
Antiplatelet Activity of a Newly Synthesized Novel Ruthenium (II): A Potential Role for Akt/JNK Signaling



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
2.1. Results
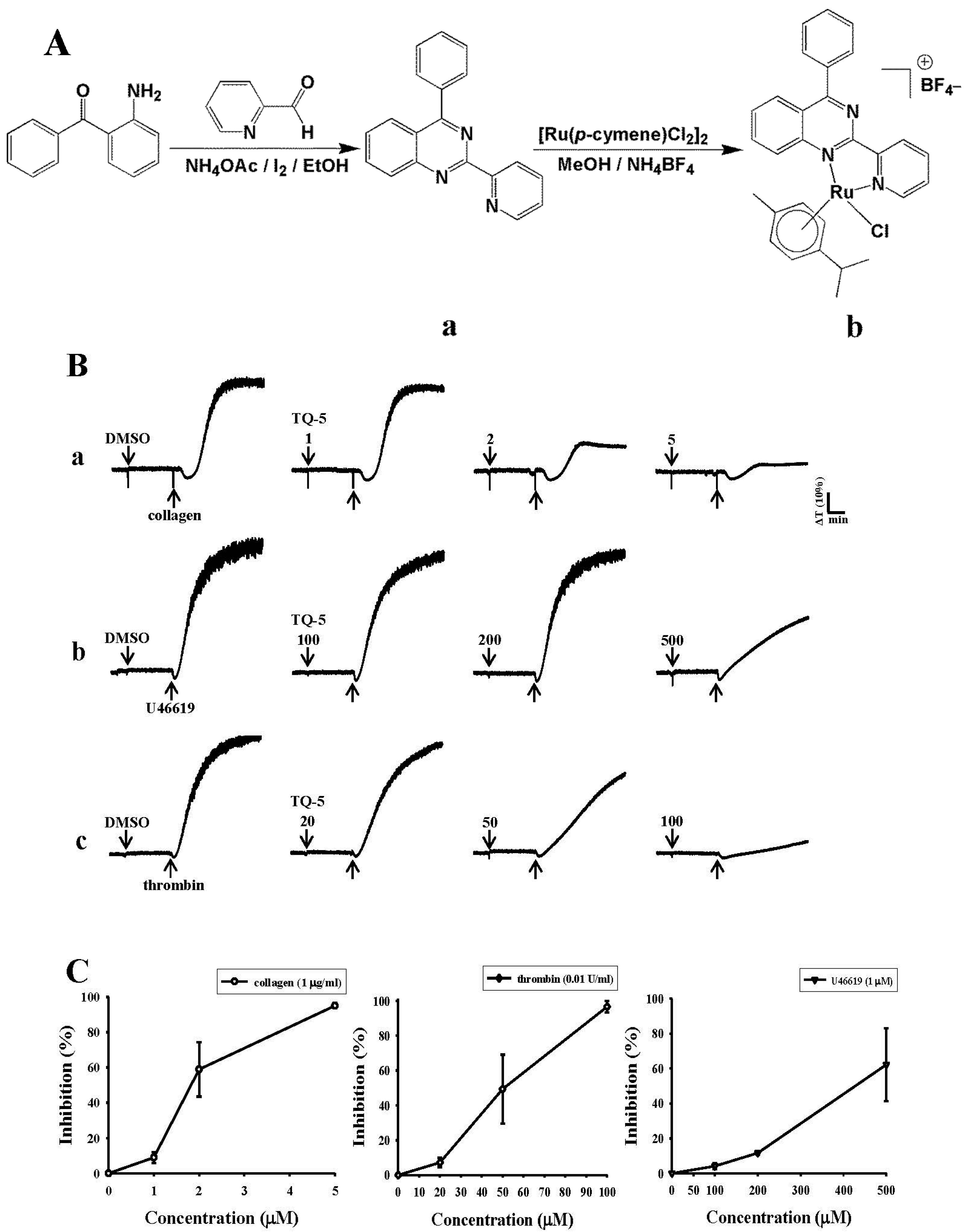
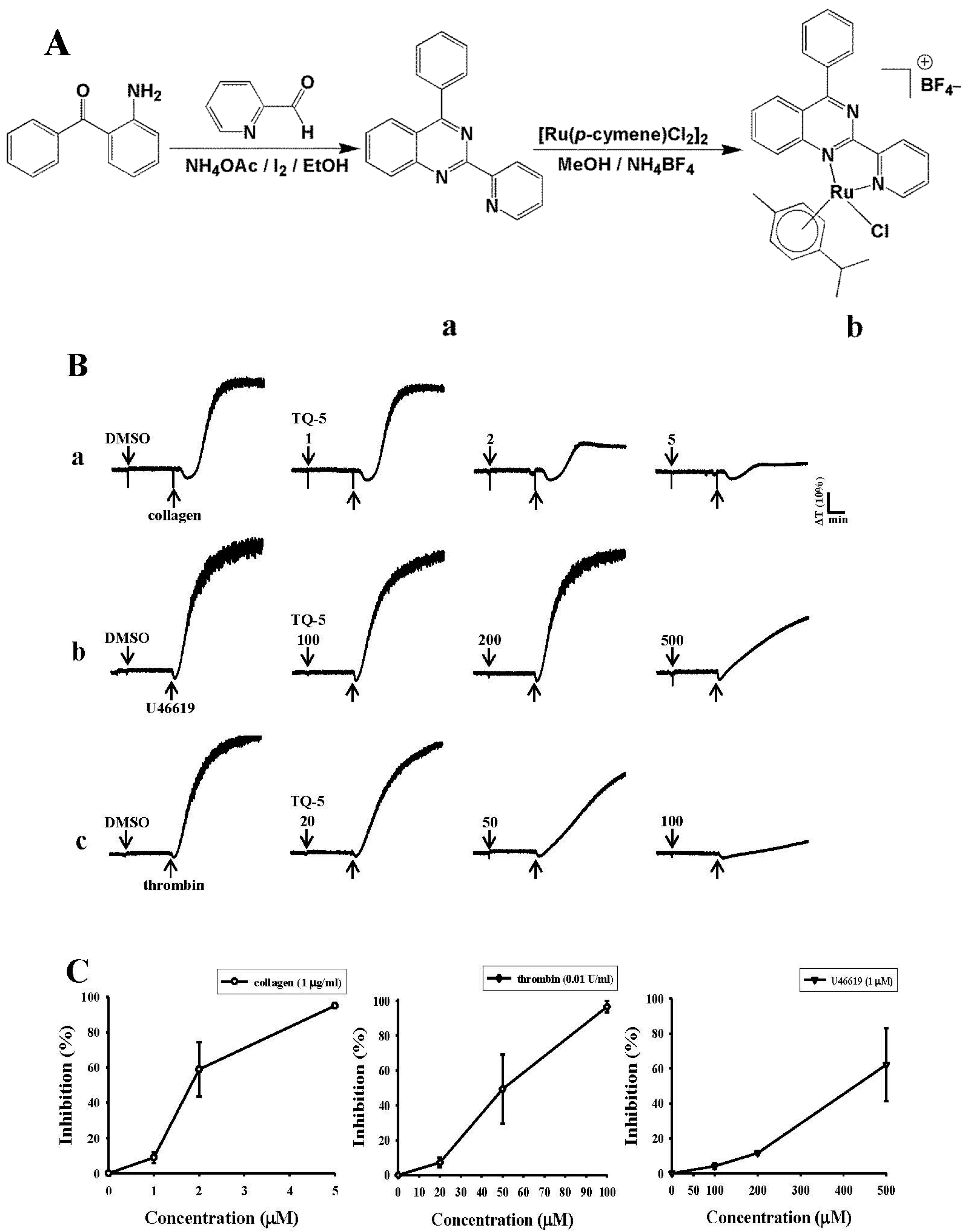
2.1.1. Nuclear Magnetic Resonance (NMR) Spectroscopy
2.1.2. TQ5 Inhibits Agonists Induced Platelet Aggregation in Washed Human Platelets
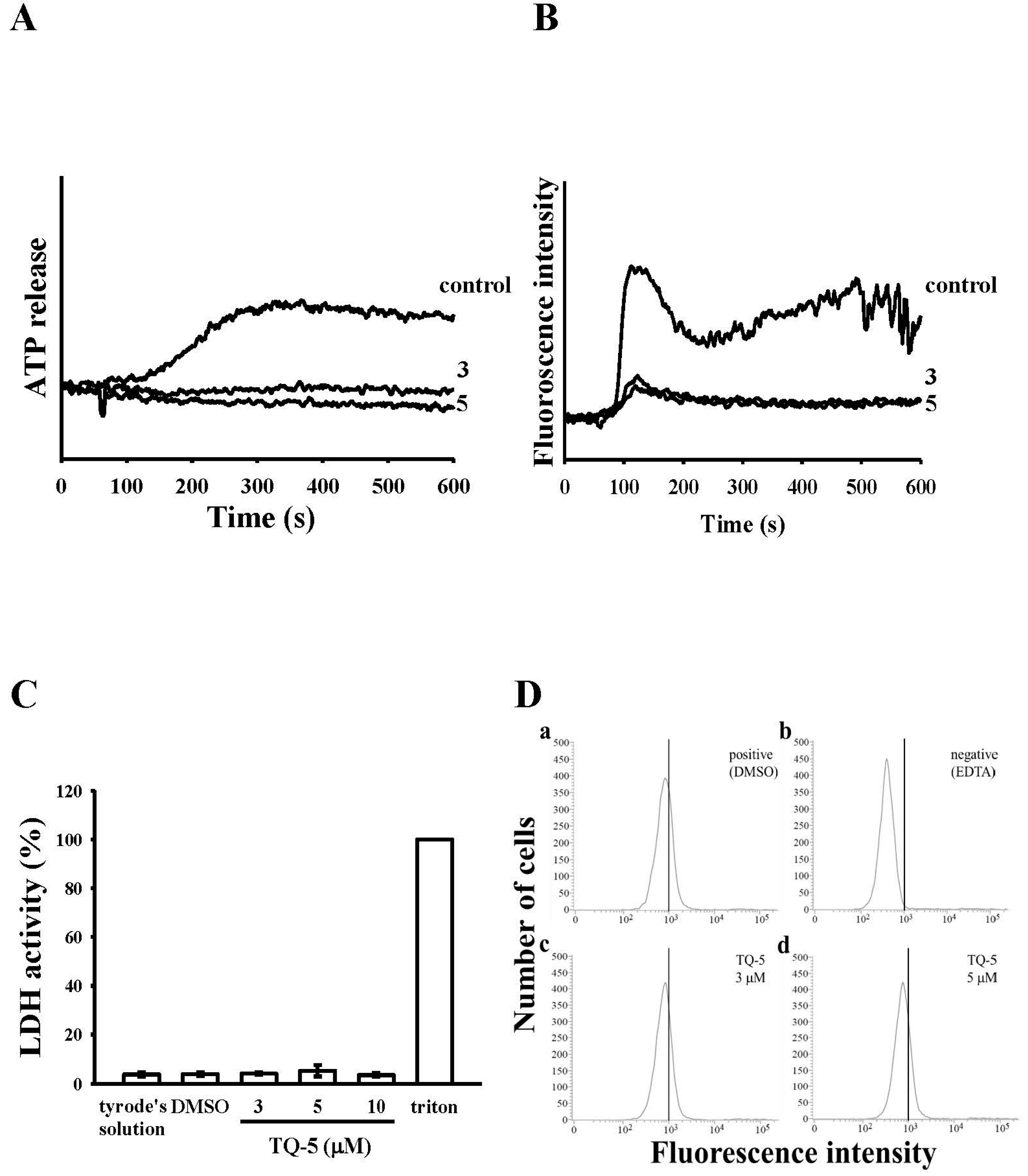
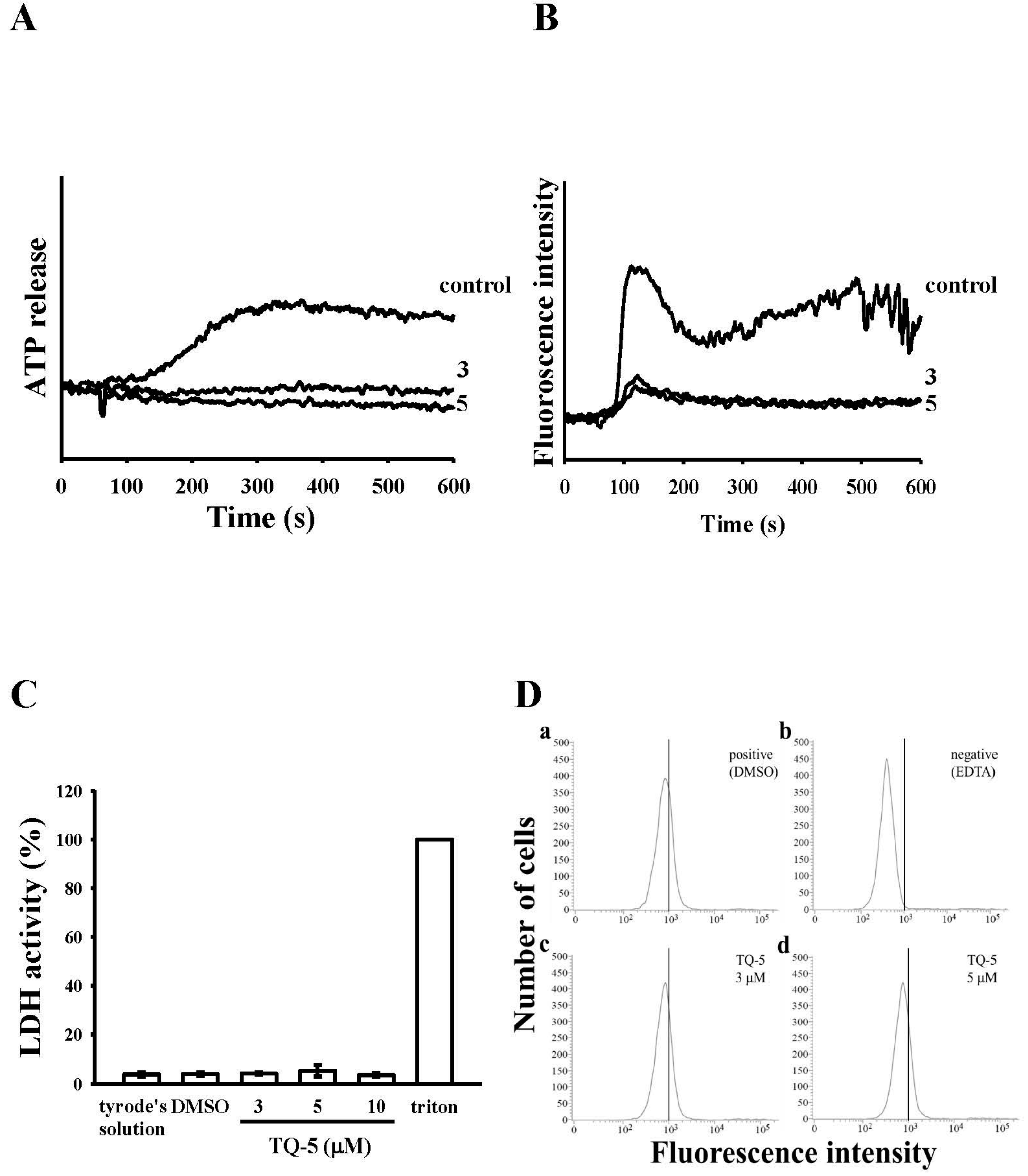
2.1.3. Effect of TQ5 on Adenosine Triphosphate (ATP) Release and [Ca2+]i Mobilization in Human Platelets
2.1.4. TQ5 Either Not Induced Cytotoxicity or Not Directly Binds to the Platelet αIIbβ3Integrin in Platelets
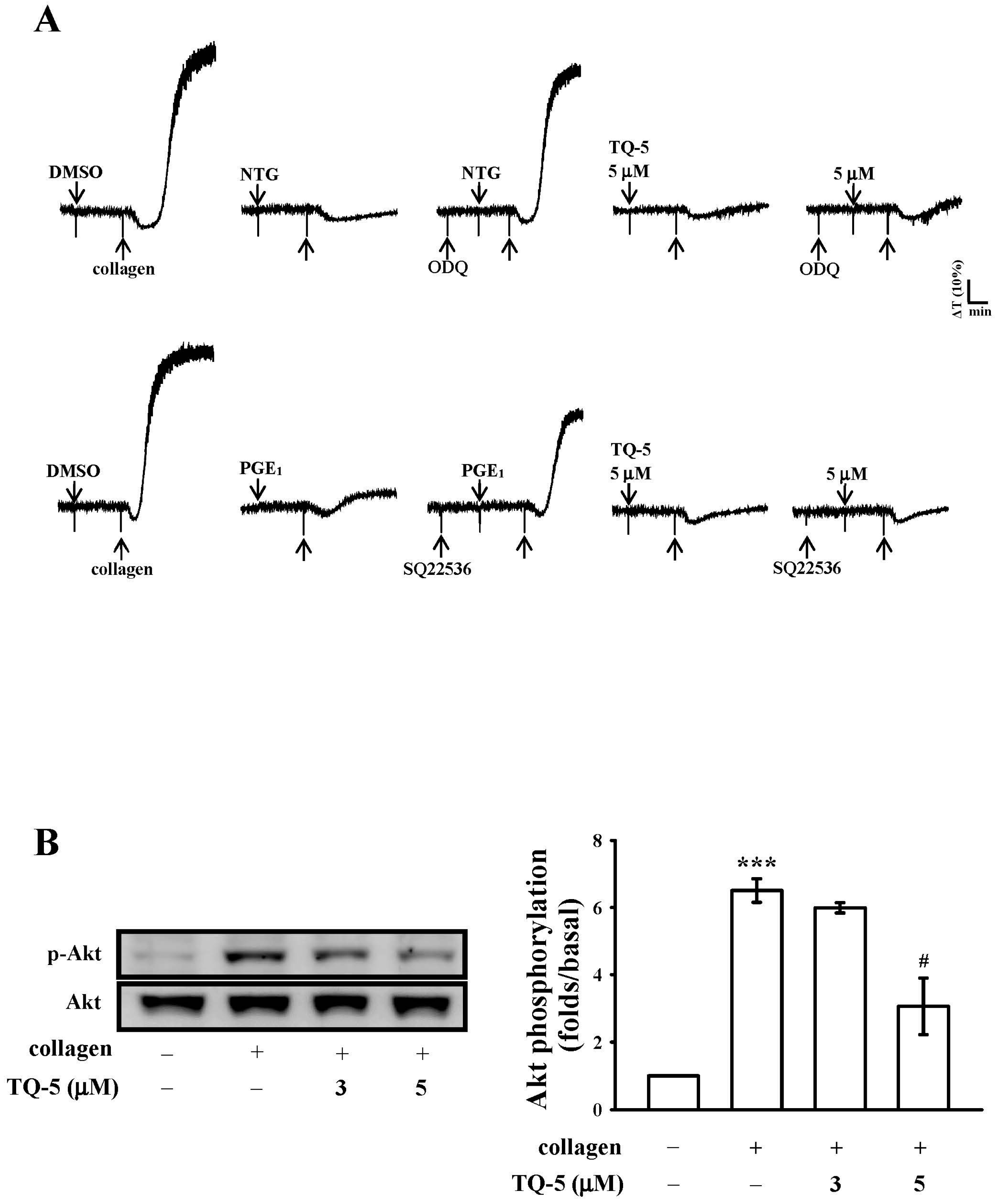
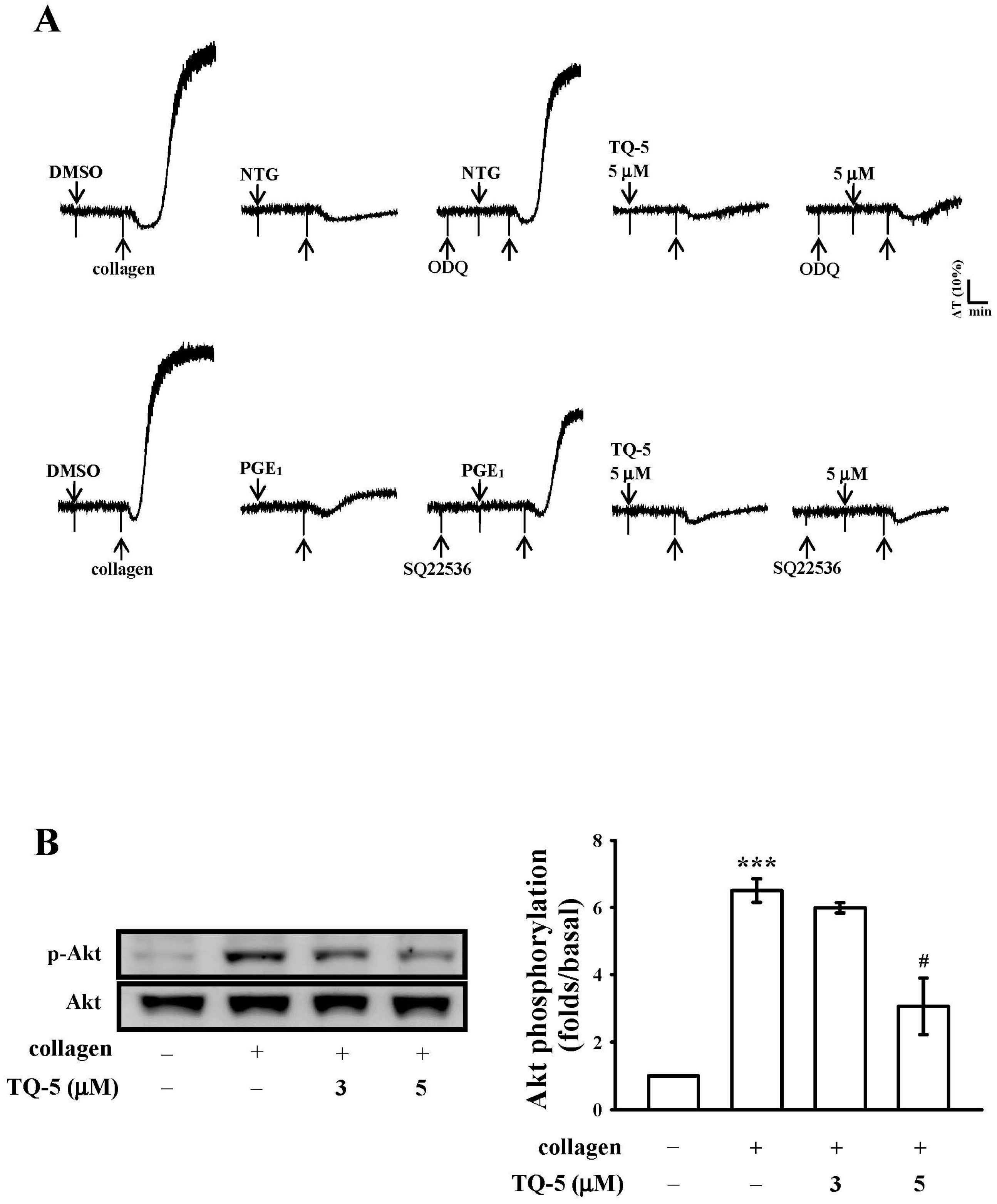
2.1.5. TQ5 on Cyclic Nucleotides Formation
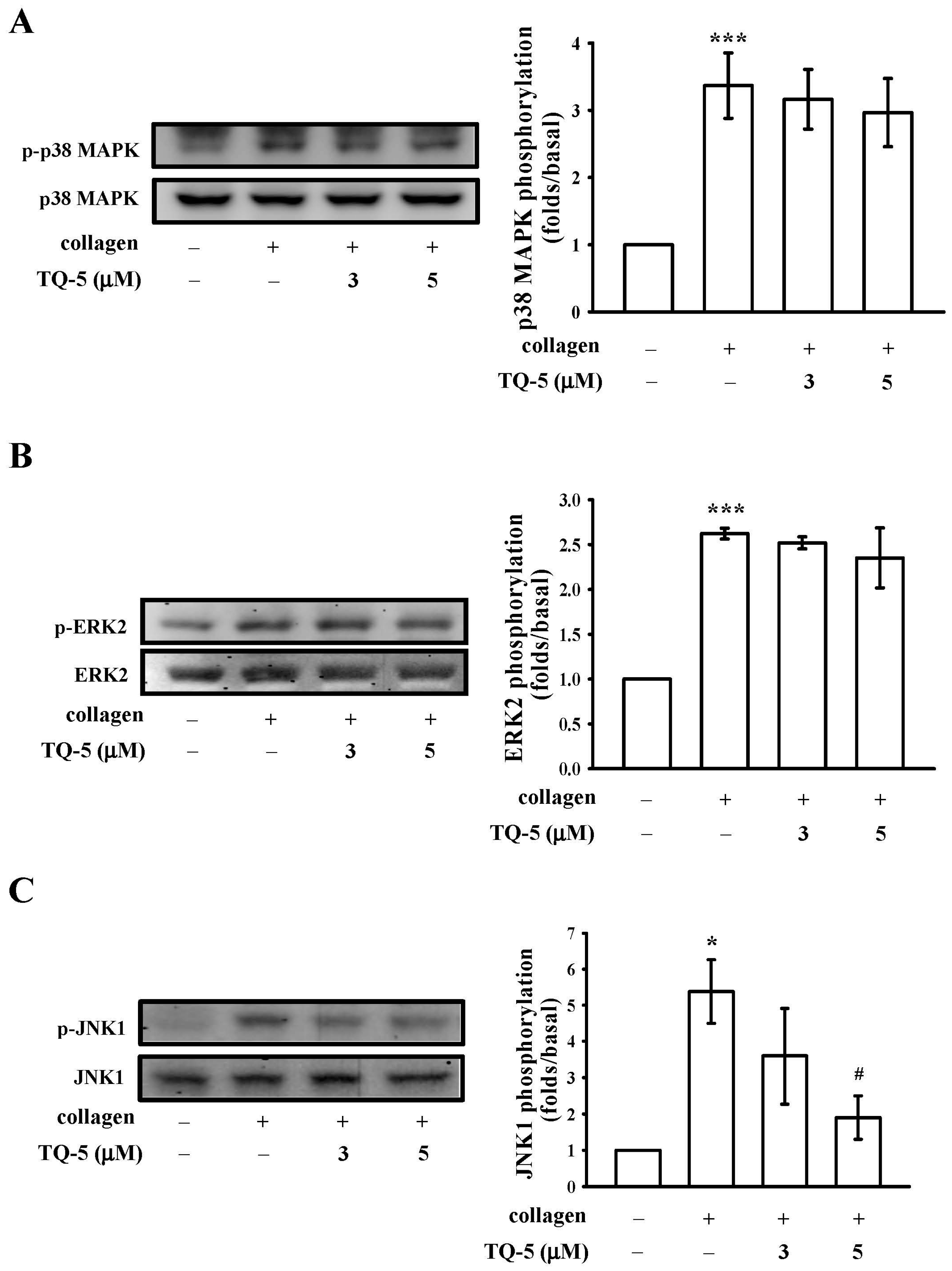
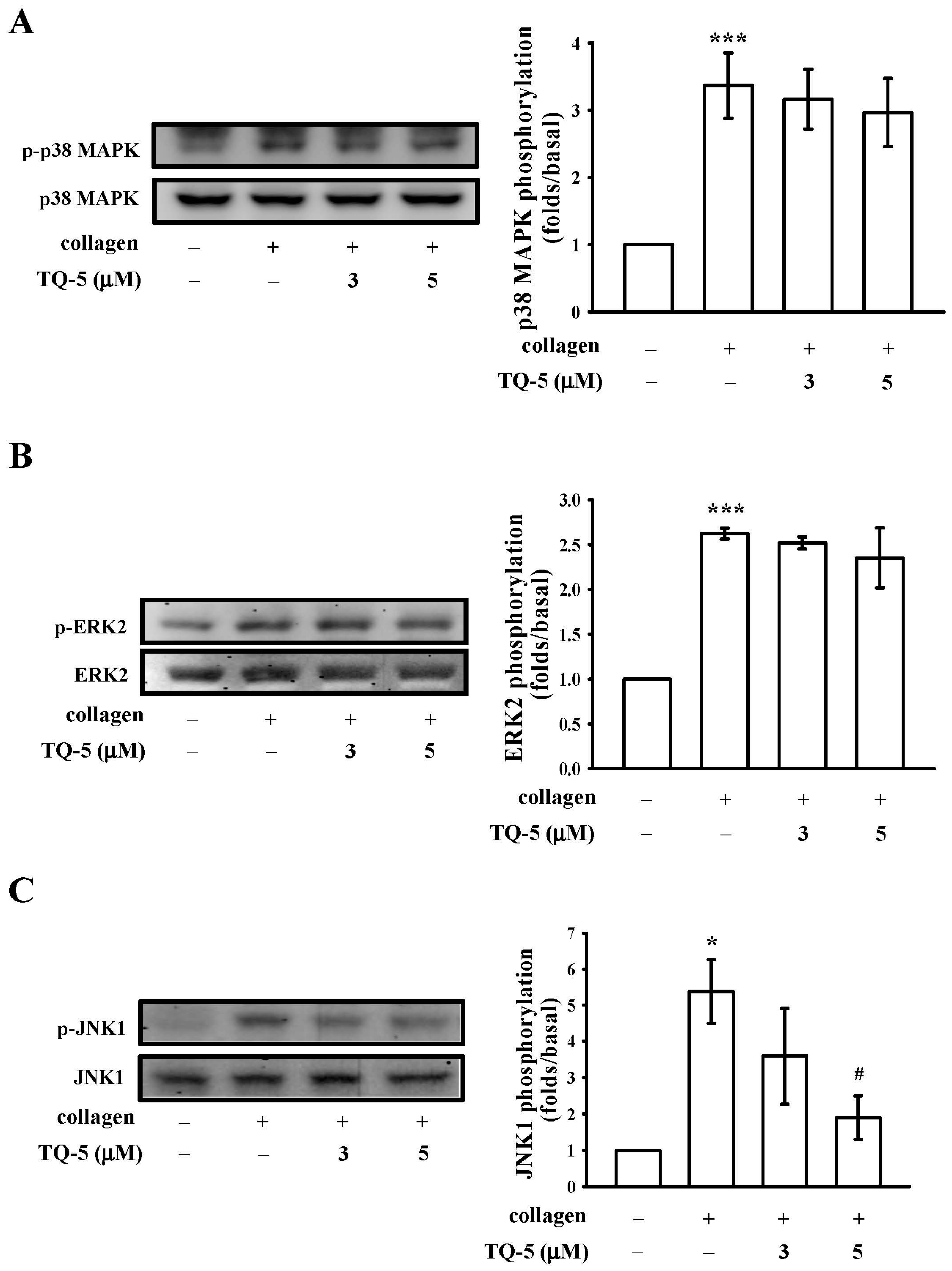
2.1.6. TQ5 Attenuated Protein Kinase B (Akt) and c-Jun N-Terminal Kinase (JNK) Phosphorylation in Collagen-Induced Human Platelets
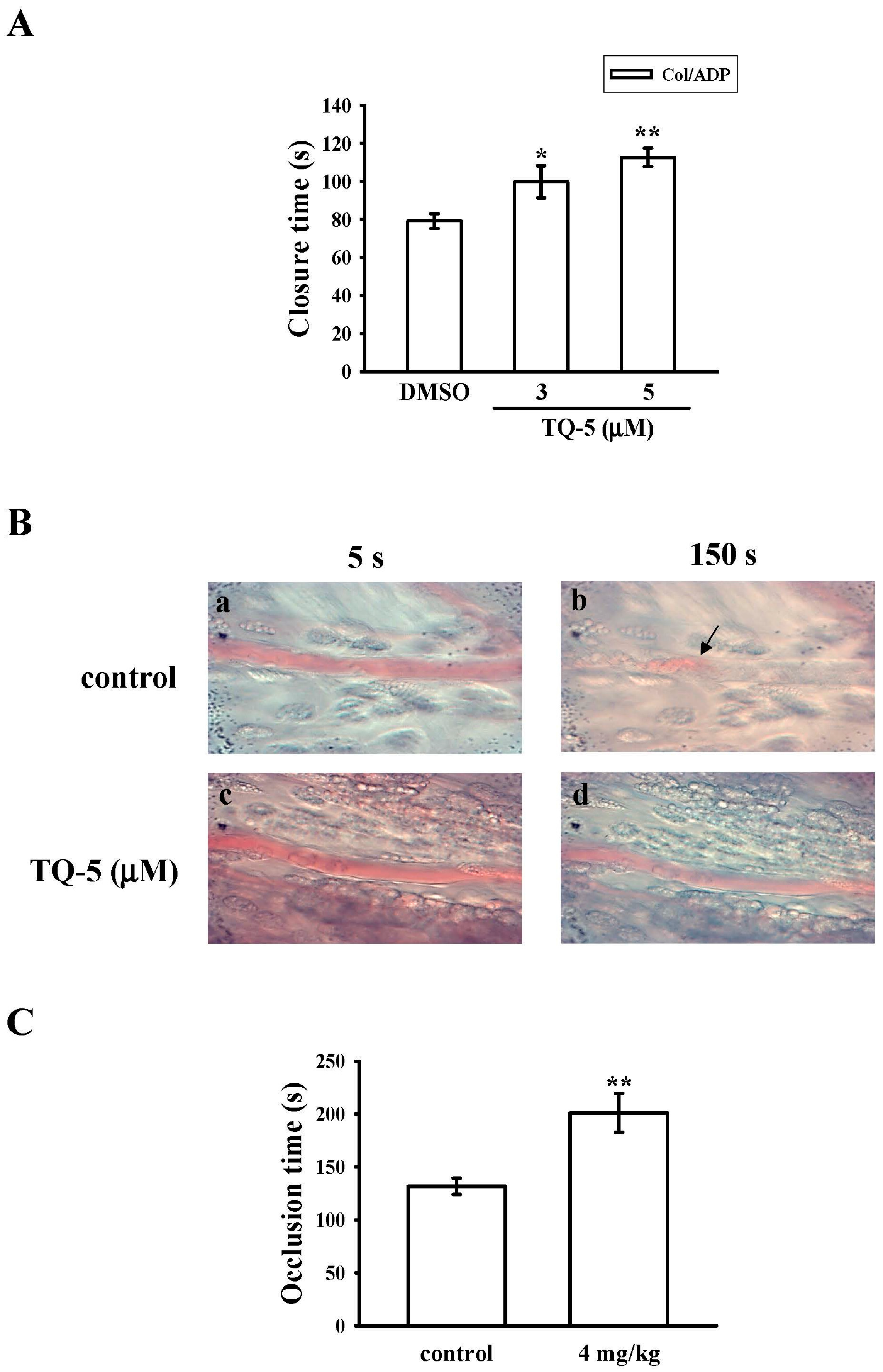
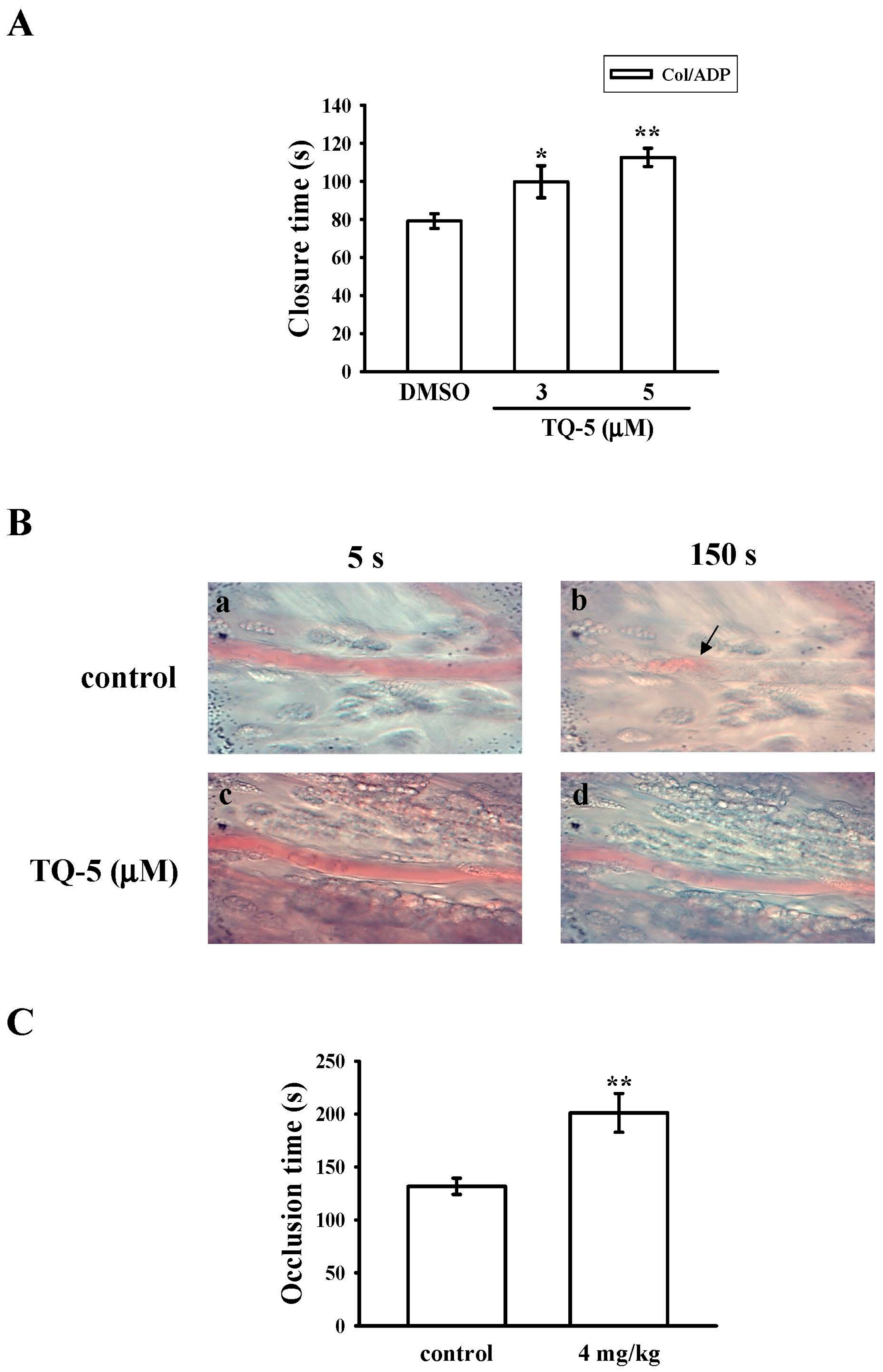
2.1.7. Ex Vivo and In Vivo Studies of TQ5 in Antithrombotic Activity
2.2. Discussion
3. Experimental Section
3.1. Materials
3.2. Synthesis of Ligand 4-Phenyl-2-pyridin-2-yl-quinazoline (L)
3.3. Synthesis of [Ru(η6-Cymene)(L)Cl]BF4 (TQ5)
3.4. Platelet Aggregation and ATP Release
3.5. Measurement of Relative Ca2+ Mobilization by Fura 2-AM Fluorescence
3.6. Immunoblotting
3.7. Analysis of Platelet Function in Whole Blood
3.8. Fluorescein-Induced Thrombus Formation in the Microvessels of Mouse Mesentery
3.9. Statistical Analysis
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Sierko, E.; Wojtukiewicz, M.Z. Platelets and angiogenesis in malignancy. Semin. Thromb. Hemost. 2004, 30, 95–108. [Google Scholar] [PubMed]
- Belloc, C.; Lu, H.; Soria, C.; Fridman, R.; Legrand, Y.; Menashi, S. The effect of platelets on invasiveness and protease production of human mammary tumor cells. Int. J. Cancer 1995, 60, 413–417. [Google Scholar] [CrossRef] [PubMed]
- Felding-Habermann, B.; Ooole, T.E.; Smith, J.W.; Fransvea, E.; Ruggeri, Z.M.; Ginsberg, M.H.; Hughes, P.E.; Pampori, N.; Shattil, S.J.; Saven, A.; et al. Integrin activation controls metastasis in human breast cancer. Proc. Natl. Acad. Sci. USA 2001, 98, 1853–1858. [Google Scholar] [CrossRef] [PubMed]
- Holmes, C.E.; Levis, J.E.; Ornstein, D.L. Activated platelets enhance ovarian cancer cell invasion in a cellular model of metastasis. Clin. Exp. Metastasis 2009, 26, 653–661. [Google Scholar] [CrossRef] [PubMed]
- Boucharaba, A.; Serre, C.M.; Gres, S.; Saulnier-Blache, J.S.; Bordet, J.C.; Guglielmi, J.; Clezardin, P.; Peyruchaud, O. Platelet-derived lysophosphatidic acid supports the progression of osteolytic bone metastases in breast cancer. J. Clin. Investig. 2004, 114, 1714–1725. [Google Scholar] [CrossRef] [PubMed]
- Haralabopoulos, G.C.; Grant, D.S.; Kleinman, H.K.; Maragoudakis, M.E. Thrombin promotes endothelial cell alignment in Matrigel in vitro and angiogenesis in vivo. Am. J. Physiol. 1997, 273, C239–C245. [Google Scholar] [PubMed]
- Zacharski, L.R.; Memoli, V.A.; Ornstein, D.L.; Rousseau, S.M.; Kisiel, W.; Kudryk, B.J. Tumor cell procoagulant and urokinase expression in carcinoma of the ovary. J. Natl. Cancer Inst. 1993, 85, 1225–1230. [Google Scholar] [CrossRef] [PubMed]
- Zucchella, M.; Dezza, L.; Pacchiarini, L.; Meloni, F.; Tacconi, F.; Bonomi, E.; Grignani, G.; Notario, A. Human tumor cells cultured “in vitro” activate platelet function by producing ADP or thrombin. Haematologica 1989, 74, 541–545. [Google Scholar] [PubMed]
- Ikeda, M.; Furukawa, H.; Imamura, H.; Shimizu, J.; Ishida, H.; Masutani, S.; Tatsuta, M.; Satomi, T. Poor prognosis associated with thrombocytosis in patients with gastric cancer. Ann. Surg. Oncol. 2002, 9, 287–291. [Google Scholar] [CrossRef] [PubMed]
- Monreal, M.; Fernandez-Llamazares, J.; Piñol, M.; Julian, J.F.; Broggi, M.; Escola, D.; Abad, A. Platelet count and survival in patients with colorectal cancer—A preliminary study. Thromb. Haemost. 1998, 79, 916–918. [Google Scholar] [PubMed]
- Symbas, N.P.; Townsend, M.F.; El-Galley, R.; Keane, T.E.; Graham, S.D.; Petros, J.A. Poor prognosis associated with thrombocytosis in patients with renal cell carcinoma. BJU Int. 2000, 86, 203–207. [Google Scholar] [CrossRef] [PubMed]
- Nierodzik, M.L.; Klepfish, A.; Karpatkin, S. Role of platelets, thrombin, integrin IIb-IIIa, fibronectin and von Willebrand factor on tumor adhesion in vitro and metastasis in vivo. Thromb. Haemost. 1995, 74, 282–290. [Google Scholar] [PubMed]
- Wenzel, J.; Zeisig, R.; Fichtner, I. Inhibition of metastasis in a murine 4T1 breast cancer model by liposomes preventing tumor cell-platelet interactions. Clin. Exp. Metastasis 2010, 27, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Tello-Montoliu, A.; Jover, E.; Rivera, J.; Valdes, M.; Angiolillo, D.J.; Marin, F. New perspectives in antiplatelet therapy. Curr. Med. Chem. 2012, 19, 406–427. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Stojanovic, A.; Hay, N.; Leibovitch, S.A. The role of Akt in the signaling pathway of the lycoprotein Ib-IX induced platelet activation. Blood 2008, 111, 658–665. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.J.; Han, J.H.; Jung, S.H.; Lee, S.G.; Kim, I.S.; Cuong, N.M.; Huong, T.T.; Khanh, P.N.; Kim, Y.H.; Yun, Y.P.; et al. Antiplatelet action of indirubin-3′-monoxime through suppression of glycoprotein VI-mediated signal transduction: A possible role for ERK signaling in platelets. Vascul. Pharmacol. 2014, 63, 182–192. [Google Scholar] [CrossRef] [PubMed]
- Bruijnincx, P.C.; Sadler, P.J. New trends for metal complexes with anticancer activity. Curr. Opin. Chem. Biol. 2008, 12, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Kelland, L. The resurgence of platinum-based cancer chemotherapy. Nat. Rev. Cancer 2007, 7, 573–584. [Google Scholar] [CrossRef] [PubMed]
- Habtemariam, A.; Melchart, M.; Fernandez, R.; Parsons, S.; Oswald, I.D.H.; Parkin, A.; Fabbiani, F.P.A.; Davidson, J.E.; Dawson, A.; Aird, R.E.; et al. Structure-activity relationships for cytotoxic ruthenium(II) arene complexes containing N,N-, N,O-, and O,O-chelating ligands. J. Med. Chem. 2006, 49, 6858–6868. [Google Scholar] [CrossRef] [PubMed]
- Gismondi, A.; Reina, G.; Orlanducci, S.; Mizzoni, F.; Gay, S.; Terranova, M.L.; Canini, A. Nanodiamonds coupled with plant bioactive metabolites: A nanotech approach for cancer therapy. Biomaterials 2015, 38, 22–35. [Google Scholar] [CrossRef] [PubMed]
- Asif, M. Chemical characteristics, synthetic methods, and biological potential of quinazoline and quinazolinone derivatives. Int. J. Med. Chem. 2014, 2014, 395637. [Google Scholar] [CrossRef] [PubMed]
- Gawad, N.M.A.; Georgey, H.H.; Youssef, R.M.; El-Sayed, N.A. Synthesis and antitumor activity of some 2, 3-disubstituted quinazolin-4(3H)-ones and 4, 6-disubstituted-1,2,3,4-tetrahydroquinazolin-2H-ones. Eur. J. Med. Chem. 2010, 45, 6058–6067. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Liao, S.Y.; Tan, C.P.; Ye, R.R.; Xu, Y.W.; Zhao, M.; Ji, L.N.; Mao, Z.W. Ruthenium-arene-β-carboline complexes as potent inhibitors of cyclin dependent kinase 1: Synthesis, characterization and anticancer mechanism studies. Chem. Eur. J. 2013, 19, 12152–12160. [Google Scholar] [CrossRef] [PubMed]
- Davies, D.L.; Duaij, O.A.; Fawcett, J.; Giardiello, M.; Hilton, S.T.; Russell, D.R. Room-temperature cyclometallation of amines, imines and oxazolines with [MCl2Cp*]2 (M = Rh, Ir) and [RuCl2( p-cymene)]2. Dalton Trans. 2003, 21, 4132–4138. [Google Scholar] [CrossRef]
- Jilma, B. Platelet function analyzer (PFA-100): A tool to quantify congenital or acquired platelet dysfunction. J. Lab. Clin. Med. 2001, 138, 152–163. [Google Scholar] [CrossRef] [PubMed]
- Lip, G.Y.; Chin, B.S.; Blann, A.D. Cancer and the prothrombotic state. Lancet Oncol. 2002, 3, 27–34. [Google Scholar] [CrossRef]
- Park, J.Y.; Hong, M.; Jia, Q.; Lee, Y.C.; Yayeh, T.; Hyun, E.; Kwak, D.M.; Cho, J.Y.; Rhee, M.H. Pistacia chinensis Methanolic Extract Attenuated MAPK and Akt Phosphorylations in ADP Stimulated Rat Platelets In Vitro. Evid. Based Complement. Altern. Med. 2012, 2012, 895729. [Google Scholar] [CrossRef] [PubMed]
- Ferroni, P.; Della-Morte, D.; Palmirotta, R.; McClendon, M.; Testa, G.; Abete, P.; Rengo, F.; Rundex, T.; Guadagni, F.; Roselli, M. Platinum-based compounds and risk for cardiovascular toxicity in the elderly: Role of the antioxidants in chemoprevention. Rejuvenation Res. 2011, 14, 293–308. [Google Scholar] [CrossRef] [PubMed]
- Jafri, M.; Protheroe, A. Cisplatin-associated thrombosis. Anticancer Drugs 2008, 19, 927–929. [Google Scholar] [CrossRef] [PubMed]
- Barni, S.; Labianca, R.; Agnelli, G.; Bonizzoni, E.; Verso, M.; Mandala, M.; Brighenti, M.; Petrelli, F.; Bianchini, C.; Perrone, T.; et al. Chemotherapy-associated thromboembolic risk in cancer outpatients and effect of nadroparin thromboprophylaxis: Results of a retrospective analysis of the PROTECHT study. J. Transl. Med. 2011, 9, 179. [Google Scholar] [CrossRef] [PubMed]
- Dasanu, C.A. Gemcitabine: Vascular toxicity and prothrombotic potential. Expert Opin. Drug Saf. 2008, 7, 703–716. [Google Scholar] [CrossRef] [PubMed]
- Jennings, L.K. Role of platelets in atherothrombosis. Am. J. Cardiol. 2009, 103, 4A–10A. [Google Scholar] [CrossRef] [PubMed]
- Kaibuchi, K.; Sano, K.; Hoshijima, M.; Takai, Y.; Nishizuka, Y. Phosphatidylinositol turnover in platelet activation; calcium mobilization and protein phosphorylation. Cell Calcium 1982, 3, 323–335. [Google Scholar] [CrossRef]
- Walter, U.; Eigenthaler, M.; Geiger, J.; Reinhard, M. Role of cyclic nucleotide dependent protein kinases and their common substrate VASP in the regulation of human platelets. Adv. Exp. Med. Biol. 1993, 344, 237–249. [Google Scholar] [PubMed]
- Lu, W.J.; Lin, K.C.; Liu, C.P.; Lin, C.Y.; Wu, H.C.; Chou, D.S.; Geraldine, P.; Huang, S.Y.; Hsieh, C.Y.; Sheu, J.R. Prevention of arterial thrombosis by nobiletin: In vitro and in vivo studies. J. Nutr. Biochem. 2016, 28, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.P.; Yap, C.L.; Anderson, K.E. Phosphoinositide 3-kinases and the regulation of platelet function. Biochem. Soc. Trans. 2004, 32, 387–392. [Google Scholar] [CrossRef] [PubMed]
- Moore, S.F.; van den Bosch, M.T.; Hunter, R.W.; Sakamoto, K.; Poole, A.W.; Hers, I. Dual regulation of glycogen synthase kinase 3 (GSK3)α/β by protein kinase C (PKC)α and Akt promotes thrombin-mediated integrin αIIbβ3 activation and granule secretion in platelets. J. Biol. Chem. 2013, 288, 3918–3928. [Google Scholar] [CrossRef] [PubMed]
- Dangelmaier, C.; Manne, B.K.; Liverani, E.; Jin, J.; Bray, P.; Kunapuli, S.P. PDK1 selectively phosphorylates Thr(308) on AKT and contributes to human platelet functional responses. Thromb. Haemost. 2014, 111, 508–517. [Google Scholar] [CrossRef] [PubMed]
- Deb, T.B.; Coticchia, C.M.; Dickson, R.B. Calmodulin-mediated activation of Akt regulates survival of c-Myc-overexpressing mouse mammary carcinoma cells. J. Biol. Chem. 2004, 279, 38903–38911. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; De, S.; Damron, D.S.; Chen, W.S.; Hay, N.; Byzova, T.V. Impaired platelet responses to thrombin and collagen in AKT-1-deficient mice. Blood 2004, 104, 1703–1710. [Google Scholar] [CrossRef] [PubMed]
- Adam, F.; Kauskot, A.; Nurden, P.; Sulpice, E.; Hoylaerts, M.F.; Davis, R.J.; Rosa, J.P.; Bryckaert, M. Platelet JNK1 is involved in secretion and thrombus formation. Blood 2010, 115, 4083–4092. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.; Karin, M. Mammalian MAP kinase signalling cascades. Nature 2001, 410, 37–40. [Google Scholar] [CrossRef] [PubMed]
- Hughes, P.E.; Renshaw, M.W.; Pfaff, M.; Forsyth, J.; Keivens, V.M.; Schwartz, M.A.; Ginsberg, M.H. Suppression of integrin activation: A novel function of a Ras/Raf-initiated MAP kinase pathway. Cell 1997, 88, 521–530. [Google Scholar] [CrossRef]
- Ager, D.J.; de Vries, A.H.M.; de Vries, J.G. Phosphoramidite-controlled asymmetric hydrogenation with rhodium catalysts. Platin. Met. Rev. 2006, 50, 54–63. [Google Scholar] [CrossRef]
- Levina, A.; Mitra, A.; Lay, P.A. Recent developments in ruthenium anticancer drugs. Metallomics 2009, 1, 458–470. [Google Scholar] [CrossRef] [PubMed]
- Zacharski, L.R.; Donati, M.B.; Rickles, F.R. Registry of clinical trials of antithrombotic drugs in cancer: Second report. The Scientific and Standardization Committee of the International Society on Thrombosis and Haemostasis Subcommittee on Hemostasis and Malignancy. Thromb. Haemost. 1993, 70, 357–360. [Google Scholar] [PubMed]
- Sheu, J.R.; Lee, C.R.; Lin, C.H.; Hsiao, G.; Ko, W.C.; Chen, Y.C.; Yen, M.H. Mechanisms involved in the antiplatelet activity of Staphylococcus aureus lipoteichoic acid in human platelets. Thromb. Haemost. 2000, 83, 777–784. [Google Scholar] [PubMed]
- Lin, K.H.; Kuo, J.R.; Lu, W.J.; Chung, C.L.; Chou, D.S.; Huang, S.Y.; Lee, H.C.; Sheu, J.R. Hinokitiol inhibits platelet activation ex vivo and thrombus formation in vivo. Biochem. Pharmacol. 2013, 85, 1478–1485. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, G.; Lin, K.H.; Chang, Y.; Chen, T.L.; Tzu, N.H.; Chou, D.S.; Sheu, J.R. Protective mechanisms of inosine in platelet activation and cerebral ischemic damage. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 1998–2004. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khamrang, T.; Hung, K.-C.; Hsia, C.-H.; Hsieh, C.-Y.; Velusamy, M.; Jayakumar, T.; Sheu, J.-R. Antiplatelet Activity of a Newly Synthesized Novel Ruthenium (II): A Potential Role for Akt/JNK Signaling. Int. J. Mol. Sci. 2017, 18, 916. https://doi.org/10.3390/ijms18050916
Khamrang T, Hung K-C, Hsia C-H, Hsieh C-Y, Velusamy M, Jayakumar T, Sheu J-R. Antiplatelet Activity of a Newly Synthesized Novel Ruthenium (II): A Potential Role for Akt/JNK Signaling. International Journal of Molecular Sciences. 2017; 18(5):916. https://doi.org/10.3390/ijms18050916
Chicago/Turabian StyleKhamrang, Themmila, Kuo-Chen Hung, Chih-Hsuan Hsia, Cheng-Ying Hsieh, Marappan Velusamy, Thanasekaran Jayakumar, and Joen-Rong Sheu. 2017. "Antiplatelet Activity of a Newly Synthesized Novel Ruthenium (II): A Potential Role for Akt/JNK Signaling" International Journal of Molecular Sciences 18, no. 5: 916. https://doi.org/10.3390/ijms18050916
APA StyleKhamrang, T., Hung, K. -C., Hsia, C. -H., Hsieh, C. -Y., Velusamy, M., Jayakumar, T., & Sheu, J. -R. (2017). Antiplatelet Activity of a Newly Synthesized Novel Ruthenium (II): A Potential Role for Akt/JNK Signaling. International Journal of Molecular Sciences, 18(5), 916. https://doi.org/10.3390/ijms18050916