
SCF/C-Kit/JNK/AP-1 Signaling Pathway Promotes Claudin-3 Expression in Colonic Epithelium and Colorectal Carcinoma

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
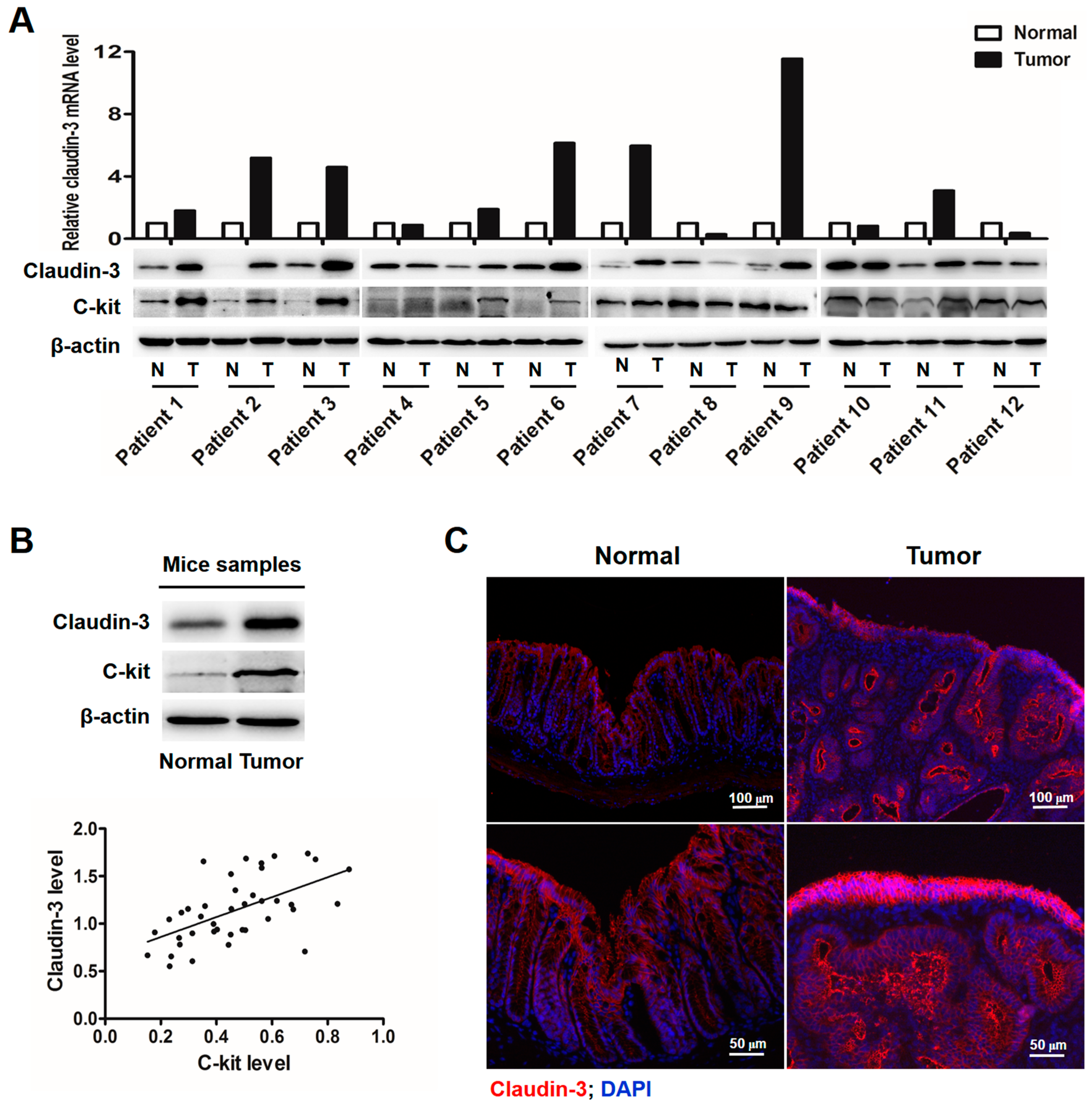
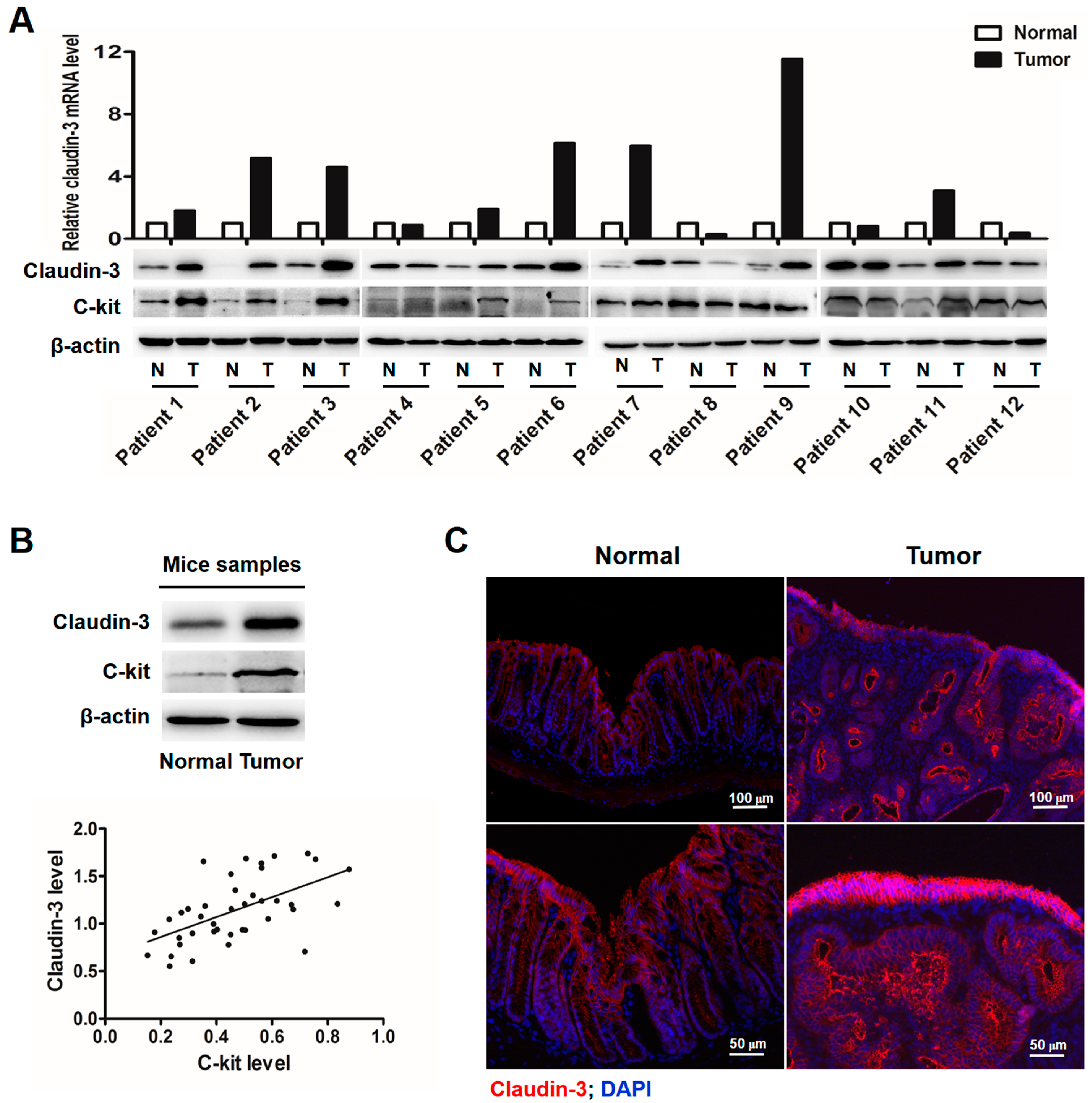
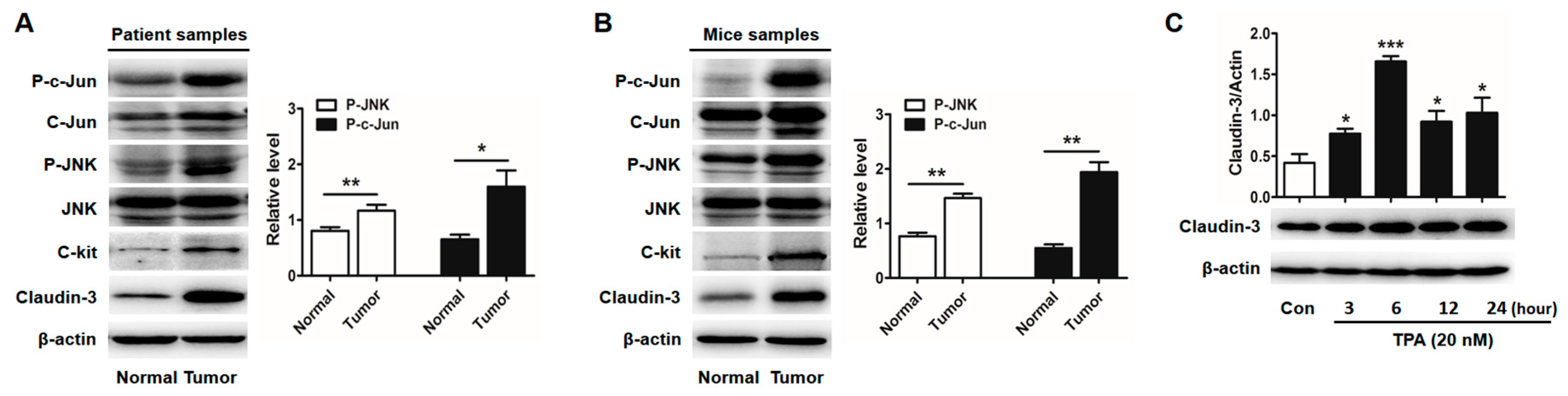
2.1. Clauin-3 Level Is Positively Correlated with C-Kit Expression in Colorectal Cancer (CRC) Tissues
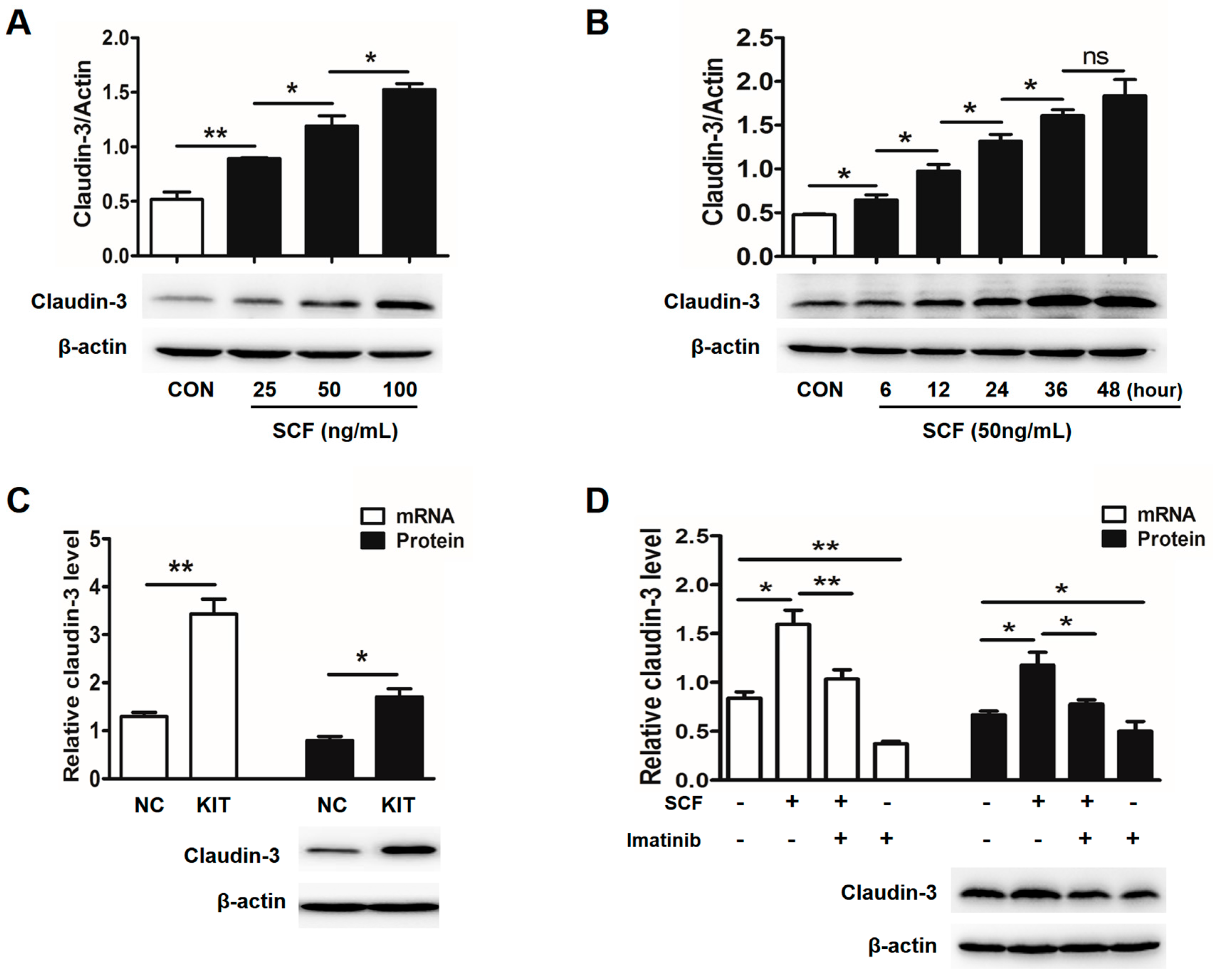
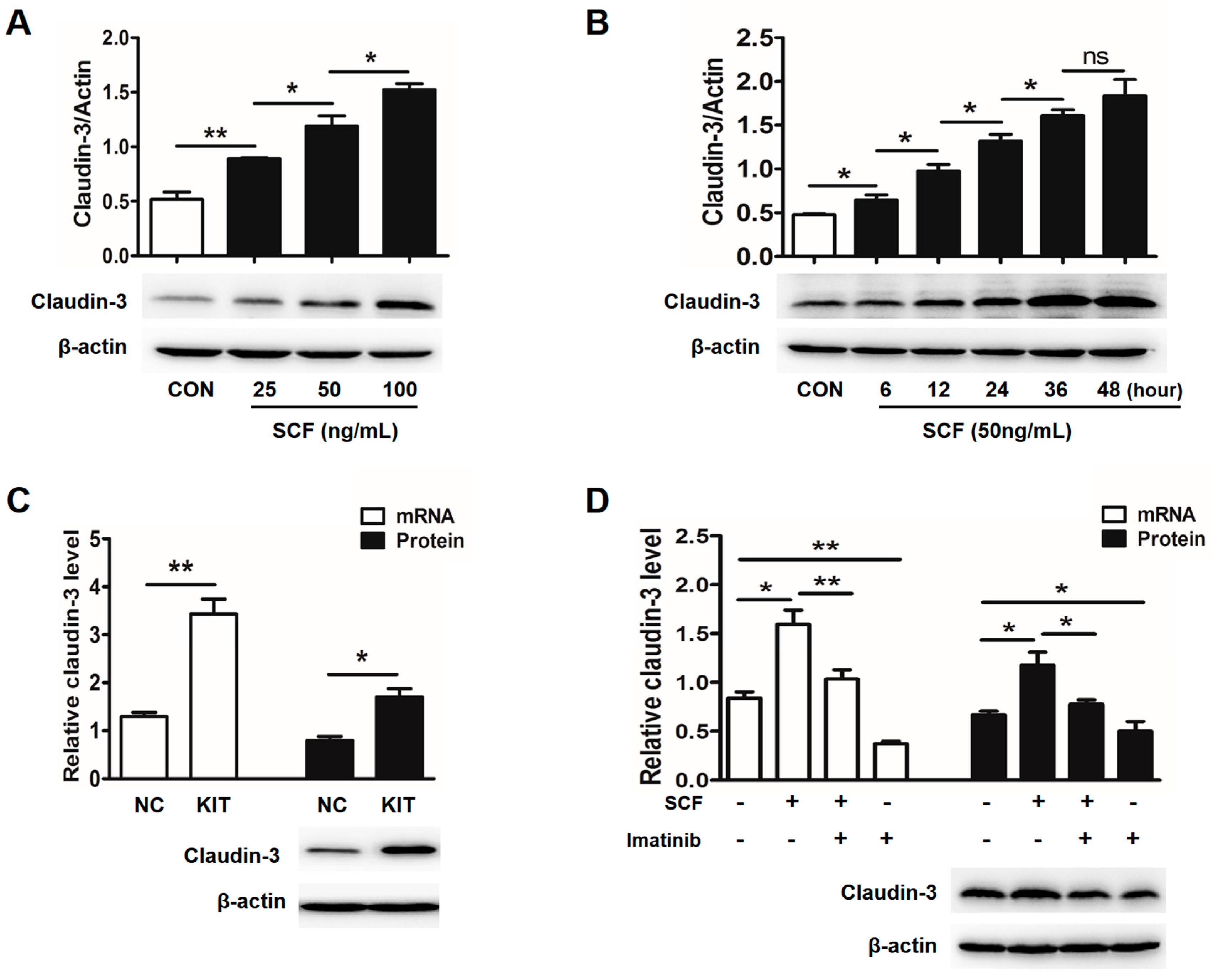
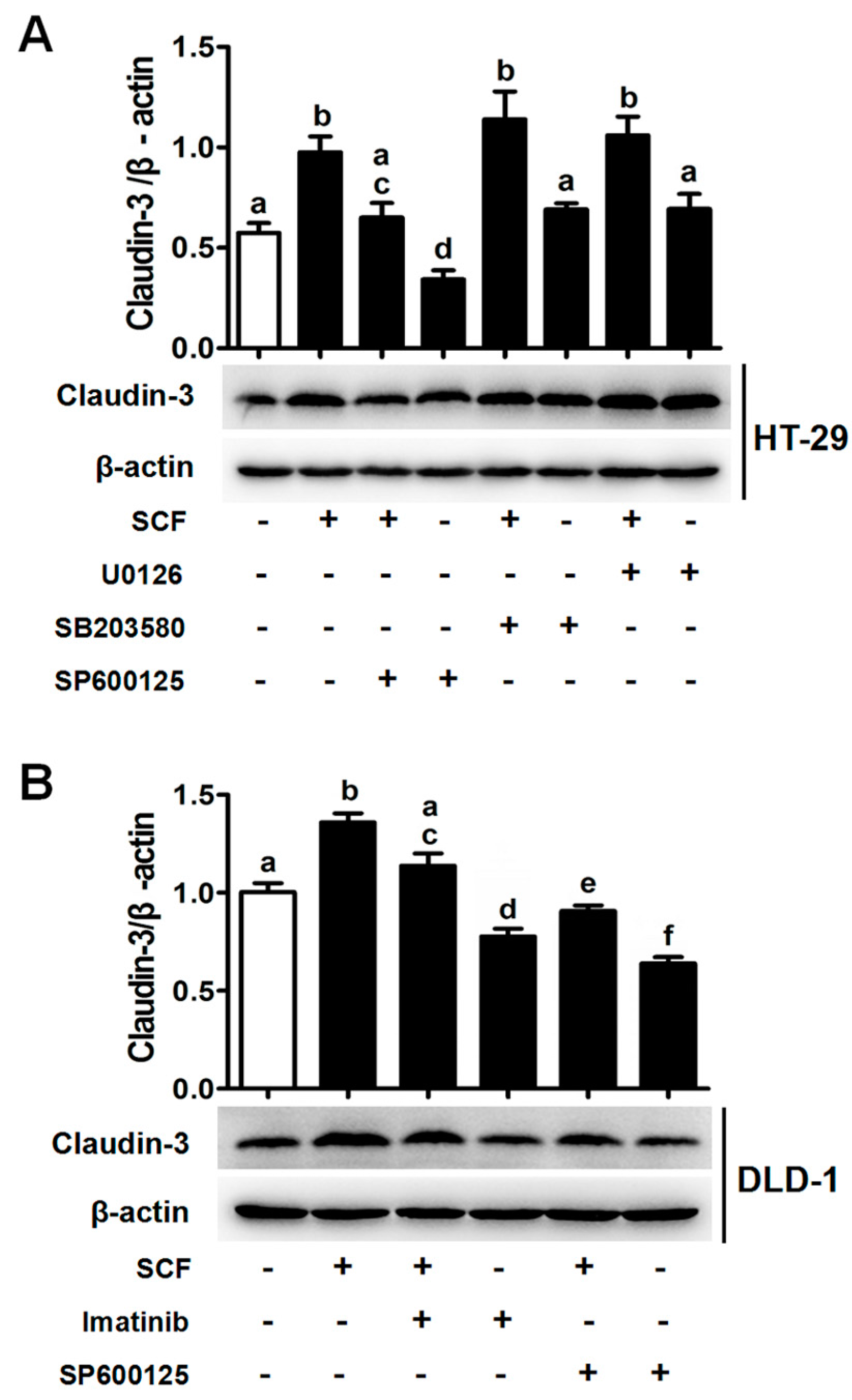
2.2. Stem Cell Factor (SCF)/C-Kit Signaling Significantly Increases Claudin-3 Expression
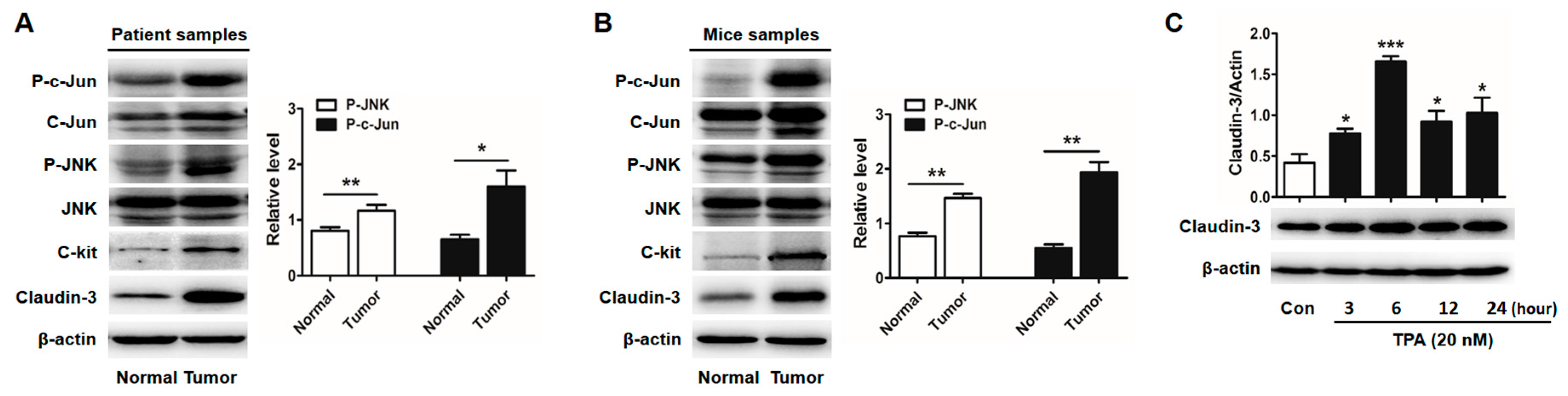
2.3. Activator Protein-1 (AP-1) Is Activated by c-Jun N-Terminal Kinase (JNK) in CRC Tissues
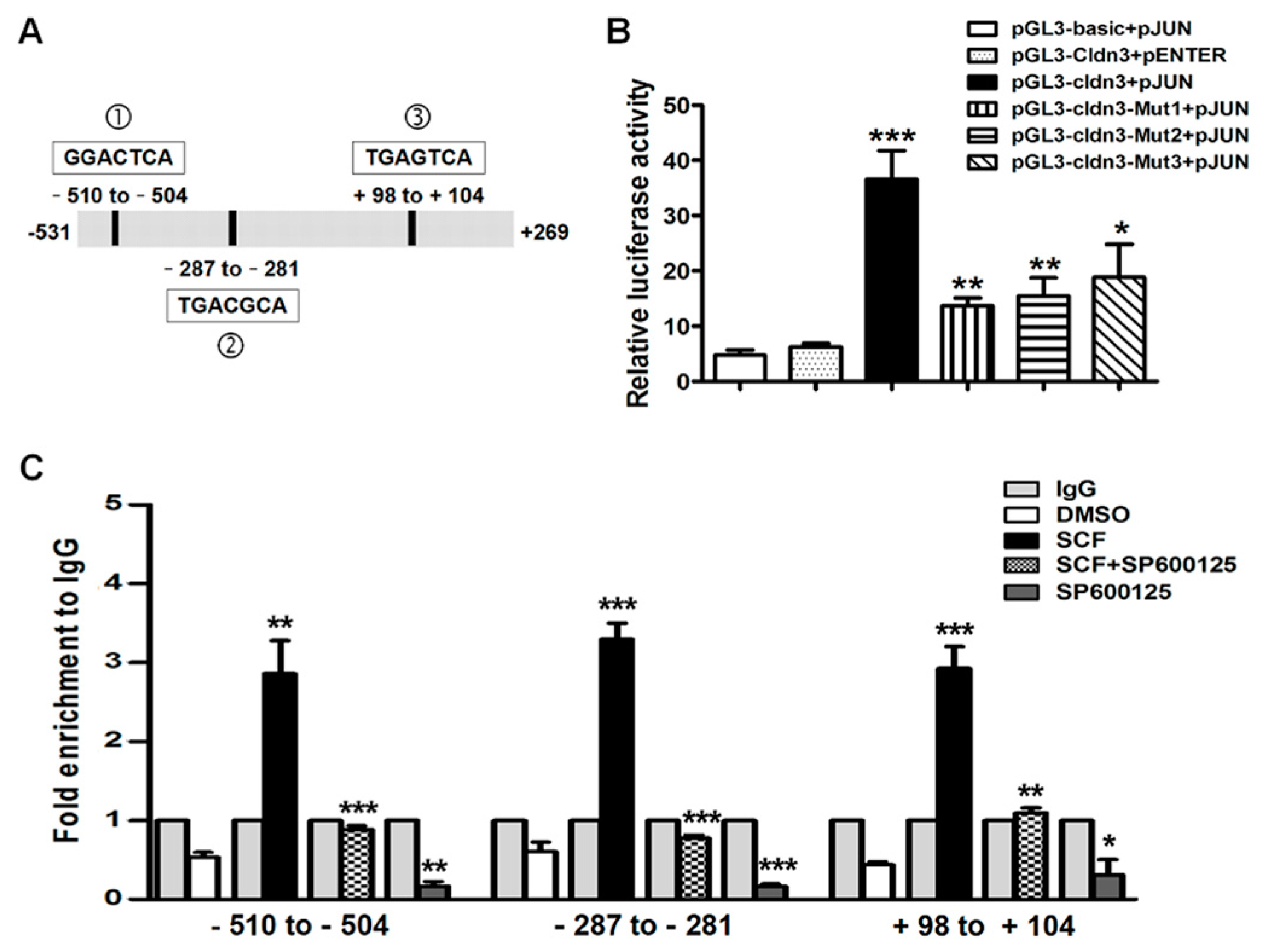
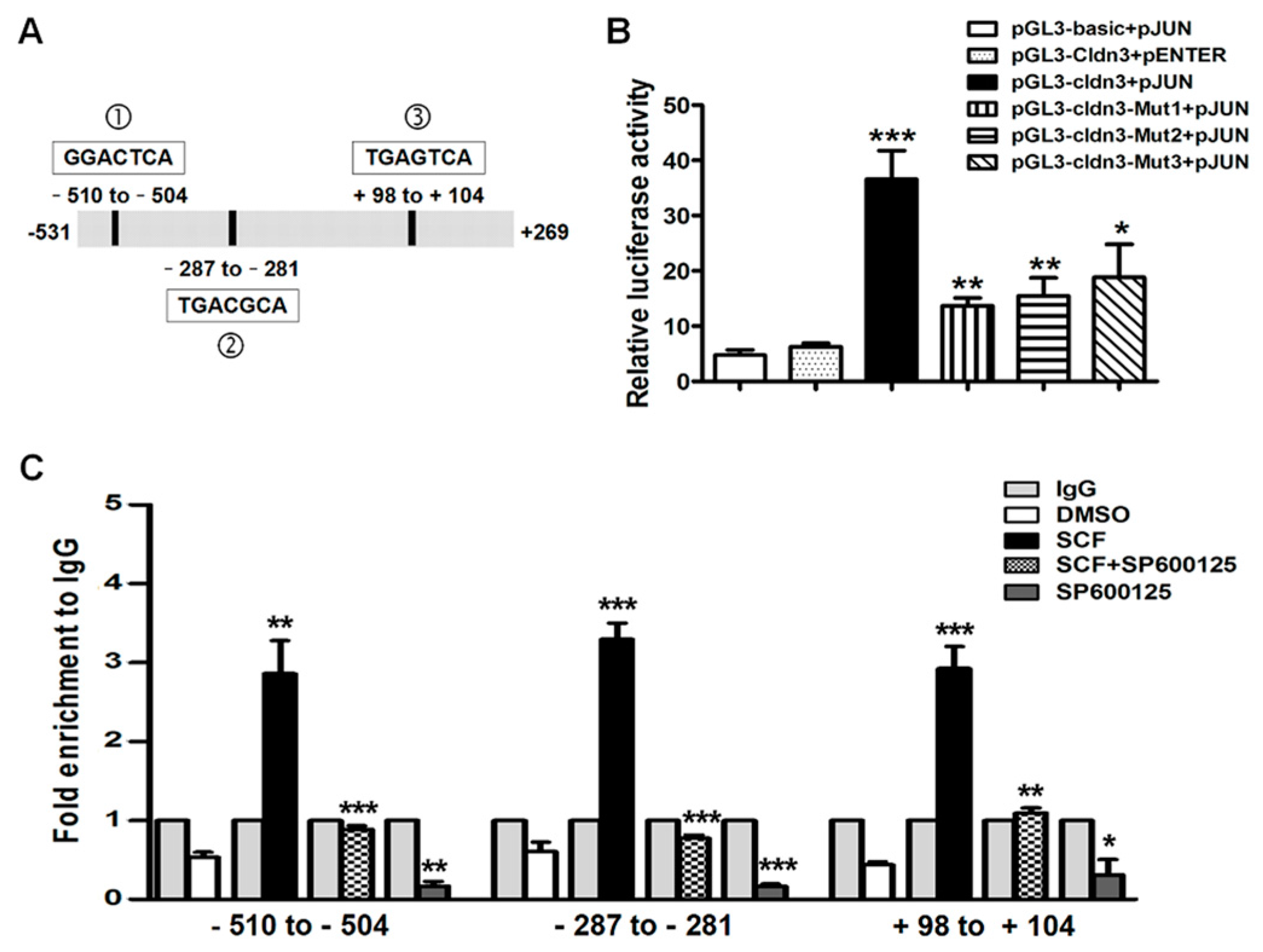
2.4. AP-1/c-Jun Enhances CLDN-3 Promoter Activity
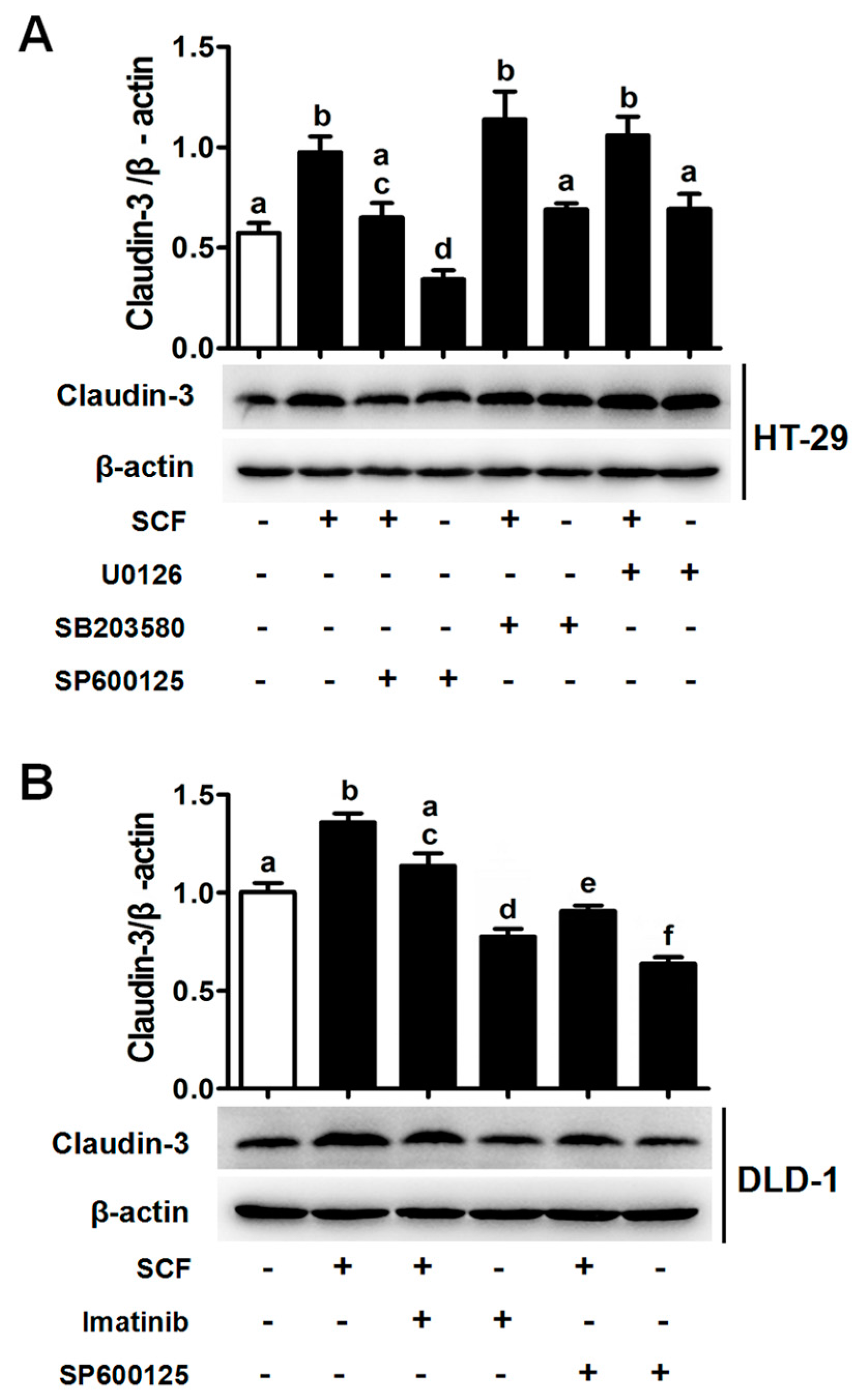
2.5. SCF/C-Kit/JNK Signaling Promotes the Binding of AP-1/c-Jun with CLDN-3 Promoter
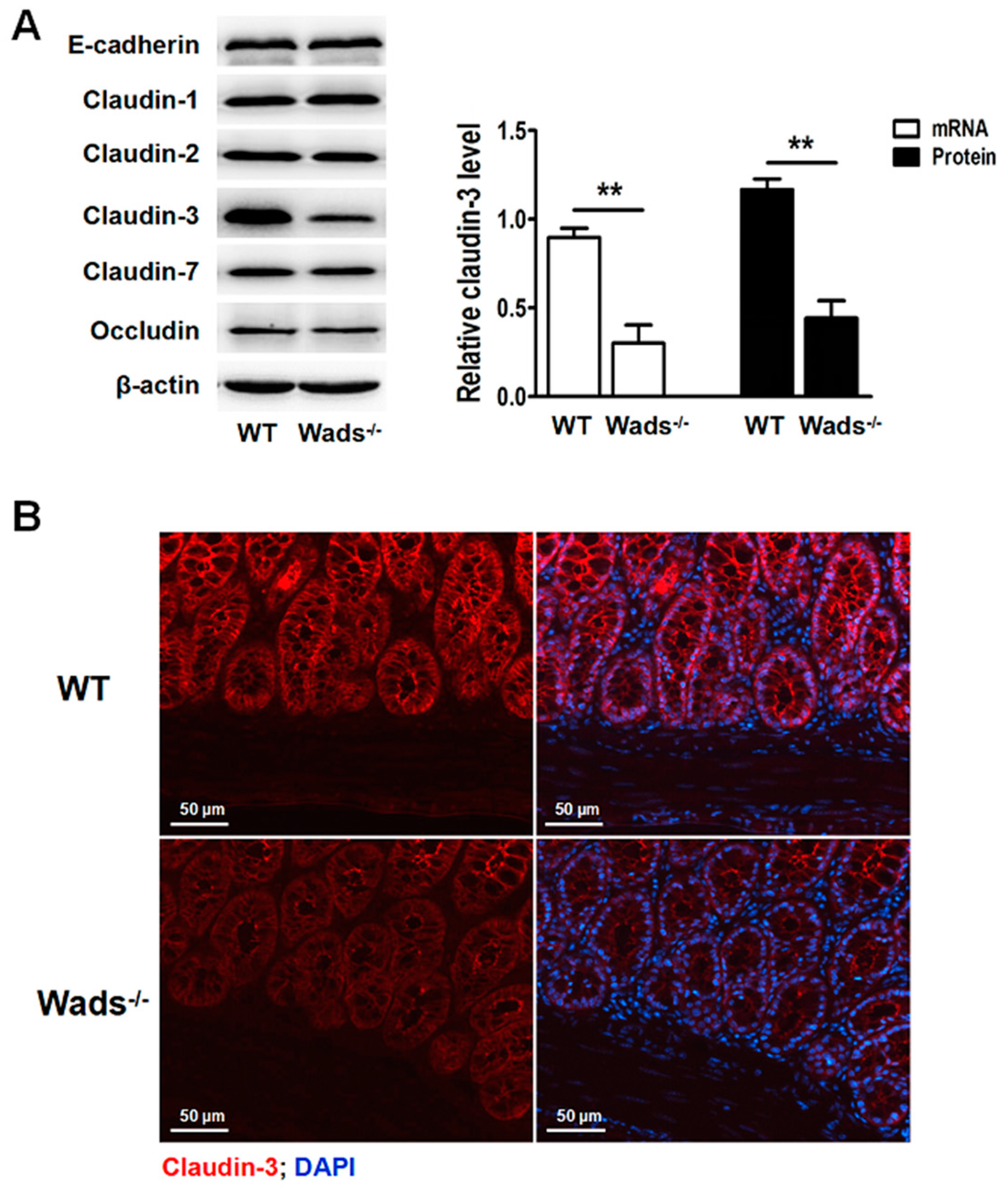
2.6. Claudin-3 Decreases in Colonic Mucosa of c-Kit Loss-of-Function Mutant Mice
3. Discussion
4. Materials and Methods
4.1. Ethics Statement
4.2. Patient Samples
4.3. Establishment of CRC Murine Model
4.4. RNA Exraction and Real-Time PCR
4.5. Western Blot
4.6. Immunofluorescence Staining
4.7. Cell Culture
4.8. Lentiviral Vector Construction and Infection
4.9. Construction of Plasmids
4.10. Luciferase Reporter Assay
4.11. Chromatin Immunoprecipitation (ChIP)
4.12. Animals
4.13. Statistics
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| TJs | Tight junctions |
| CRC | Colorectal cancer |
| JNK | C-Jun N-terminal kinase |
| ChIP | Chromatin immunoprecipitation |
| AP-1 | Activator protein-1 |
| RTK | Receptor tyrosine kinases |
| EGFR | Epidermal growth factor receptor |
| MAPK | Mitogen-activated protein kinase |
| SCF | Stem cell factor |
| rhSCF | Recombinant human SCF |
| ERK | Extracellular signal-regulated kinases |
| IGF-1 | Insulin-like growth factor 1 |
| VEGFR | Vascular endothelial growth factor |
References
- Singh, A.B.; Dhawan, P. Claudins and cancer: Fall of the soldiers entrusted to protect the gate and keep the barrier intact. Semin. Cell Dev. Biol. 2015, 42, 58–65. [Google Scholar] [CrossRef] [PubMed]
- Cereijido, M.; Contreras, R.G.; Shoshani, L. Cell Adhesion, Polarity, and Epithelia in the Dawn of Metazoans. Physiol. Rev. 2004, 84, 1229–1262. [Google Scholar] [CrossRef] [PubMed]
- Lal-Nag, M.; Morin, P.J. The claudins. Genome Biol. 2009, 10, 235. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Blanco, J.A.; Estevez, J.; Shea-Donohue, T.; Martinez, V.; Vergara, P. Changes in Epithelial Barrier Function in Response to Parasitic Infection: Implications for IBD Pathogenesis. J. Crohns Colitis 2015, 9, 463–476. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.L.; Kim, J.; Kwon, M.J.; Choi, J.S.; Kim, T.J.; Bae, D.S.; Koh, S.S.; In, Y.H.; Park, Y.W.; Kim, S.H.; et al. Expression profile of tight junction protein claudin-3 and claudin-4 in ovarian serous adenocarcinoma with prognostic correlation. Histol. Histopathol 2007, 22, 1185–1195. [Google Scholar] [PubMed]
- Cocco, E.; Casagrande, F.; Bellone, S.; Richter, C.E.; Bellone, M.; Todeschini, P.; Holmberg, J.C.; Fu, H.H.; Montagna, M.K.; Mor, G.; et al. Clostridium perfringens enterotoxin carboxy-terminal fragment is a novel tumor-homing peptide for human ovarian cancer. BMC Cancer 2010, 10, 349. [Google Scholar] [CrossRef] [PubMed]
- Kominsky, S.L.; Vali, M.; Korz, D.; Gabig, T.G.; Weitzman, S.A.; Argani, P.; Sukumar, S. Clostridium perfringens enterotoxin elicits rapid and specific cytolysis of breast carcinoma cells mediated through tight junction proteins claudin-3 and -4. Am. J. Pathol. 2004, 164, 1627–1633. [Google Scholar] [CrossRef]
- Long, H.; Crean, C.D.; Lee, W.H.; Cummings, O.W.; Gabig, T.G. Expression of Clostridium perfringens enterotoxin receptors claudin-3 and claudin-4 in prostate cancer epithelium. Cancer Res. 2001, 61, 7878–7881. [Google Scholar] [PubMed]
- Montgomery, E.; Mamelak, A.J.; Gibson, M.; Maitra, A.; Sheikh, S.; Amr, S.S.; Yang, S.; Brock, M.; Forastiere, A.; Zhang, S.; et al. Overexpression of claudin proteins in esophageal adenocarcinoma and its precursor lesions. Appl. Immunohistochem. Mol. Morphol. 2006, 14, 24–30. [Google Scholar] [CrossRef] [PubMed]
- Eftang, L.L.; Esbensen, Y.; Tannaes, T.M.; Blom, G.P.; Bukholm, I.R.; Bukholm, G. Up-regulation of CLDN1 in gastric cancer is correlated with reduced survival. BMC Cancer 2013, 13, 586. [Google Scholar] [CrossRef] [PubMed]
- Kinugasa, T.; Huo, Q.; Higashi, D.; Shibaguchi, H.; Kuroki, M.; Tanaka, T.; Futami, K.; Yamashita, Y.; Hachimine, K.; Maekawa, S.; et al. Selective up-regulation of claudin-1 and claudin-2 in colorectal cancer. Anticancer Res. 2007, 27, 3729–3734. [Google Scholar] [CrossRef]
- Tabaries, S.; Siegel, P.M. The role of claudins in cancer metastasis. Oncogene 2017, 36, 1176–1190. [Google Scholar] [CrossRef] [PubMed]
- Dhawan, P.; Ahmad, R.; Chaturvedi, R.; Smith, J.J.; Midha, R.; Mittal, M.K.; Krishnan, M.; Chen, X.; Eschrich, S.; Yeatman, T.J.; et al. Claudin-2 expression increases tumorigenicity of colon cancer cells: Role of epidermal growth factor receptor activation. Oncogene 2011, 30, 3234–3247. [Google Scholar] [CrossRef] [PubMed]
- Ikari, A.; Sato, T.; Watanabe, R.; Yamazaki, Y.; Sugatani, J. Increase in claudin-2 expression by an EGFR/MEK/ERK/c-Fos pathway in lung adenocarcinoma A549 cells. Biochim. Biophys. Acta 2012, 1823, 1110–1118. [Google Scholar] [CrossRef] [PubMed]
- Fujii, N.; Matsuo, Y.; Matsunaga, T.; Endo, S.; Sakai, H.; Yamaguchi, M.; Yamazaki, Y.; Sugatani, J.; Ikari, A. Hypotonic Stress-induced Down-regulation of Claudin-1 and -2 Mediated by Dephosphorylation and Clathrin-dependent Endocytosis in Renal Tubular Epithelial Cells. J. Biol. Chem. 2016, 291, 24787–24799. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.; Breton, S. The MAPK/ERK-Signaling pathway regulates the expression and distribution of tight junction proteins in the mouse proximal epididymis. Biol. Reprod. 2016, 94, 22. [Google Scholar] [CrossRef] [PubMed]
- Ikari, A.; Atomi, K.; Yamazaki, Y.; Sakai, H.; Hayashi, H.; Yamaguchi, M.; Sugatani, J. Hyperosmolarity-induced up-regulation of claudin-4 mediated by NADPH oxidase-dependent H2O2 production and Sp1/c-Jun cooperation. Biochim. Biophys. Acta 2013, 1833, 2617–2627. [Google Scholar] [CrossRef] [PubMed]
- Bellone, G.; Smirne, C.; Carbone, A.; Buffolino, A.; Scirelli, T.; Prati, A.; Solerio, D.; Pirisi, M.; Valente, G.; Nano, M.; et al. KIT/stem cell factor expression in premalignant and malignant lesions of the colon mucosa in relationship to disease progression and outcomes. Int. J. Oncol. 2006, 29, 851–859. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.S.; Chen, K.T.; Fan, C.W.; Han, C.L.; Chen, Y.J.; Yu, J.S.; Chang, Y.S.; Chien, C.W.; Wu, C.P.; Hung, R.P.; et al. Comparison of membrane fraction proteomic profiles of normal and cancerous human colorectal tissues with gel-assisted digestion and iTRAQ labeling mass spectrometry. FEBS J. 2010, 277, 3028–3038. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.; Yang, S.; Shen, P.; Sun, H.; Xiao, J.; Wang, Y.; Wu, B.; Ji, F.; Yan, J.; Xue, H.; et al. C-kit signaling promotes proliferation and invasion of colorectal mucinous adenocarcinoma in a murine model. Oncotarget 2015, 6, 27037–27048. [Google Scholar] [CrossRef] [PubMed]
- De Souza, W.F.; Fortunato-Miranda, N.; Robbs, B.K.; de Araujo, W.M.; de-Freitas-Junior, J.C.; Bastos, L.G.; Viola, J.P.; Morgado-Diaz, J.A. Claudin-3 overexpression increases the malignant potential of colorectal cancer cells: Roles of ERK1/2 and PI3K-Akt as modulators of EGFR signaling. PLoS ONE 2013, 8, e74994. [Google Scholar] [CrossRef] [PubMed]
- Ashida, R.; Tominaga, K.; Sasaki, E.; Watanabe, T.; Fujiwara, Y.; Oshitani, N.; Higuchi, K.; Mitsuyama, S.; Iwao, H.; Arakawa, T. AP-1 and colorectal cancer. Inflammopharmacology 2005, 13, 113–125. [Google Scholar] [CrossRef] [PubMed]
- Rauhavirta, T.; Lindfors, K.; Koskinen, O.; Laurila, K.; Kurppa, K.; Saavalainen, P.; Maki, M.; Collin, P.; Kaukinen, K. Impaired epithelial integrity in the duodenal mucosa in early stages of celiac disease. Transl. Res. 2014, 164, 223–231. [Google Scholar] [CrossRef] [PubMed]
- Komljenovic, D.; Sandhoff, R.; Teigler, A.; Heid, H.; Just, W.W.; Gorgas, K. Disruption of blood-testis barrier dynamics in ether-lipid-deficient mice. Cell Tissue Res. 2009, 337, 281–299. [Google Scholar] [CrossRef] [PubMed]
- Morin, P.J. Claudin proteins in human cancer: Promising new targets for diagnosis and therapy. Cancer Res. 2005, 65, 9603–9606. [Google Scholar] [CrossRef] [PubMed]
- De Oliveira, S.S.; de Oliveira, I.M.; de Souza, W.; Morgado-Diaz, J.A. Claudins upregulation in human colorectal cancer. FEBS Lett. 2005, 579, 6179–6185. [Google Scholar] [CrossRef] [PubMed]
- Kwon, M.J.; Kim, S.S.; Choi, Y.L.; Jung, H.S.; Balch, C.; Kim, S.H.; Song, Y.S.; Marquez, V.E.; Nephew, K.P.; Shin, Y.K. Derepression of CLDN3 and CLDN4 during ovarian tumorigenesis is associated with loss of repressive histone modifications. Carcinogenesis 2010, 31, 974–983. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.B.; Harris, R.C. Epidermal growth factor receptor activation differentially regulates claudin expression and enhances transepithelial resistance in Madin-Darby canine kidney cells. J. Biol. Chem. 2004, 279, 3543–3552. [Google Scholar] [CrossRef] [PubMed]
- Hatakeyama, N.; Kojima, T.; Iba, K.; Murata, M.; Thi, M.M.; Spray, D.C.; Osanai, M.; Chiba, H.; Ishiai, S.; Yamashita, T.; Sawada, N. IGF-I regulates tight-junction protein claudin-1 during differentiation of osteoblast-like MC3T3-E1 cells via a MAP-kinase pathway. Cell Tissue Res. 2008, 334, 243–254. [Google Scholar] [CrossRef] [PubMed]
- Laakkonen, J.P.; Lappalainen, J.P.; Theelen, T.L.; Toivanen, P.I.; Nieminen, T.; Jauhiainen, S.; Kaikkonen, M.U.; Sluimer, J.C.; Yla-Herttuala, S. Differential regulation of angiogenic cellular processes and claudin-5 by histamine and VEGF via PI3K-signaling, transcription factor SNAI2 and interleukin-8. Angiogenesis 2017, 20, 109–124. [Google Scholar] [CrossRef] [PubMed]
- Wisdom, R.; Johnson, R.S.; Moore, C. C-Jun regulates cell cycle progression and apoptosis by distinct mechanisms. EMBO J. 1999, 18, 188–197. [Google Scholar] [CrossRef] [PubMed]
- Ikari, A.; Watanabe, R.; Sato, T.; Taga, S.; Shimobaba, S.; Yamaguchi, M.; Yamazaki, Y.; Endo, S.; Matsunaga, T.; Sugatani, J. Nuclear distribution of claudin-2 increases cell proliferation in human lung adenocarcinoma cells. Biochim. Biophys. Acta 2014, 1843, 2079–2088. [Google Scholar] [CrossRef] [PubMed]
- Van Itallie, C.M.; Tietgens, A.J.; Anderson, J.M. Visualizing the dynamic coupling of claudin strands to the actin cytoskeleton through ZO-1. Mol. Biol. Cell 2017, 28, 524–534. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Wu, B.; Sun, H.; Ji, F.; Sun, T.; Zhao, Y.; Zhou, D. Interrupted E2F1-miR-34c-SCF negative feedback loop by hyper-methylation promotes colorectal cancer cell proliferation. Biosci. Rep. 2015, 36, e00293. [Google Scholar] [CrossRef] [PubMed]

© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Sun, T.; Sun, H.; Yang, S.; Li, D.; Zhou, D. SCF/C-Kit/JNK/AP-1 Signaling Pathway Promotes Claudin-3 Expression in Colonic Epithelium and Colorectal Carcinoma. Int. J. Mol. Sci. 2017, 18, 765. https://doi.org/10.3390/ijms18040765
Wang Y, Sun T, Sun H, Yang S, Li D, Zhou D. SCF/C-Kit/JNK/AP-1 Signaling Pathway Promotes Claudin-3 Expression in Colonic Epithelium and Colorectal Carcinoma. International Journal of Molecular Sciences. 2017; 18(4):765. https://doi.org/10.3390/ijms18040765
Chicago/Turabian StyleWang, Yaxi, Tingyi Sun, Haimei Sun, Shu Yang, Dandan Li, and Deshan Zhou. 2017. "SCF/C-Kit/JNK/AP-1 Signaling Pathway Promotes Claudin-3 Expression in Colonic Epithelium and Colorectal Carcinoma" International Journal of Molecular Sciences 18, no. 4: 765. https://doi.org/10.3390/ijms18040765