synthetic oligodeoxyribonucleotides for the isolation of specific cloned DNA sequences. Dev. Biol. Using Purif. Genes 1981, 1981, 11. [Google Scholar] [CrossRef]
- Dean, F.B.; Nelson, J.R.; Giesler, T.L.; Lasken, R.S. Rapid amplification of plasmid and phage DNA using φ29 DNA polymerase and multiply-primed rolling circle amplification. Genome Res. 2001, 11, 1095–1099. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.Z.; Adalsteinsson, V.A.; Francis, J.; Cornils, H.; Jung, J.; Maire, C.; Ligon, K.L.; Meyerson, M.; Love, J.C. Calibrating genomic and allelic coverage bias in single-cell sequencing. Nat. Commun. 2015, 6, 6822. [Google Scholar] [CrossRef] [PubMed]
- Geigl, J.B.; Obenauf, A.C.; Waldispuehl-Geigl, J.; Hoffmann, E.M.; Auer, M.; Hormann, M.; Fischer, M.; Trajanoski, Z.; Schenk, M.A.; Baumbusch, L.O.; et al. Identification of small gains and losses in single cells after whole genome amplification on tiling oligo arrays. Nucleic Acids Res. 2009, 37, e105. [Google Scholar] [CrossRef] [PubMed]
- Pinard, R.; de Winter, A.; Sarkis, G.J.; Gerstein, M.B.; Tartaro, K.R.; Plant, R.N.; Egholm, M.; Rothberg, J.M.; Leamon, J.H. Assessment of whole genome amplification-induced bias through high-throughput, massively parallel whole genome sequencing. BMC Genom. 2006, 7, 216. [Google Scholar] [CrossRef] [PubMed]
- Gole, J.; Gore, A.; Richards, A.; Chiu, Y.J.; Fung, H.L.; Bushman, D.; Chiang, H.I.; Chun, J.; Lo, Y.H.; Zhang, K. Massively parallel polymerase cloning and genome sequencing of single cells using nanoliter microwells. Nat. Biotechnol. 2013, 31, 1126–1132. [Google Scholar] [CrossRef] [PubMed]
- Picher, A.J.; Budeus, B.; Wafzig, O.; Kruger, C.; Garcia-Gomez, S.; Martinez-Jimenez, M.I.; Diaz-Talavera, A.; Weber, D.; Blanco, L.; Schneider, A. TruePrime is a novel method for whole-genome amplification from single cells based on TthPrimPol. Nat. Commun. 2016, 7, 13296. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, Y.; Hosokawa, M.; Maruyama, T.; Yamagishi, K.; Mori, T.; Takeyama, H. Monodisperse Picoliter Droplets for Low-Bias and Contamination-Free Reactions in Single-Cell Whole Genome Amplification. PLoS ONE 2015, 10, e0138733. [Google Scholar] [CrossRef] [PubMed]
- Rudner, R.; Karkas, J.D.; Chargaff, E. Separation of B. subtilis DNA into complementary strands. 3. Direct analysis. Proc. Natl. Acad. Sci. USA 1968, 60, 921–922. [Google Scholar] [CrossRef] [PubMed]
- Von Ahsen, N.; Wittwer, C.T.; Schutz, E. Oligonucleotide melting temperatures under PCR conditions: Nearest-neighbor corrections for Mg2+, deoxynucleotide triphosphate, and dimethyl sulfoxide concentrations with comparison to alternative empirical formulas. Clin. Chem. 2001, 47, 1956–1961. [Google Scholar] [PubMed]
- Wetmur, J.G. DNA probes: Applications of the principles of nucleic acid hybridization. Crit. Rev. Biochem. Mol. Biol. 1991, 26, 227–259. [Google Scholar] [CrossRef] [PubMed]
- Chavali, S.; Mahajan, A.; Tabassum, R.; Maiti, S.; Bharadwaj, D. Oligonucleotide properties determination and primer designing: A critical examination of predictions. Bioinformatics 2005, 21, 3918–3925. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Overlap Length (nt) | Number of Chimeras | Average GC Content | Td (°C) |
|---|---|---|---|
| 1 | 11,992 | 36.77% | 2.74 |
| 2 | 22,292 | 44.40% | 5.78 |
| 3 | 71,262 | 49.88% | 8.99 |
| 4 | 208,729 | 45.31% | 11.62 |
| 5 | 336,932 | 44.26% | 14.43 |
| 6 | 476,409 | 42.84% | 17.14 |
| 7 | 547,136 | 41.82% | 19.86 |
| 8 | 528,555 | 40.53% | 22.49 |
| 9 | 459,454 | 39.19% | 25.05 |
| 10 | 356,470 | 37.82% | 27.56 |
| 11 | 253,356 | 36.17% | 29.96 |
| 12 | 163,502 | 34.35% | 32.24 |
| 13 | 97,992 | 32.71% | 34.51 |
| 14 | 54,711 | 31.08% | 36.70 |
| 15 | 28,218 | 29.93% | 38.98 |
| 16 | 14,630 | 28.59% | 41.15 |
| 17 | 7429 | 28.05% | 43.54 |
| 18 | 4189 | 29.79% | 46.73 |
| 19 | 2310 | 32.13% | 50.21 |
| 20 | 1661 | 33.99% | 53.60 |
| 21 | 1200 | 35.80% | 57.04 |
| 22 | 932 | 37.78% | 60.62 |
| 23 | 740 | 40.11% | 64.45 |
| 24 | 587 | 38.96% | 66.70 |
| 25 | 480 | 40.10% | 70.05 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tu, J.; Lu, N.; Duan, M.; Huang, M.; Chen, L.; Li, J.; Guo, J.; Lu, Z. Hotspot Selective Preference of the Chimeric Sequences Formed in Multiple Displacement Amplification. Int. J. Mol. Sci. 2017, 18, 492. https://doi.org/10.3390/ijms18030492
Tu J, Lu N, Duan M, Huang M, Chen L, Li J, Guo J, Lu Z. Hotspot Selective Preference of the Chimeric Sequences Formed in Multiple Displacement Amplification. International Journal of Molecular Sciences. 2017; 18(3):492. https://doi.org/10.3390/ijms18030492
Chicago/Turabian StyleTu, Jing, Na Lu, Mengqin Duan, Mengting Huang, Liang Chen, Junji Li, Jing Guo, and Zuhong Lu. 2017. "Hotspot Selective Preference of the Chimeric Sequences Formed in Multiple Displacement Amplification" International Journal of Molecular Sciences 18, no. 3: 492. https://doi.org/10.3390/ijms18030492