
Interplay between Oxidative Stress and Nutrient Sensing Signaling in the Developmental Origins of Cardiovascular Disease



1
Department of Pediatrics, Kaohsiung Chang Gung Memorial Hospital and Chang Gung University College of Medicine, Kaohsiung 833, Taiwan
2
Institute for Translational Research in Biomedicine, Kaohsiung Chang Gung Memorial Hospital and Chang Gung University College of Medicine, Kaohsiung 833, Taiwan
3
Department of Pharmacy, Kaohsiung Chang Gung Memorial Hospital, Kaohsiung 833, Taiwan
4
School of Pharmacy, Kaohsiung Medical University, Kaohsiung 807, Taiwan
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2017, 18(4), 841; https://doi.org/10.3390/ijms18040841
Submission received: 4 March 2017
/
Revised: 4 April 2017
/
Accepted: 13 April 2017
/
Published: 15 April 2017
(This article belongs to the Special Issue Correlation between Nutrition, Oxidative Stress and Disease)
Abstract
:Cardiovascular disease (CVD) presents a global health burden, despite recent advances in management. CVD can originate from early life by so-called “developmental origins of health and disease” (DOHaD). Epidemiological and experimental evidence supports that early-life insults can induce programming of later CVD. Underlying the DOHaD concept, early intervention may offset programming process to prevent the development of CVD, namely reprogramming. Oxidative stress and nutrient sensing signals have been considered to be major mechanisms of cardiovascular programming, while the interplay between these two mechanisms have not been examined in detail. This review summarizes current evidence that supports the link between oxidative stress and nutrient sensing signaling to cardiovascular programming, with an emphasis on the l-arginine–asymmetric dimethylarginine (ADMA)–nitric oxide (NO) pathway. This review provides an overview of evidence from human studies supporting fetal programming of CVD, insight from animal models of cardiovascular programming and oxidative stress, impact of the l-arginine–ADMA–NO pathway in cardiovascular programming, the crosstalk between l-arginine metabolism and nutrient sensing signals, and application of reprogramming interventions to prevent the programming of CVD. A greater understanding of the mechanisms underlying cardiovascular programming is essential to developing early reprogramming interventions to combat the globally growing epidemic of CVD.

1. Introduction
Cardiovascular disease (CVD) remains a crucial challenge for public health. Despite advances in medical and surgical treatment, CVD still accounts for most noncommunicable disease (NCD) deaths all over the world [1]. CVD can originate from early life, referred to as the developmental origins of health and disease (DOHaD) [2]. A growing body of epidemiological and experimental evidence supports that adverse environments on pregnancy (including nutrition) lead to permanent alterations of function and structure in specific organs that are vulnerable to develop CVD in later life, namely cardiovascular programming [3,4,5]. On the other hand, the DOHaD concept opens a new window in a way that helps to offset the programming process in early life in order to prevent the development of CVD in the later lifespan, through so-called reprogramming [3,6].
So far, a number of hypothetical mechanisms, including oxidative stress [7,8], dysregulation of nutrient-sensing signals [9], activation of renin-angiotensin system (RAS) [10], low nephron endowment [11], glucocorticoid effect [12], epigenetic regulation [13], and sex differences [4,5], have been examined in relation to cardiovascular programming and its comorbid illness [3,4,5]. For details, please refer to previous reviews [3,4,5,6,7,8,9,10,11,12,13]. Although oxidative stress and nutrient sensing signals have been extensively studied individually, the emerging link between these two mechanisms in cardiovascular programming have not been studied in detail.
Oxidative stress is an oxidative shift characterized by an imbalance between pro-oxidant molecules and antioxidant defenses, mainly related to dysregulation of reactive oxygen species (ROS) and nitric oxide (NO). Although ROS are by-products of physiological respiration, they are major players in the pathogenesis of all sorts of diseases. NO is a free radical that is considered to be toxic or protective depending on its concentration, subcellular localization, and reaction with ROS. Oxidative stress is frequently associated with in utero programming in a number of pregnancy-associated diseases such as pregnancy-induced hypertension, preeclampsia, gestational diabetes, malnutrition, premature labor, preterm prelabor rupture of the membranes, intrauterine growth restriction, maternal stress, and placental insufficiency [5,8,14]. Moreover, diseases originating in the perinatal and neonatal periods such as bronchopulmonary dysplasia, retinopathy of prematurity, necrotizing enterocolitis, and periventricular leukomalacia are closely related to oxidative stress [15]. Newborns, especially if preterm, are particularly vulnerable to oxidative stress because they exhibit accelerated production of ROS and limited antioxidant protection, which increases susceptibility to disease later in life. Oxidative stress-mediated programming may act directly through epigenetic regulation of gene expression or indirectly via the effects of certain free radical signals [5,8,14]. However, which particular ROS or redox-sensitive signaling is responsible for cardiovascular programming remains unclear. So far, there remains a lack of data on how and when to reprogram oxidative stress-related programmed diseases [16].
Works that haven been published in recent years support that NO–ROS imbalance is important for the developmental programming of CVD [4,5,6,7,8]. Among the reasons for NO–ROS imbalance, increasing attention has been centered on asymmetric dimethylarginine (ADMA) [17]. ADMA can compete with l-arginine (the substrate for NO synthase) to reduce the synthesis of NO, but on the other hand, it induces superoxide production by uncoupling nitric oxide synthase (NOS). Thus, cellular ADMA concentrations tightly regulate the local NO–ROS balance [17,18,19]. A growing body of evidence from clinical trials and animal studies suggest an impact of ADMA related NO–ROS imbalance on the developmental programming of CVD and cardiovascular outcome [14,19,20,21,22].
Next, the important role of l-arginine in normal pregnancy, fetal growth and development, and cardiovascular health has been reviewed elsewhere [23,24,25,26]. l-arginine and other amino acids, sugars, and fatty acids are important cellular nutrients. Specific nutrients have been recognized as signaling molecules transmitting and translating dietary signals into changes in gene expression via the appropriate sensing mechanisms, also known as nutrient sensing signaling pathway [27]. Increasing evidence supports that the l-arginine–NO pathway regulates the metabolism of energy substrates and nutrient sensing signals [28].
This review aims to summarize evidence linking the l-arginine–ADMA–NO pathway to the development of CVD, with a focus on the interplay between oxidative stress and nutrient sensing signaling, and provide various early manipulations targeting the above mechanisms as a reprogramming approach to prevent the cardiovascular programming and its related comorbidities across the later lifespan. A schematic summarizing the links between the l-arginine–ADMA–NO pathway, oxidative stress, nutrient sensing signals, and cardiovascular programming is presented in Figure 1.
2. Evidence for Programming of Cardiovascular Disease in the Human
Important support for developmental origins of CVD came from observations on the severe famines of 1944–1945 in The Netherlands [29], of 1941–1944 in Saint Petersburg [30], and of 1967–1971 in the Biafra [31]. Offspring exposed to famine during pregnancy are prone to develop a number of chronic diseases, including hypertension and coronary artery disease [29,30,31,32,33,34]. Another line of evidence comes from studies of twin pregnancies. In twins, there was a positive association between birth weight and blood pressure (BP) in infants [35]. The lighter twins are prone to die from heart disease [36]. However, it is practically impossible to test prospectively for critical windows in human development, and to gather such information from most epidemiological studies.
As reviewed elsewhere [6], the risk of programmed hypertension has been assessed in a number of mother-child cohorts. Several risks affecting early-life BP of offspring in these cohorts include undernutrition, smoking, low vitamin D intake, gestational hypertension, maternal obesity, short-term breastfeeding, and excessive postnatal weight gain [6]. However, these cohorts cannot yet per se directly establish a causal relationship between the specific insult and phenotypes of CVD. Some DOHaD theories have been developed to explain these epidemiological observations, such as thrifty phenotype [37], catch-up growth hypothesis [38], and predictive adaptive responses [39]. Nevertheless, these hypotheses do not advocate molecular mechanisms by which way the phenotype is generated. As a consequence of ethical considerations concerning what is feasible or not in human studies, animal models are of critical importance. It stands to reason that much of our knowledge seems to come mainly from animal models, which identify how types of early life insults may program cardiovascular phenotypes, which developmental window is critical for cardiovascular programming, and what reprogramming strategy can be used.
3. Impact of Oxidative Stress on Cardiovascular Programming in Animal Models
Mounting evidence from animal studies confirms the link between adverse conditions in early life, cardiovascular programming, and oxidative stress. The most common species used in DOHaD field are rodents, as well as rabbits, sheep, pigs, and non-human primates and will depend on their similarity to the human being like developmental window, length of gestation, placental structure, and litter size [40,41,42]. Since cardiovascular phenotypes often develop after a prolonged asymptomatic phase in childhood and for the sake of brevity, we have restricted this review to data obtained from adult offspring. Here we summarize in Table 1 studies documenting cardiovascular programming related to oxidative stress [43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60,61,62,63,64,65,66,67,68].
A number of early-life insults have been reported to cause cardiovascular programming related to oxidative stress, including undernutrition [43,48,51,54,55,61,63,64], streptozotocin (STZ)-induced diabetes [44,62], preeclampsia [45,46,47], prenatal hypoxia [49,56], maternal nicotine exposure [59,60], ethanol consumption [50], maternal inflammation [57], prenatal glucocorticoid exposure [52,53,65,67,68], and maternal high-fat intake [58,66]. Limited information is available using large animals to study oxidative stress and cardiovascular programming simultaneously [67,68]. Despite early-life insults induced cardiovascular outcome and oxidative stress having been reported individually in young nonhuman primate offspring [69,70], there remains a lack of data regarding their long-term interrelationship. As shown in Table 1, the rat is the most widely used laboratory mammal in this research field. Rats grow rapidly during their childhood and become sexually mature at approximately the sixth week. In adulthood, one rat month is comparable to three human years [42]. Female rats enter menopause between the ages of 15 and 20 months. Thus, Table 1 lists cardiovascular outcomes evaluated at different ages, which allows calculations to refer to humans of a specific age group.
Since embryo development occurs in a relatively low-oxygen environment [7], the developing fetus is highly vulnerable to oxidant injury. Oxidative stress can develop from birth through adulthood to old age in different experimental models of cardiovascular programming [7]. Insults need only last for a brief moment during pre- or peri-natal periods, as little as a few days [57], to cause long-term cardiovascular consequences. Also, early postnatal environment can influence redox balance to elicit cardiovascular programming. In an early postnatal overfeeding model [71], elevation in BP and cardiac hypertrophy are observed at weaning. In addition, impaired heart contractility, cardiac fibrosis, and increased vulnerability to ischemia-reperfusion injury have been observed in adulthood. These observations suggest that there are a number of critical windows during prenatal and postnatal life, and that a range of insults for cardiovascular programming should be taken into account. Studies listed in Table 1 indicate that cardiovascular programming can be attributed to multiple hits [49,53], although oxidative stress is unlikely to constitute the sole programmed mechanism that increases the vulnerability to later superimposed cardiovascular injury. Therefore, it is important to consider the potential interactions between the pre- and post-natal environment and between oxidative stress and other programmed mechanisms in determining final cardiovascular outcomes.
Although the common pathogenic mechanisms of cardiovascular programming are still inconclusive, animal models have provided certain mechanisms, including, but not limited to, renin-angiotensin system (RAS), endothelial dysfunction, epigenetics, glucocorticoid effects, low nephron endowment, oxidative stress, and dysregulated nutrient sensing signals [3,4,5,6,7,8,9,10,11,12,13]. Importantly, among these hypothetical mechanisms, the l-arginine–ADMA–NO pathway is closely interrelated to the others in determining the programming process.
4. Impact of ADMA Induced NO–ROS Imbalance in Cardiovascular Programming
Emerging evidence demonstrates that ADMA is involved in the development of CVD [19,20,21,22]. As we reviewed before [14], many studies support that ADMA related NO-ROS imbalance plays a major role in compromised pregnancy and fetal programming. ADMA is an l-arginine analogue, which can compete with l-arginine to inhibit the activity of NOS, resulting in the reduction of NO (Figure 2). Protein-incorporated ADMA is a methylated arginine derivative generated by the addition of methyl groups in arginine residue in proteins through the type I protein arginine methyltransferase (PRMT) family. Free ADMA is then released after protein degradation [17,18,19]. Free ADMA can be transported to other organs by cationic amino acid transporter or excreted into the urine. Nearly 80% of ADMA is metabolized by dimethylarginine dimethylaminohydrolase-1 (DDAH-1) and -2 (DDAH-2), and alanine-glyoxylate aminotransferase 2 (AGXT2) [17,18,19]. On the other hand, our body can use l-citrulline to make l-arginine via the argininosuccinate (AS) pathway. l-arginine can also be metabolized by arginase to generate ornithine, which can be further converted to l-arginine by ornithine carbamoyltransferase (OCT). Therefore, there is a close interplay between l-arginine and ADMA to control the synthesis and metabolism of each other. Another isomer of ADMA methylated by PRMTs are symmetric dimethylarginine (SDMA). Unlike enzymatically metabolized ADMA, SDMA is eliminated primarily by renal excretion. High plasma ADMA or SDMA concentrations not only predict all-cause mortality and CVD events, but are also relevant to a broad range of diseases [19].
ROS have been shown to increase PRMT and inhibit DDAH activity, leading to an increase in ADMA [72,73]. Further, high levels of ADMA can uncouple NOS isoenzymes to produce superoxide instead of NO, contributing to the burden of oxidative stress [74]. Thus, ADMA per se can lead to the production of ROS and reduction of NO. Indeed, increasing evidence indicates that ADMA-related NO–ROS imbalance is critical in the development of hypertension and CVD, as reviewed elsewhere [19,20,21,22].
Several lines of evidence indicate that the ADMA induced NO–ROS imbalance interacts with other mechanisms to incite cardiovascular programming. First, a growing body of evidence indicates that angiotensin II-induced increases of ADMA and oxidative stress contribute to the development of CVD and hypertension [75,76], whereas early blockade of the RAS has been shown to reduce ADMA and prevent the development of hypertension [77,78]. Second, ADMA competes with l-arginine to inhibit NOS activity and is involved in endothelial dysfunction [17,18]. It has been well established that endothelial dysfunction is central to the development of CVD [24]. Third, there are studies that show that redox signaling and NO control epigenetic regulation, contributing to cardiovascular programming [79]. Fourth, there are many reports that NO-ROS imbalance plays a crucial role in several models of glucocorticoid-induced programmed hypertension [52,53,80,81,82]. Last, increasing evidence supports that the l-arginine–NO pathway regulates the metabolism of energy substrates and nutrient sensing signals [28]. All of these observations provide a close link between the l-arginine–ADMA–NO pathway and other important mechanisms involved in cardiovascular programming.
5. Metabolic Crosstalk between Arginine Metabolism and Nutrient Sensing Signaling
Maternal nutrition plays a critical role in placental function, fetal growth, organogenesis, and development. As shown in Table 1, a number of nutritional insults can induce cardiovascular programming [43,48,51,54,55,58,61,63,64,66]. Since altered maternal nutrition causes disturbed nutrient-sensing signals [9], nutrient-sensing signaling might therefore be a common mechanism underlying nutritional programming of CVD. Nutrient-sensing signaling pathways orchestrate fetal metabolism and development in response to maternal nutritional insults. Accordingly, a number of these signaling pathways exist in the cardiovascular system, including silent information regulator transcript (SIRT), cyclic adenosine monophosphate (AMP)-activated protein kinase (AMPK), peroxisome proliferator-activated receptors (PPARs), PPARγ coactivator-1α (PGC-1α), and mammalian target of rapamycin (mTOR) pathway. Among them, PGC-1α acts as a hub for a nutrient-sensing cluster. As shown in Figure 3, activation of AMPK by increased NAD+/NADH ratio, activation of SIRT1 by increased mitochondrial AMP/adenosine triphosphate (ATP) ratio, or NO can affect PGC-1α activity to promote mitochondria biogenesis [83,84]. AMPK induces mitochondrial biogenesis by activating PGC-1α either directly or through SIRT1 [83,85]. In addition, SIRT1 and AMPK can mediate deacetylation and phosphorylation of PGC-1α, respectively [85], to regulate the expression of PPAR target genes. Early-life nutritional insults might drive nutrient sensing signals to regulate PPARs and their target genes and thereby go through cardiovascular programming [86,87]. A recent review by us demonstrated that several PPAR target genes contribute to programmed hypertension, such as Nos2, Nos3, Sod2, Nrf2, Sirt7, Ren, and Sgk1 [88]. Furthermore, our reports demonstrated that the PPAR signaling pathway is significantly regulated in a variety of models of programmed hypertension, such as maternal caloric restriction [89], STZ-induced maternal diabetes [89], prenatal dexamethasone exposure [81], and maternal high-fructose consumption [90]. In addition to PPARs, PGC-1α can activate other nuclear receptors, like estrogen receptor related receptor (ERR) and nuclear respiratory factor (NRF) to regulate energy metabolism [85].
PGC-1α not only interacts with nutrient sensing signals to regulate mitochondrial biogenesis, but also results in the degradation of mitochondria via the autophagy-lysosome machinery. Autophagy is a cellular catabolic process in which key organelles, such as mitochondria, are transported to lysosomes for degradation [91]. PGC-1α can regulate transcription factor EB (TFEB) to mediate autophagy. The mTOR is another key autophagy regulator integrating amino acid starvation and increases of ROS to the autophagy pathway activities [92]. Autophagy is inhibited by the mTOR, while the negative regulator of mTOR (e.g., AMPK) promotes it. Both mTOR and AMPK can oppositely regulate unc-51-like kinase 1/2 (ULK1/2) activity by phosphorylation. In addition to activating ULK1/2, AMPK can promote autophagy through SIRT1. SIRT1 can deacetylate and activate several autophagy-related (Atg) proteins, such as Atg5, Atg7, and Atg8 [93]. Importantly, these regulatory pathways of autophagy are mainly activated upon ROS overproduction, NO deficiency, or nutrient deprivation [91,92]. Because mitochondria are a major source of ROS within most mammalian cells, these observations suggest that there are close interconnections among autophagy, mitochondrial ROS, NO production, and cell apoptosis vs. survival [94]. Given the essential role of endothelial dysfunction in the pathogenesis of CVD, disturbed autophagy in response to early-life nutritional insults in endothelial cells are likely to have significant contributions to cardiovascular programming.
Four lines of evidence suggest that the l-arginine–ADMA–NO pathway mediates nutrient sensing mechanisms to drive programming of CVD. First, recent reports showed that endothelial NOS is needed to couple AMPK activation to mammalian target of rapamycin complex 1 (mTORC1) [95], which is required for embryonic cardiovascular development and for postnatal maintenance of cardiac structure and function [96]. The second line of evidence consistent with an important role for l-arginine in nutrient sensing and fetal programming is that multiple catabolic fates enable l-arginine to have multi-faced functions in the development of the cardiovascular system [26]. In addition to NO, l-arginine is a nitrogenous precursor for synthesis of ornithine, polyamines, proline, glutamine, creatine, and agmatine. Many products of its catabolism are essential for a healthy pregnancy and fetal development. Indeed, many reports have demonstrated the beneficial effects of l-arginine supplementation in fetal outcome and CVD [23,26]. Third, there are studies that indicate that placental mTOR signaling regulates amino acid transporter and l-arginine regulates the mTOR signaling pathway [97,98]. These observations demonstrate a crosstalk between the l-arginine–NO pathway and the nutrient sensing signaling pathway in the placenta. Thus, placental nutrient sensing plays a critical role in modulating maternal-fetal resource allocation, affecting fetal programming and life-long CV health [99]. Fourth, our recent study showed that NO depletion in pregnancy induced programmed hypertension in adult offspring, which was associated with massive alterations in renal transcriptome [46]. By using next-generation sequencing (NGS) techniques, we observed that more than 2000 genes of renal transcriptome were altered by maternal NG-nitro-l-arginine-methyl ester (l-NAME) administration (a NOS inhibitor). Furthermore, our NGS data indicated that early redox imbalance programs a diverse range of pathways in later life, including the PPAR signaling pathway [46]. Additionally, we identified that several genes related to the nutrient sensing signaling pathway were significantly differential expressed, such as Ppargcia (encoding for the protein PGC-1α, fold change = 2.98), Prkag3 (encoding for the γ3 chain of AMPK, fold change = 0.48), Slc7a12 (encoding for amino acid transporter, y+ system, member 12, fold change = 3.66), and Fabp4 (encoding for fatty acid binding protein 4, fold change = 2.73). These observations suggest a possible mechanism that early NO deficiency programs nutrient sensing signals, leading to the development of CVD in later life.
6. Reprograming Strategy via Targeting NO-ROS Balance and Nutrient Sensing Signaling
As we previously reviewed, numerous interventions have been reported to lower ADMA levels and restore NO-ROS balance in human and experimental studies, such as angiotensin-converting enzyme inhibitors, angiotensin receptor blockers, aliskiren, vitamin E, l-citrulline, melatonin, aminoguanidine, pioglitazone, probucol, farnesoid X receptor agonists, resveratrol, N-acetylcysteine, fenofibrate, folic acid, metformin, oral contraceptives, and α-lipoic acid [6,18,19,100,101]. However, to date, a specific ADMA-lowering agent remains inaccessible in clinical practice. Because PRMTs control ADMA production, whereas DDAHs and AGXT2 regulate its metabolism, the discovery of specific PRMT inhibitors, DDAHs, and AGXT2 activators might help in developing a therapeutic approach to lower ADMA in the near future.
Given that oxidative stress is a major mechanism contributing to cardiovascular programming, it is a reasonable assumption that antioxidant supplementation would be part of potential therapy strategies for CVD. However, currently the cardiovascular benefits of antioxidant have not yet been proven [102]. Because CVD is a multifactorial disorder, there remains a lack of data on which organ-specific redox-sensitive signals are responsible for oxidative stress-related programming processes and which developmental window (e.g., in utero or at the pre-weaning stage) is appropriate for reprogramming [16]. Therefore, the identification of specific antioxidants targeting restoration of NO–ROS imbalance in early life may constitute a potential intervention aimed at reprogramming the development of CVD in adulthood.
Targeting nutrient sensing signals is another potential reprogramming strategy to prevent the development of CVD. Several dietary phytonutrients have been reported to decrease the risk for CVD [103]. In addition to their antioxidant properties, such nature-derived phytonutrients, like quercetin, fisetin, genistein, epigallocatechins, and resveratrol have been shown to act as AMPK activators [104,105]. Additionally, several kinds of AMPK activators have been studied in CVD in adulthood [106], including metformin, thiazolidinediones (TZDs), polyphenols, ginsenoside, α-lipoic acid, and 5-aminoimidazole-4carboxamide riboside (AICAR). However, the ability of early interventions with AMPK activators to prevent the development of CVD in later life still await further elucidation. Next, practically all sirtuin activators have been described only for SIRT1 [107]. Resveratrol is a natural phytonutrient that activates SIRT1, and which may help in the prevention of fetal programming [49,108]. Despite other SIRT1 activators that are structurally unrelated to resveratrol having also been developed, their roles in preventing the developmental programming of CVD remain unclear. Moreover, increasing evidence has shown that all three PPARs are involved in the pathogenesis of CVD and its comorbidities, and their ligands may be considered for therapeutic use to prevent the development of CVD [86]. To date, however, only a few studies have explored the impact of early intervention by PPAR modulators to prevent cardiovascular programming, as reviewed previously [88]. There remains a long road ahead to establish the particular PPAR modulator, the right developmental window, and organ-specific effects, to prevent cardiovascular programming in different models of fetal programming.
7. Conclusions
Despite advances in therapy, CVD kills millions of people every year all over the world. The burden of CVD continues to increase globally, including many children who are at risk at an early age. Extensive experimental animal studies have shown that the interplay among the l-arginine–ADMA–NO pathway, oxidative stress, and nutrient sensing signals contributes to cardiovascular programming. Although major progress has been made in research on cardiovascular programming, many challenges still lie ahead. Future programming research should aim to bridge the translational gap between animal models and human therapeutics. Underlying the DOHaD concept, research into effective reprogramming strategies for cardiovascular programming that begin early in life will have a profound impact on economic burden of CVD over the next few decades.
Acknowledgments
This work was supported by Grant MOST 104-2314-B-182-056-MY3 from the Ministry of Science and Technology (Taipei, Taiwan), and Grants CMRPG8F0021 and CMRPG8F0022 from Chang Gung Memorial Hospital (Kaohsiung, Taiwan).
Author Contributions
You-Lin Tain: contributed to concept generation, data interpretation, drafting of the manuscript, critical revision of the manuscript, and approval of the article; Chien-Ning Hsu: contributed to concept generation, data interpretation, critical revision of the manuscript, and approval of the article.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| ADMA | Asymmetric dimethylarginine |
| AGXT2 | Alanine-glyoxylate aminotransferase 2 |
| CAT | Cationic amino acid transporter |
| CVD | Cardiovascular disease |
| DDAH | Dimethylarginine dimethylaminohydrolase |
| DOHaD | Developmental origins of health and disease |
| l-NAME | NG-nitro-l-arginine-methyl ester |
| mTOR | Mammalian target of rapamycin |
| NOS | Nitric oxide synthase |
| OCT | Ornithine carbamoyltransferase |
| PGC-1α | PPARγ coactivator-1α |
| PPAR | Peroxisome proliferator-activated receptor |
| PRMT | Protein arginine methyltransferase |
| RAS | Renin-angiotensin system |
| SDMA | Symmetric dimethylarginine |
| SIRT | Silent information regulator transcript |
| STZ | Streptozotocin |
References
- McAloon, C.J.; Boylan, L.M.; Hamborg, T.; Stallard, N.; Osman, F.; Lim, P.B.; Hayat, S.A. The changing face of cardiovascular disease 2000–2012: An analysis of the world health organisation global health estimates data. Int. J. Cardiol. 2016, 224, 256–264. [Google Scholar] [CrossRef] [PubMed]
- Hanson, M.; Gluckman, P. Developmental origins of noncommunicable disease: Population and public health implications. Am. J. Clin. Nutr. 2011, 94, 1754S–1758S. [Google Scholar] [CrossRef] [PubMed]
- Santos, M.S.; Joles, J.A. Early determinants of cardiovascular disease. Best Pract. Res. Clin. Endocrinol. Metab. 2012, 26, 581–597. [Google Scholar] [CrossRef] [PubMed]
- Alexander, B.T.; Dasinger, J.H.; Intapad, S. Fetal programming and cardiovascular pathology. Compr Physiol. 2015, 5, 997–1025. [Google Scholar] [PubMed]
- Blackmore, H.L.; Ozanne, S.E. Programming of cardiovascular disease across the life-course. J. Mol. Cell Cardiol. 2015, 83, 122–130. [Google Scholar] [CrossRef] [PubMed]
- Tain, Y.L.; Joles, J.A. Reprogramming: A preventive strategy in hypertension focusing on the kidney. Int. J. Mol. Sci. 2015, 17, 23. [Google Scholar] [CrossRef] [PubMed]
- Dennery, P.A. Oxidative stress in development: Nature or nurture? Free Radic. Biol. Med. 2010, 49, 1147–1151. [Google Scholar] [CrossRef] [PubMed]
- Thompson, L.P.; Al-Hasan, Y. Impact of oxidative stress in fetal programming. J. Pregnancy 2012, 2012, 582748. [Google Scholar] [CrossRef] [PubMed]
- Jansson, T.; Powell, T.L. Role of placental nutrient sensing in developmental programming. Clin. Obstet. Gynecol. 2013, 56, 591–601. [Google Scholar] [CrossRef] [PubMed]
- Vehaskari, V.M.; Stewart, T.; Lafont, D.; Soyez, C.; Seth, D.; Manning, J. Kidney angiotensin and angiotensin receptor expression in prenatally programmed hypertension. Am. J. Physiol. Ren. Physiol. 2004, 287, F262–F267. [Google Scholar] [CrossRef] [PubMed]
- Bagby, S.P. Maternal nutrition, low nephron number, and hypertension in later life: Pathways of nutritional programming. J. Nutr. 2007, 137, 1066–1072. [Google Scholar] [PubMed]
- Cottrell, E.C.; Seckl, J.R. Prenatal stress, glucocorticoids and the programming of adult disease. Front. Behav. Neurosci. 2009, 3, 19. [Google Scholar] [CrossRef] [PubMed]
- Napoli, C.; Infante, T.; Casamassimi, A. Maternal-foetal epigenetic interactions in the beginning of cardiovascular damage. Cardiovasc. Res. 2011, 92, 367–374. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.T.; Hsieh, C.S.; Chang, K.A.; Tain, Y.L. Roles of nitric oxide and asymmetric dimethylarginine in pregnancy and fetal programming. Int. J. Mol. Sci. 2012, 13, 14606–14622. [Google Scholar] [CrossRef] [PubMed]
- Marseglia, L.; D’Angelo, G.; Manti, S.; Arrigo, T.; Barberi, I.; Reiter, R.J.; Gitto, E. Oxidative stress-mediated aging during the fetal and perinatal periods. Oxid. Med. Cell Longev. 2014, 2014, 358375. [Google Scholar] [CrossRef] [PubMed]
- Tain, Y.L. Targeting redox balance to deprogramme obesity: Are we starting early enough? J. Physiol. 2015, 593, 4689–4690. [Google Scholar] [CrossRef] [PubMed]
- Teerlink, T.; Luo, Z.; Palm, F.; Wilcox, C.S. Cellular ADMA: Regulation and action. Pharmacol. Res. 2009, 60, 448–460. [Google Scholar] [CrossRef] [PubMed]
- Tain, Y.L.; Huang, L.T. Asymmetric dimethylarginine: Clinical applications in pediatric medicine. J. Formos. Med. Assoc. 2011, 110, 70–77. [Google Scholar] [CrossRef]
- Tain, Y.L.; Hsu, C.N. Toxic dimethylarginines: Asymmetric dimethylarginine (ADMA) and symmetric dimethylarginine (SDMA). Toxins 2017, 9, 92. [Google Scholar] [CrossRef] [PubMed]
- Böger, R.H.; Maas, R.; Schulze, F.; Schwedhelm, E. Asymmetric dimethylarginine (ADMA) as a prospective marker of cardiovascular disease and mortality—An update on patient populations with a wide range of cardiovascular risk. Pharmacol. Res. 2009, 60, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Alpoim, P.N.; Sousa, L.P.; Mota, A.P.; Rios, D.R.; Dusse, L.M. Asymmetric Dimethylarginine (ADMA) in cardiovascular and renal disease. Clin. Chim. Acta 2015, 440, 36–39. [Google Scholar] [CrossRef] [PubMed]
- Franceschelli, S.; Ferrone, A.; Pesce, M.; Riccioni, G.; Speranza, L. Biological functional relevance of asymmetric dimethylarginine (ADMA) in cardiovascular disease. Int. J. Mol. Sci. 2013, 14, 24412–24421. [Google Scholar] [CrossRef] [PubMed]
- Sudar-Milovanovic, E.; Obradovic, M.; Jovanovic, A.; Zaric, B.; Zafirovic, S.; Panic, A.; Radak, D.; Isenovic, E.R. Benefits of l-Arginine on Cardiovascular System. Mini Rev. Med. Chem. 2016, 16, 94–103. [Google Scholar] [CrossRef] [PubMed]
- Khalil, A.; Hardman, L.; O Brien, P. The role of arginine, homoarginine and nitric oxide in pregnancy. Amino Acids 2015, 47, 1715–1727. [Google Scholar] [CrossRef] [PubMed]
- Bassareo, P.P.; Mussap, M.; Bassareo, V.; Flore, G.; Mercuro, G. Nitrergic system and plasmatic methylarginines: Evidence of their role in the perinatal programming of cardiovascular diseases. Clin. Chim. Acta 2015, 451, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Bazer, F.W.; Satterfield, M.C.; Li, X.; Wang, X.; Johnson, G.A.; Burghardt, R.C.; Dai, Z.; Wang, J.; Wu, Z. Impacts of arginine nutrition on embryonic and fetal development in mammals. Amino Acids 2013, 45, 241–256. [Google Scholar] [CrossRef] [PubMed]
- Efeyan, A.; Comb, W.C.; Sabatini, D.M. Nutrient-sensing mechanisms and pathways. Nature 2015, 517, 302–310. [Google Scholar] [CrossRef] [PubMed]
- Dai, Z.; Wu, Z.; Yang, Y.; Wang, J.; Satterfield, M.C.; Meininger, C.J.; Bazer, F.W.; Wu, G. Nitric oxide and energy metabolism in mammals. Biofactors 2013, 39, 383–391. [Google Scholar] [CrossRef] [PubMed]
- Schulz, L.C. The Dutch Hunger Winter and the Developmental Origins of Health and Disease. Proc. Natl. Acad. Sci. USA 2010, 107, 16757–16758. [Google Scholar] [CrossRef] [PubMed]
- Stanner, S.A.; Yudkin, J.S. Fetal programming and the Leningrad Siege study. Twin Res. 2001, 4, 287–292. [Google Scholar] [CrossRef] [PubMed]
- Hult, M.; Tornhammar, P.; Ueda, P.; Chima, C.; Bonamy, A.K.; Ozumba, B.; Norman, M. Hypertension, diabetes and overweight: Looming legacies of the Biafran famine. PLoS ONE 2010, 5, e13582. [Google Scholar] [CrossRef] [PubMed]
- Painter, R.C.; de Rooij, S.R.; Bossuyt, P.M.; Phillips, D.I.; Osmond, C.; Barker, D.J.; Bleker, O.P.; Roseboom, T.J. Blood pressure response to psychological stressors in adults after prenatal exposure to the Dutch famine. J. Hypertens. 2006, 24, 1771–1778. [Google Scholar] [CrossRef] [PubMed]
- Painter, R.C.; de Rooij, S.R.; Bossuyt, P.M.; Simmers, T.A.; Osmond, C.; Barker, D.J.; Bleker, O.P.; Roseboom, T.J. Early onset of coronary artery disease after prenatal exposure to the Dutch famine. Am. J. Clin. Nutr. 2006, 84, 322–327. [Google Scholar] [PubMed]
- Roseboom, T.J.; van derMeulen, J.H.; Osmond, C.; Barker, D.J.; Ravelli, A.C.; Schroeder-Tanka, J.M.; van Montfrans, G.A.; Michels, R.P.; Bleker, O.P. Coronary heart disease after prenatal exposure to the Dutch famine, 1944–1945. Heart 2000, 84, 595–598. [Google Scholar] [CrossRef] [PubMed]
- Levine, R.S.; Hennekens, C.H.; Jesse, M.J. Blood pressure in prospective population based cohort of newborn and infant twins. BMJ 1994, 308, 298–302. [Google Scholar] [CrossRef] [PubMed]
- Vågerö, D.; Leon, D. Ischaemic heart disease and low birth weight: A test of the fetal-origins hypothesis from the Swedish Twin Registry. Lancet 1994, 343, 260–263. [Google Scholar] [CrossRef]
- Hales, C.N.; Barker, D.J. The thrifty phenotype hypothesis. Br. Med. Bull. 2001, 60, 5–20. [Google Scholar] [CrossRef] [PubMed]
- Cianfarani, S.; Germani, D.; Branca, F. Low birthweight and adult insulin resistance: The “catch-up growth” hypothesis. Arch. Dis. Child. Fetal Neonatal. 1999, 81, F71–F73. [Google Scholar] [CrossRef]
- Gluckman, P.D.; Hanson, M.A. Living with the past: Evolution, development, and patterns of disease. Science 2004, 305, 1733–1736. [Google Scholar] [CrossRef] [PubMed]
- McMullen, S.; Mostyn, A. Animal models for the study of the developmental origins of health and disease. Proc. Nutr. Soc. 2009, 68, 306–320. [Google Scholar] [CrossRef] [PubMed]
- Chavatte-Palmer, P.; Tarrade, A.; Rousseau-Ralliard, D. Diet before and during Pregnancy and Offspring Health: The Importance of Animal Models and What Can Be Learned from Them. Int. J. Environ. Res. Public Health 2016, 13, 586. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, P. The Laboratory Rat: Relating Its Age With Human’s. Int. J. Prev. Med. 2013, 4, 624–630. [Google Scholar] [PubMed]
- Cambonie, G.; Comte, B.; Yzydorczyk, C.; Ntimbane, T.; Germain, N.; Lê, N.L.; Pladys, P.; Gauthier, C.; Lahaie, I.; Abran, D.; et al. Antenatal antioxidant prevents adult hypertension, vascular dysfunction, and microvascular rarefaction associated with in utero exposure to a low-protein diet. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2007, 292, R1236–R1245. [Google Scholar] [CrossRef] [PubMed]
- Tain, Y.Y.; Lee, W.C.; Hsu, C.N.; Lee, W.C.; Huang, L.T.; Lee, C.T.; Lin, C.Y. Asymmetric dimethylarginine is associated with developmental programming of adult kidney disease and hypertension in offspring of streptozotocin-treated mothers. PLoS ONE 2013, 8, e55420. [Google Scholar] [CrossRef] [PubMed]
- Tain, Y.L.; Huang, L.T.; Lee, C.T.; Chan, J.Y.; Hsu, C.N. Maternal citrulline supplementation prevents prenatal NG-nitro-l-arginine-methyl ester (l-NAME)-induced programmed hypertension in rats. Biol. Reprod. 2015, 92, 7. [Google Scholar] [CrossRef] [PubMed]
- Tain, Y.L.; Lee, C.T.; Chan, J.Y.; Hsu, C.N. Maternal melatonin or N-acetylcysteine therapy regulates hydrogen sulfide-generating pathway and renal transcriptome to prevent prenatal N(G)-Nitro-l-arginine-methyl ester (l-NAME)-induced fetal programming of hypertension in adult male offspring. Am. J. Obstet. Gynecol. 2016, 215, 636. [Google Scholar] [CrossRef] [PubMed]
- Tain, Y.L.; Hsu, C.N.; Lee, C.T.; Lin, Y.J.; Tsai, C.C. N-Acetylcysteine Prevents Programmed Hypertension in Male Rat Offspring Born to Suramin-Treated Mothers. Biol. Reprod. 2016, 95, 8. [Google Scholar] [CrossRef] [PubMed]
- Tain, Y.L.; Huang, L.T.; Hsu, C.N.; Lee, C.T. Melatonin therapy prevents programmed hypertension and nitric oxide deficiency in offspring exposed to maternal caloric restriction. Oxid. Med. Cell Longev. 2014, 2014, 283180. [Google Scholar] [CrossRef] [PubMed]
- Shah, A.; Reyes, L.M.; Morton, J.S.; Fung, D.; Schneider, J.; Davidge, S.T. Effect of resveratrol on metabolic and cardiovascular function in male and female adult offspring exposed to prenatal hypoxia and a high-fat diet. J. Physiol. 2016, 594, 1465–1482. [Google Scholar] [CrossRef] [PubMed]
- Shirpoor, A.; Nemati, S.; Ansari, M.H.; Ilkhanizadeh, B. The protective effect of vitamin E against prenatal and early postnatal ethanol treatment-induced heart abnormality in rats: A 3-month follow-up study. Int. Immunopharmacol. 2015, 26, 72–79. [Google Scholar] [CrossRef] [PubMed]
- Nascimento, L.; Freitas, C.M.; Silva-Filho, R.; Leite, A.C.; Silva, A.B.; da Silva, A.I.; Ferreira, D.S.; Pedroza, A.A.; Maia, M.B.; Fernandes, M.P.; et al. The effect of maternal low-protein diet on the heart of adult offspring: Role of mitochondria and oxidative stress. Appl. Physiol. Nutr. Metab. 2014, 39, 880–887. [Google Scholar] [CrossRef] [PubMed]
- Tain, Y.L.; Sheen, J.M.; Chen, C.C.; Yu, H.R.; Tiao, M.M.; Kuo, H.C.; Huang, L.T. Maternal citrulline supplementation prevents prenatal dexamethasone-induced programmed hypertension. Free Radic. Res. 2014, 48, 580–586. [Google Scholar] [CrossRef] [PubMed]
- Tai, I.H.; Sheen, J.M.; Lin, Y.J.; Yu, H.R.; Tiao, M.M.; Chen, C.C.; Huang, L.T.; Tain, Y.L. Maternal N-acetylcysteine therapy regulates hydrogen sulfide-generating pathway and prevents programmed hypertension in male offspring exposed to prenatal dexamethasone and postnatal high-fat diet. Nitric Oxide 2016, 53, 6–12. [Google Scholar] [CrossRef] [PubMed]
- Franco Mdo, C.; Akamine, E.H.; Aparecida de Oliveira, M.; Fortes, Z.B.; Tostes, R.C.; Carvalho, M.H.; Nigro, D. Vitamins C and E improve endothelial dysfunction in intrauterine undernourished rats by decreasing vascular superoxide anion concentration. J. Cardiovasc. Pharmacol. 2003, 42, 211–217. [Google Scholar] [CrossRef] [PubMed]
- Franco Mdo, C.; Ponzio, B.F.; Gomes, G.N.; Gil, F.Z.; Tostes, R.; Carvalho, M.H.; Fortes, Z.B. Micronutrient prenatal supplementation prevents the development of hypertension and vascular endothelial damage induced by intrauterine malnutrition. Life Sci. 2009, 85, 327–333. [Google Scholar] [CrossRef] [PubMed]
- Giussani, D.A.; Camm, E.J.; Niu, Y.; Richter, H.G.; Blanco, C.E.; Gottschalk, R.; Blake, E.Z.; Horder, K.A.; Thakor, A.S.; Hansell, J.A.; et al. Developmental programming of cardiovascular dysfunction by prenatal hypoxia and oxidative stress. PLoS ONE 2012, 7, e31017. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, J.; Luo, H.; Chen, C.; Pei, F.; Cai, Y.; Yang, X.; Wang, N.; Fu, J.; Xu, Z.; et al. Prenatal lipopolysaccharide exposure causes mesenteric vascular dysfunction through the nitric oxide and cyclic guanosine monophosphate pathway in offspring. Free Radic. Biol. Med. 2015, 86, 322–330. [Google Scholar] [CrossRef] [PubMed]
- Resende, A.C.; Emiliano, A.F.; Cordeiro, V.S.; de Bem, G.F.; de Cavalho, L.C.; de Oliveira, P.R.; Neto, M.L.; Costa, C.A.; Boaventura, G.T.; de Moura, R.S. Grape skin extract protects against programmed changes in the adult rat offspring caused by maternal high-fat diet during lactation. J. Nutr. Biochem. 2013, 24, 2119–2126. [Google Scholar] [CrossRef] [PubMed]
- Xiao, D.; Wang, L.; Huang, X.; Li, Y.; Dasgupta, C.; Zhang, L. Protective Effect of Antenatal Antioxidant on Nicotine-Induced Heart Ischemia-Sensitive Phenotype in Rat Offspring. PLoS ONE 2016, 11, e0150557. [Google Scholar] [CrossRef] [PubMed]
- Xiao, D.; Huang, X.; Li, Y.; Dasgupta, C.; Wang, L.; Zhang, L. Antenatal Antioxidant Prevents Nicotine-Mediated Hypertensive Response in Rat Adult Offspring. Biol. Reprod. 2015, 93, 66. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Rodríguez, P.; López de Pablo, A.L.; García-Prieto, C.F.; Somoza, B.; Quintana-Villamandos, B.; Gómez de Diego, J.J.; Gutierrez-Arzapalo, P.Y.; Ramiro-Cortijo, D.; González, M.C.; Arribas, S.M. Long term effects of fetal undernutrition on rat heart. Role of hypertension and oxidative stress. PLoS ONE 2017, 12, e0171544. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Zhao, Y.C.; Liang, Y.; Lin, X.H.; Tan, Y.J.; Wu, D.D.; Li, X.Z.; Ye, B.Z.; Kong, F.Q.; Sheng, J.Z.; et al. The impaired myocardial ischemic tolerance in adult offspring of diabetic pregnancy is restored by maternal melatonin treatment. J. Pineal Res. 2016, 61, 340–352. [Google Scholar] [CrossRef] [PubMed]
- Chisaka, T.; Mogi, M.; Nakaoka, H.; Kan-No, H.; Tsukuda, K.; Wang, X.L.; Bai, H.Y.; Shan, B.S.; Kukida, M.; Iwanami, J.; et al. Low-Protein Diet-Induced Fetal Growth Restriction Leads to Exaggerated Proliferative Response to Vascular Injury in Postnatal Life. Am. J. Hypertens. 2016, 29, 54–62. [Google Scholar] [CrossRef] [PubMed]
- Blackmore, H.L.; Piekarz, A.V.; Fernandez-Twinn, D.S.; Mercer, J.R.; Figg, N.; Bennett, M.; Ozanne, S.E. Poor maternal nutrition programmes a pro-atherosclerotic phenotype in ApoE−/− mice. Clin. Sci. 2012, 123, 251–257. [Google Scholar] [CrossRef] [PubMed]
- Roghair, R.D.; Wemmie, J.A.; Volk, K.A.; Scholz, T.D.; Lamb, F.S.; Segar, J.L. Maternal antioxidant blocks programmed cardiovascular and behavioural stress responses in adult mice. Clin. Sci. 2011, 121, 427–436. [Google Scholar] [CrossRef] [PubMed]
- Torrens, C.; Ethirajan, P.; Bruce, K.D.; Cagampang, F.R.; Siow, R.C.; Hanson, M.A.; Byrne, C.D.; Mann, G.E.; Clough, G.F. Interaction between maternal and offspring diet to impair vascular function and oxidative balance in high fat fed male mice. PLoS ONE 2012, 7, e50671. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.; Bi, J.; Pulgar, V.M.; Figueroa, J.; Chappell, M.; Rose, J.C. Antenatal glucocorticoid treatment alters Na+ uptake in renal proximal tubule cells from adult offspring in a sex-specific manner. Am. J. Physiol. Renal Physiol. 2015, 308, F1268–F1275. [Google Scholar] [CrossRef] [PubMed]
- Gwathmey, T.M.; Shaltout, H.A.; Rose, J.C.; Diz, D.I.; Chappell, M.C. Glucocorticoid-induced fetal programming alters the functional complement of angiotensin receptor subtypes within the kidney. Hypertension 2011, 57, 620–626. [Google Scholar] [CrossRef] [PubMed]
- Westbrook, D.G.; Anderson, P.G.; Pinkerton, K.E.; Ballinger, S.W. Perinatal tobacco smoke exposure increases vascular oxidative stress and mitochondrial damage in non-human primates. Cardiovasc. Toxicol. 2010, 10, 216–226. [Google Scholar] [CrossRef] [PubMed]
- Fan, L.; Lindsley, S.R.; Comstock, S.M.; Takahashi, D.L.; Evans, A.E.; He, G.W.; Thornburg, K.L.; Grove, K.L. Maternal high-fat diet impacts endothelial function in nonhuman primate offspring. Int. J. Obes. 2013, 37, 254–262. [Google Scholar] [CrossRef] [PubMed]
- Habbout, A.; Li, N.; Rochette, L.; Vergely, C. Postnatal overfeeding in rodents by litter size reduction induces major short- and long-term pathophysiological consequences. J. Nutr. 2013, 143, 553–562. [Google Scholar] [CrossRef] [PubMed]
- Wilcox, C.S. Asymmetric dimethylarginine and reactive oxygen species: Unwelcome twin visitors to the cardiovascular and kidney disease tables. Hypertension 2012, 59, 375–381. [Google Scholar] [CrossRef] [PubMed]
- Tain, Y.L.; Kao, Y.H.; Hsieh, C.S.; Chen, C.C.; Sheen, J.M.; Lin, I.C.; Huang, L.T. Melatonin blocks oxidative stress-induced increased asymmetric dimethylarginine. Free Radic. Biol. Med. 2010, 49, 1088–1098. [Google Scholar] [CrossRef] [PubMed]
- Cardounel, A.J.; Cui, H.; Samouilov, A.; Johnson, W.; Kearns, P.; Tsai, A.L.; Berka, V.; Zweier, J.L. Evidence for the pathophysiological role of endogenous methylarginines in regulation of endothelial NO production and vascular function. J. Biol. Chem. 2007, 282, 879–887. [Google Scholar] [CrossRef] [PubMed]
- Sasser, J.M.; Moningka, N.C.; Cunningham, M.W., Jr.; Croker, B.; Baylis, C. Asymmetric dimethylarginine in angiotensin II-induced hypertension. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2010, 298, R740–R746. [Google Scholar] [CrossRef] [PubMed]
- Rochette, L.; Lorin, J.; Zeller, M.; Guilland, J.C.; Lorgis, L.; Cottin, Y.; Vergely, C. Nitric oxide synthase inhibition and oxidative stress in cardiovascular diseases: Possible therapeutic targets? Pharmacol. Ther. 2013, 140, 239–257. [Google Scholar] [CrossRef] [PubMed]
- Tain, Y.L.; Hsu, C.N.; Lin, C.Y.; Huang, L.T.; Lau, Y.T. Aliskiren prevents hypertension and reduces asymmetric dimethylarginine in young spontaneously hypertensive rats. Eur. J. Pharmacol. 2011, 670, 561–565. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.N.; Lee, C.T.; Huang, L.T.; Tain, Y.L. Aliskiren in early postnatal life prevents hypertension and reduces asymmetric dimethylarginine in offspring exposed to maternal caloric restriction. J. Renin Angiotensin Aldosterone Syst. 2015, 16, 506–513. [Google Scholar] [CrossRef] [PubMed]
- Ávila, J.G.; Echeverri, I.; de Plata, C.A.; Castillo, A. Impact of oxidative stress during pregnancy on fetal epigenetic patterns and early origin of vascular diseases. Nutr. Rev. 2015, 73, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Roghair, R.D.; Segar, J.L.; Volk, K.A.; Chapleau, M.W.; Dallas, L.M.; Sorenson, A.R.; Scholz, T.D.; Lamb, F.S. Vascular nitric oxide and superoxide anion contribute to sex-specific programmed cardiovascular physiology in mice. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2009, 296, R651–R662. [Google Scholar] [CrossRef] [PubMed]
- Sheen, J.M.; Yu, H.R.; Tiao, M.M.; Chen, C.C.; Huang, L.T.; Chang, H.Y.; Tain, Y.L. Prenatal dexamethasone-induced programmed hypertension and renal programming. Life Sci. 2015, 132, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.C.; Sheen, J.M.; Yu, H.R.; Lin, Y.J.; Chen, C.C.; Tiao, M.M.; Tsai, C.C.; Huang, L.T.; Tain, Y.L. Early postnatal treatment with soluble epoxide hydrolase inhibitor or 15-deoxy-Δ(12,14)-prostagandin J2 prevents prenatal dexamethasone and postnatal high saturated fat diet induced programmed hypertension in adult rat offspring. Prostaglandins Other Lipid Mediat. 2016, 124, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Sugden, M.C.; Caton, P.W.; Holness, M.J. PPAR control: It’s SIRTainly as easy as PGC. J. Endocrinol. 2010, 204, 93–104. [Google Scholar] [CrossRef] [PubMed]
- Lira, V.A.; Brown, D.L.; Lira, A.K.; Kavazis, A.N.; Soltow, Q.A.; Zeanah, E.H.; Criswell, D.S. Nitric oxide and AMPK cooperatively regulate PGC-1 in skeletal muscle cells. J. Physiol. 2010, 588, 3551–3566. [Google Scholar] [CrossRef] [PubMed]
- Finck, B.N.; Kelly, D.P. Peroxisome proliferator-activated receptor gamma coactivator-1 (PGC-1) regulatory cascade in cardiac physiology and disease. Circulation 2007, 115, 2540–2548. [Google Scholar] [CrossRef] [PubMed]
- Ajith, T.A.; Jayakumar, T.G. Peroxisome proliferator-activated receptors in cardiac energy metabolism and cardiovascular disease. Clin. Exp. Pharmacol. Physiol. 2016, 43, 649–658. [Google Scholar] [CrossRef] [PubMed]
- Fang, L.; Zhang, M.; Li, Y.; Liu, Y.; Cui, Q.; Wang, N. PPARgene: A Database of Experimentally Verified and Computationally Predicted PPAR Target Genes. PPAR Res. 2016, 2016, 6042162. [Google Scholar] [CrossRef] [PubMed]
- Tain, Y.L.; Hsu, C.N.; Chan, J.Y. PPARs Link Early Life Nutritional insults to later programmed hypertension and metabolic syndrome. Int. J. Mol. Sci. 2015, 17, 20. [Google Scholar] [CrossRef] [PubMed]
- Tain, Y.L.; Hsu, C.N.; Chan, J.Y.; Huang, L.T. Renal transcriptome analysis of programmed hypertension induced by maternal nutritional insults. Int. J. Mol. Sci. 2015, 16, 17826–17837. [Google Scholar] [CrossRef] [PubMed]
- Tain, Y.L.; Wu, K.L.; Lee, W.C.; Leu, S.; Chan, J.Y. Maternal fructose intake-induced renal programming in adult male offspring. J. Nutr. Biol. 2015, 26, 642–650. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J. Teaching the basics of autophagy and mitophagy to redox biologists—Mechanisms and experimental approaches. Redox Biol. 2015, 4, 242–259. [Google Scholar] [CrossRef] [PubMed]
- Filomeni, G.; de Zio, D.; Cecconi, F. Oxidative stress and autophagy: The clash between damage and metabolic needs. Cell Death Differ. 2015, 22, 377–388. [Google Scholar] [CrossRef] [PubMed]
- Dutta, D.; Calvani, R.; Bernabei, R.; Leeuwenburgh, C.; Marzetti, E. Contribution of impaired mitochondrial autophagy to cardiac aging: Mechanisms and therapeutic opportunities. Circ. Res. 2012, 110, 1125–1138. [Google Scholar] [CrossRef] [PubMed]
- Scheitlin, C.G.; Nair, D.M.; Crestanello, J.A.; Zweier, J.L.; Alevriadou, B.R. Fluid Mechanical Forces and Endothelial Mitochondria: A Bioengineering Perspective. Cell Mol. Bioeng. 2014, 7, 483–496. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Reif, M.M.; Craige, S.M.; Kant, S.; Keaney, J.F., Jr. Endothelial AMPK activation induces mitochondrial biogenesis and stress adaptation via eNOS-dependent mTORC1 signaling. Nitric Oxide 2016, 55–56, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Sciarretta, S.; Volpe, M.; Sadoshima, J. Mammalian target of rapamycin signaling in cardiac physiology and disease. Circ. Res. 2014, 114, 549–564. [Google Scholar] [CrossRef] [PubMed]
- Díaz, P.; Powell, T.L.; Jansson, T. The role of placental nutrient sensing in maternal-fetal resource allocation. Biol. Reprod. 2014, 91, 82. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Burghardt, R.C.; Romero, J.J.; Hansen, T.R.; Wu, G.; Bazer, F.W. Functional roles of arginine during the peri-implantation period of pregnancy. III. Arginine stimulates proliferation and interferon tau production by ovine trophectoderm cells via nitric oxide and polyamine-TSC2-MTOR signaling pathways. Biol. Reprod. 2015, 92, 75. [Google Scholar] [CrossRef] [PubMed]
- Dimasuay, K.G.; Boeuf, P.; Powell, T.L.; Jansson, T. Placental responses to changes in the maternal environment determine fetal growth. Front. Physiol. 2016, 29, 12. [Google Scholar] [CrossRef] [PubMed]
- Tain, Y.L.; Huang, L.T. Restoration of asymmetric dimethylarginine-nitric oxide balance to prevent the development of hypertension. Int. J. Mol. Sci. 2014, 15, 11773–11782. [Google Scholar] [CrossRef] [PubMed]
- Tain, Y.L.; Hsu, C.N. Targeting on asymmetric dimethylarginine related nitric oxide-reactive oxygen species imbalance to reprogram the development of hypertension. Int. J. Mol. Sci. 2016, 17, 2020. [Google Scholar] [CrossRef] [PubMed]
- Goszcz, K.; Deakin, S.J.; Duthie, G.G.; Stewart, D.; Leslie, S.J.; Megson, I.L. Antioxidants in Cardiovascular Therapy: Panacea or False Hope? Front. Cardiovasc. Med. 2015, 2, 29. [Google Scholar] [CrossRef] [PubMed]
- Riccioni, G.; Speranza, L.; Pesce, M.; Cusenza, S.; D’Orazio, N.; Glade, M.J. Novel phytonutrient contributors to antioxidant protection against cardiovascular disease. Nutrition 2012, 28, 605–610. [Google Scholar] [CrossRef] [PubMed]
- Gasparrini, M.; Giampieri, F.; Alvarez Suarez, J.M.; Mazzoni, L.; Y Forbes Hernandez, T.; Quiles, J.L.; Bullon, P.; Battino, M. AMPK as a new attractive therapeutic target for disease prevention: The role of dietary compounds AMPK and disease prevention. Curr. Drug Targets 2016, 17, 865–889. [Google Scholar] [CrossRef] [PubMed]
- Bjørklund, G.; Chirumbolo, S. Role of oxidative stress and antioxidants in daily nutrition and human health. Nutrition 2017, 33, 311–321. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Yang, G.; Kim, Y.; Kim, J.; Ha, J. AMPK activators: Mechanisms of action and physiological activities. Exp. Mol. Med. 2016, 48, e224. [Google Scholar] [CrossRef] [PubMed]
- Zou, T.; Chen, D.; Yang, Q.; Wang, B.; Zhu, M.J.; Nathanielsz, P.W.; Du, M. Resveratrol supplementation to high fat diet-fed pregnant mice promotes brown and beige adipocyte development and prevents obesity in male offspring. J. Physiol. 2017, 595, 1547–1562. [Google Scholar] [CrossRef] [PubMed]
- Villalba, J.M.; Alcaín, F.J. Sirtuin activators and inhibitors. Biofactors 2012, 38, 349–359. [Google Scholar] [CrossRef] [PubMed]
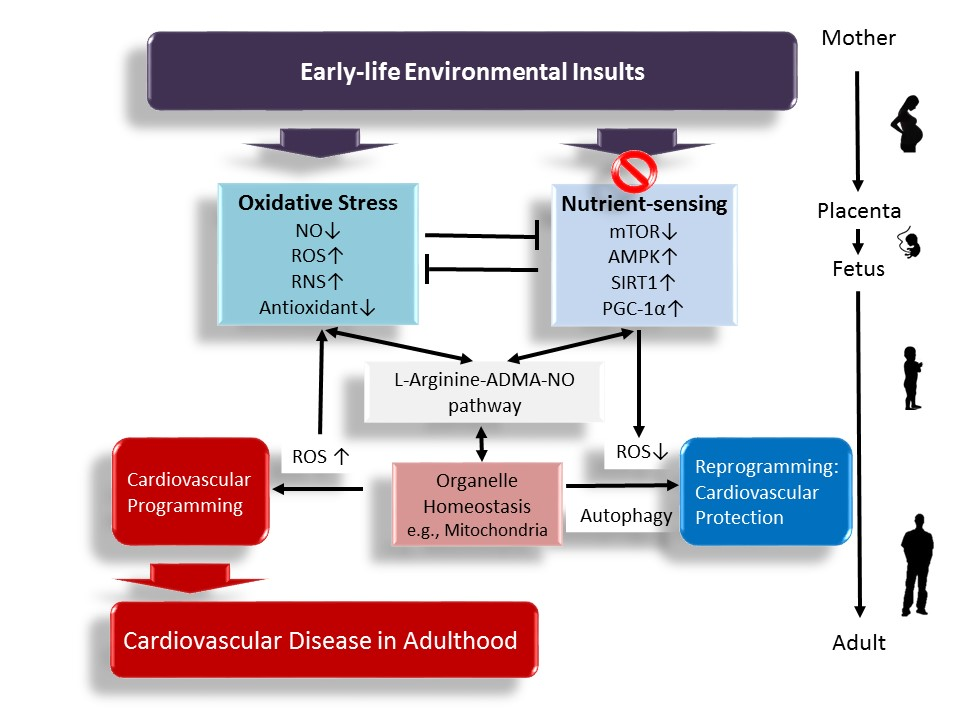
Figure 1.
A schema showing that early-life environmental insults affect the l-arginine–ADMA–NO pathway, increase oxidative stress, and dysregulate nutrient sensing signals, leading to cardiovascular programming and cardiovascular disease (CVD) in later life. Early targeting of the above mechanisms might serve as a reprogramming approach to prevent CVD and its related comorbidities in adulthood. ↑ = increased. ↓ = decreased.
Figure 1.
A schema showing that early-life environmental insults affect the l-arginine–ADMA–NO pathway, increase oxidative stress, and dysregulate nutrient sensing signals, leading to cardiovascular programming and cardiovascular disease (CVD) in later life. Early targeting of the above mechanisms might serve as a reprogramming approach to prevent CVD and its related comorbidities in adulthood. ↑ = increased. ↓ = decreased.

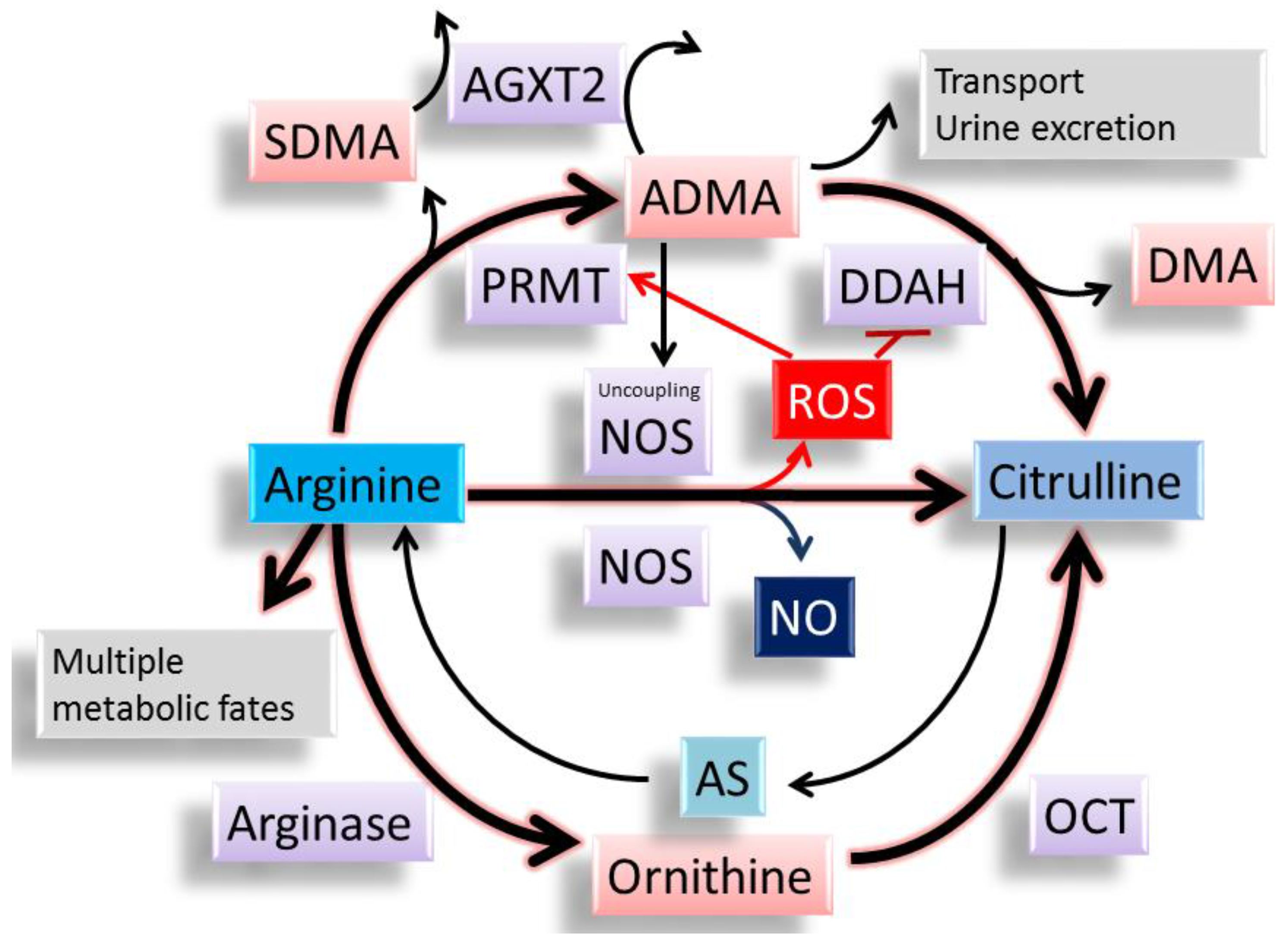
Figure 2.
The synthesis and metabolism of l-arginine, asymmetric dimethylarginine (ADMA), and l-citrulline. l-arginine has multiple metabolic fates, including metabolism by NOS, arginase, and other enzymes. ADMA can compete with l-arginine to reduce the synthesis of NO. Both ADMA and symmetric dimethylarginine (SDMA) come from methylated l-arginine by protein arginine methyltransferase (PRMT). ADMA can be transported to other organs or excreted into the urine. Unlike SDMA, only ADMA can be metabolized by dimethylarginine dimethylaminohydrolase (DDAH)-1 and -2. Alanine-glyoxylate aminotransferase 2 (AGXT2) can metabolize ADMA as well as SDMA. l-citrulline can be generated by NOS, DDAHs, and ornithine carbamoyltransferase (OCT). l-citrulline can be used to make l-arginine via the argininosuccinate (AS) pathway. ADMA can uncouple NOS to produce reactive oxygen species (ROS). ROS can induce PRMT and inhibit DDAH activity, leading to an increase in ADMA.
Figure 2.
The synthesis and metabolism of l-arginine, asymmetric dimethylarginine (ADMA), and l-citrulline. l-arginine has multiple metabolic fates, including metabolism by NOS, arginase, and other enzymes. ADMA can compete with l-arginine to reduce the synthesis of NO. Both ADMA and symmetric dimethylarginine (SDMA) come from methylated l-arginine by protein arginine methyltransferase (PRMT). ADMA can be transported to other organs or excreted into the urine. Unlike SDMA, only ADMA can be metabolized by dimethylarginine dimethylaminohydrolase (DDAH)-1 and -2. Alanine-glyoxylate aminotransferase 2 (AGXT2) can metabolize ADMA as well as SDMA. l-citrulline can be generated by NOS, DDAHs, and ornithine carbamoyltransferase (OCT). l-citrulline can be used to make l-arginine via the argininosuccinate (AS) pathway. ADMA can uncouple NOS to produce reactive oxygen species (ROS). ROS can induce PRMT and inhibit DDAH activity, leading to an increase in ADMA.

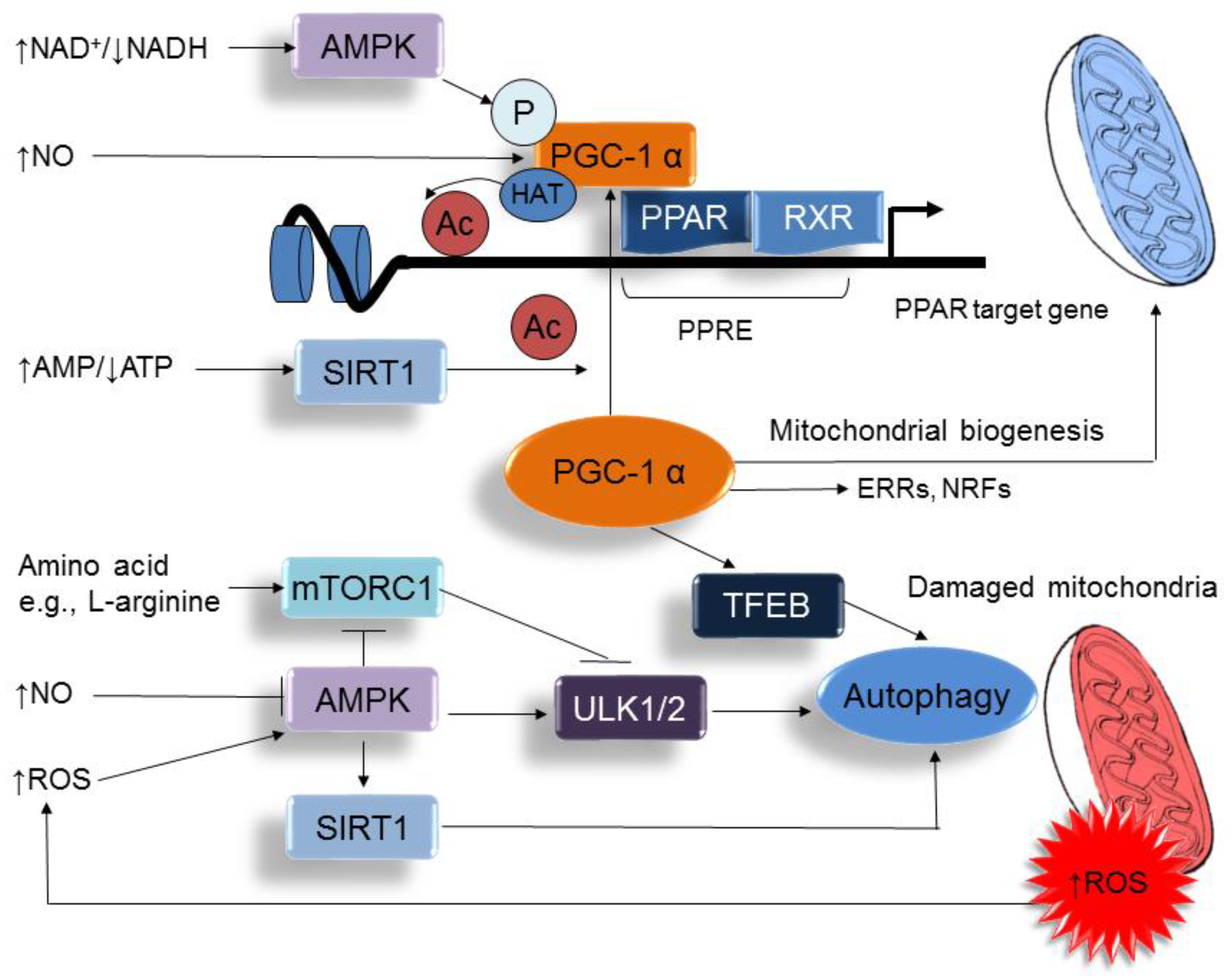
Figure 3.
A schema showing the interplay between nutrient sensing signals and oxidative stress on the regulation of PPARγ coactivator-1α (PGC-1α), peroxisome proliferator-activated receptor (PPAR) target genes, mitochondria biogenesis, and autophagy. ↑ = increased. ↓ = decreased.
Figure 3.
A schema showing the interplay between nutrient sensing signals and oxidative stress on the regulation of PPARγ coactivator-1α (PGC-1α), peroxisome proliferator-activated receptor (PPAR) target genes, mitochondria biogenesis, and autophagy. ↑ = increased. ↓ = decreased.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Cardiovascular programming related to oxidative stress in animal models.
| Animal Model | Species | Cardiovascular Phenotypes | Programming Mechanisms Related to Oxidative Stress | Age at Evaluation | Ref. |
|---|---|---|---|---|---|
| Maternal protein restriction | Rats | Hypertension, vascular dysfunction | ↑ Oxidative stress, ↓ glutathione | 12 wk | [43] |
| STZ-induced diabetes | Rats | Hypertension | ↑ ADMA, ↓ NO | 12 wk | [44] |
| Maternal l-NAME administration | Rats | Hypertension | ↑ Oxidative stress | 12 wk | [45,46] |
| Maternal suramin administration | Rats | Hypertension | ↑ ADMA, ↓ NO, ↓ H2S | 12 wk | [47] |
| Maternal caloric restriction | Rats | Hypertension | ↑ ADMA, ↓ NO | 12 wk | [48] |
| Prenatal hypoxia plus postnatal high-fat intake | Rats | Vulnerable to cardiac ischemia/reperfusion (I/R) injury | ↑ Superoxide production | 12 wk | [49] |
| Maternal ethanol consumption | Rats | Coronary tissue proliferation | ↑ Lipid peroxidation | 12 wk | [50] |
| Protein restricted diet | Rats | Hypertension | ↑ ROS production and ↓ Antioxidant capacity in heart | 14 wk | [51] |
| Prenatal dexamethasone exposure | Rats | Hypertension | ↓ NO | 16 wk | [52] |
| Prenatal dexamethasone exposure plus postnatal high-fat intake | Rats | Hypertension | ↓ NO, ↑ Oxidative stress | 16 wk | [53] |
| Maternal caloric restriction | Rats | Hypertension, vascular dysfunction | ↑ Superoxide production | 16 wk | [54,55] |
| Prenatal hypoxia exposure | Rats | Endothelial dysfunction | ↑ Oxidative stress in aorta | 16 wk | [56] |
| Prenatal LPS exposure | Rats | Hypertension, endothelial dysfunction | ↓ NO, ↓ antioxidant enzyme expression | 19 wk | [57] |
| Maternal high-fat intake | Rats | Hypertension | ↑ Lipid peroxidation, ↓ NO | 24 wk | [58] |
| Maternal nicotine exposure | Rats | Hypertension, vulnerable to cardiac I/R injury | ↑ Arterial ROS production | 8 mo | [59,60] |
| Maternal caloric restriction | Rats | Hypertension, cardiac damage | ↑ Oxidative stress in heart | 22 mo | [61] |
| STZ-induced diabetes | Mice | Myocardial ischemia/reperfusion injury | ↑ Oxidative stress | 8 wk | [62] |
| Maternal protein restriction | Mice | Vulnerable to vascular injury | ↑ Oxidative stress | 11 wk | [63] |
| Maternal protein restriction | Mice | Atherosclerosis | ↑ Oxidative stress | 6 mo | [64] |
| Prenatal 11β-HSD inhibition | Mice | Endothelial dysfunction | ↑ Oxidative stress | 6 mo | [65] |
| Maternal high-fat intake | Mice | Hypertension | ↑ Arterial ROS production, ↓ NO | 7.5 mo | [66] |
| Prenatal betamethasone exposure | Sheep | Hypertension | ↑ ROS production, ↓ NO | 18 mo | [67,68] |
Studies tabulated according to species and age at evaluation. wk = week. mo = month. STZ = Streptozotocin. l-NAME = NG-nitro-l-arginine-methyl ester. LPS = Lipopolysaccharides. 11β-HSD = 11β-hydroxysteroid dehydrogenase. ROS = reactive oxygen species. ↑ = increased. ↓ = decreased.
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Tain, Y.-L.; Hsu, C.-N. Interplay between Oxidative Stress and Nutrient Sensing Signaling in the Developmental Origins of Cardiovascular Disease. Int. J. Mol. Sci. 2017, 18, 841. https://doi.org/10.3390/ijms18040841
AMA Style
Tain Y-L, Hsu C-N. Interplay between Oxidative Stress and Nutrient Sensing Signaling in the Developmental Origins of Cardiovascular Disease. International Journal of Molecular Sciences. 2017; 18(4):841. https://doi.org/10.3390/ijms18040841
Chicago/Turabian StyleTain, You-Lin, and Chien-Ning Hsu. 2017. "Interplay between Oxidative Stress and Nutrient Sensing Signaling in the Developmental Origins of Cardiovascular Disease" International Journal of Molecular Sciences 18, no. 4: 841. https://doi.org/10.3390/ijms18040841
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.