Influence of the Periodontal Disease, the Most Prevalent Inflammatory Event, in Peroxisome Proliferator-Activated Receptors Linking Nutrition and Energy Metabolism

{kind=link}
{kind=link}
Abstract
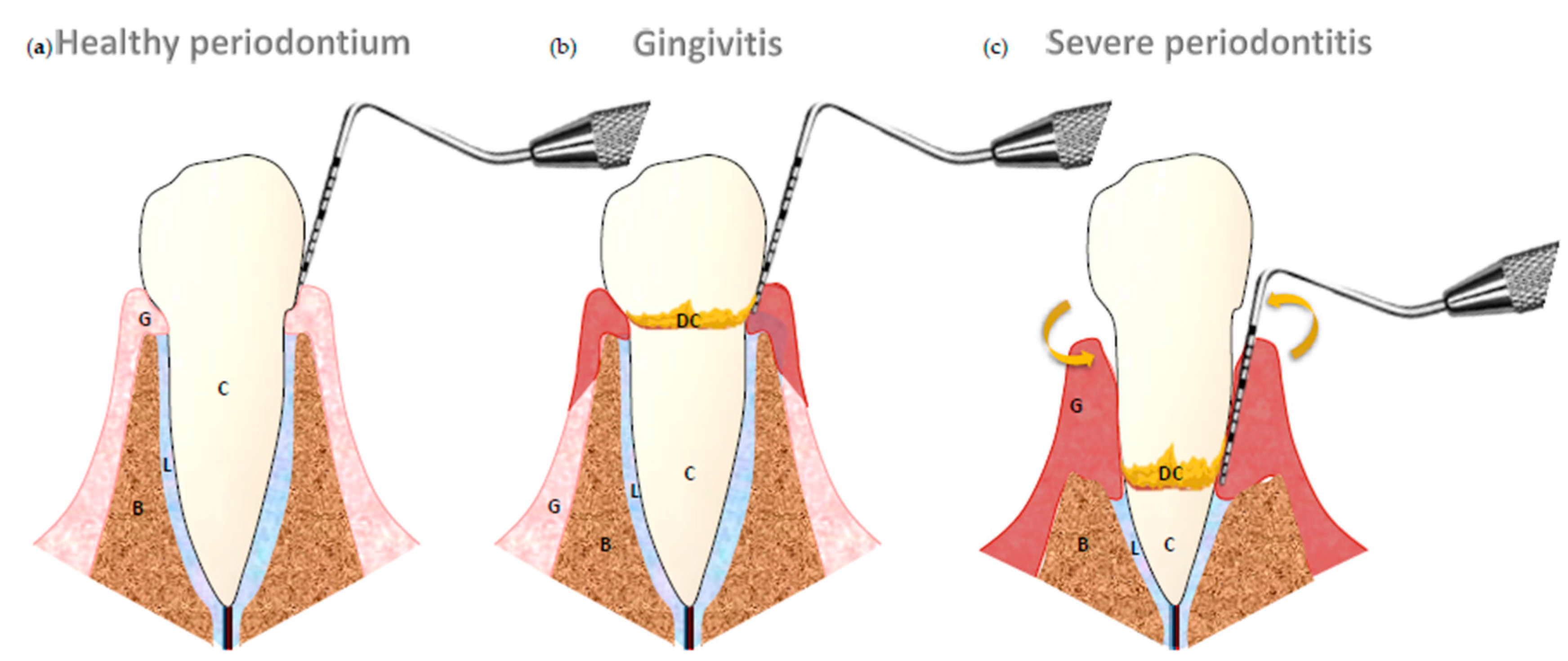
:1. Introduction
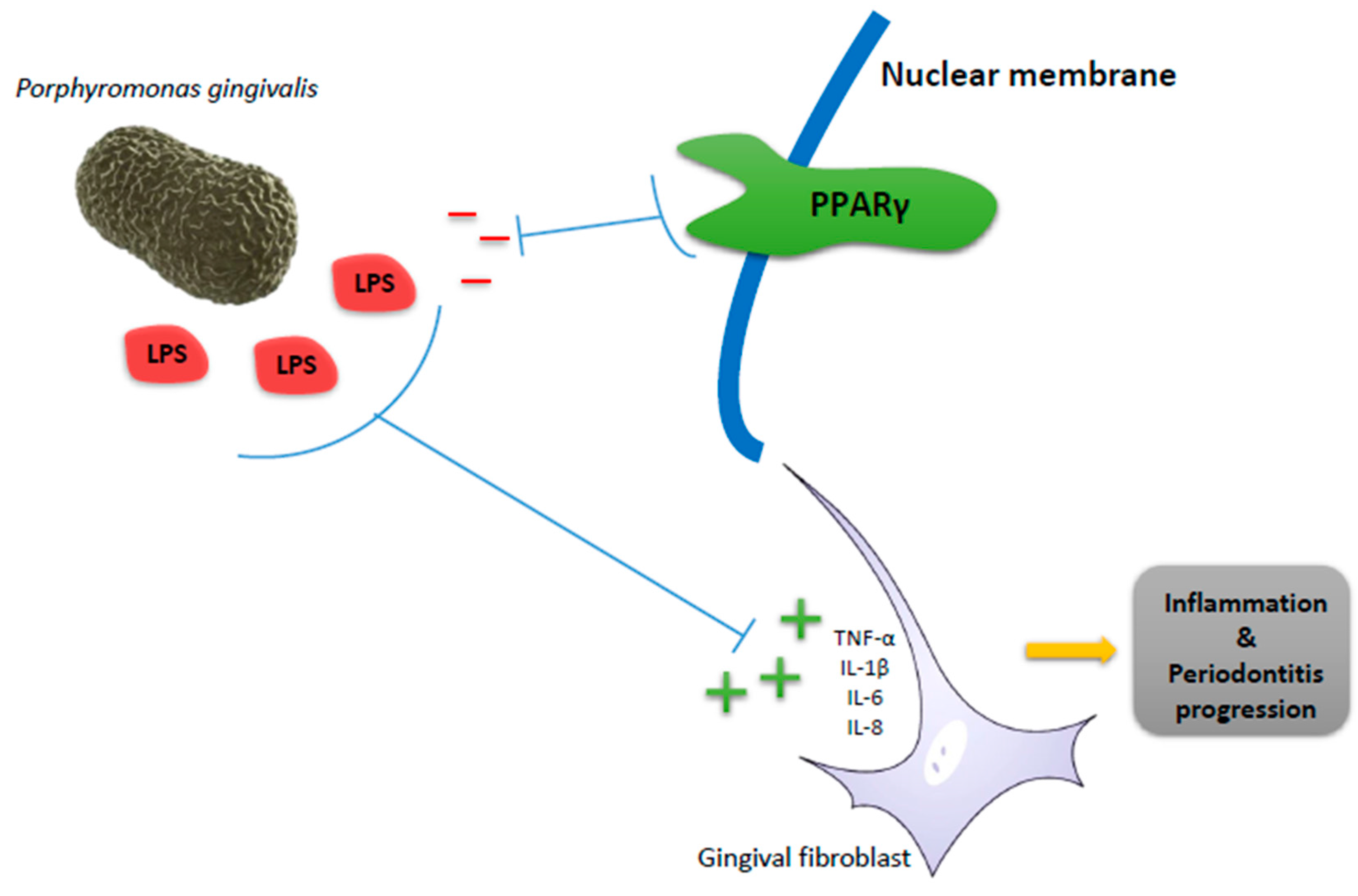
2. Peroxisome Proliferator-Activated Receptors (PPARs) in Periodontitis as Inflammatory Disease
3. PPARs in Periodontitis as Bone Disease
4. PPARs in Periodontitis and Its Related Systemic Diseases
5. Conclusions
Conflicts of Interest
Abbreviations
| PAMPs | Pathogen-Associated Molecular Patterns |
| DAMPs | Danger-Associated Molecular Patterns |
| PRRs | Pattern Recognition Receptors |
| NLRP3 | Nod Like Receptors Family, Pyrin Domain Containing 3 |
| ATP | Adenosine 5´-Triphosphate |
| ROS | Reactive Oxygen Species |
| mTOR | Mammalian Target of Rapamycin |
| IL | Interleukin |
| COX | Cyclooxygenase |
| MSC | Mesenchymal Stem Cells |
| HGF | Human Gingival Fibroblasts |
| RGZ | Rosiglitazone |
| RANKL | Receptor Activator for Nuclear Factor κ B Ligand |
| NF-κB | nuclear factor kappa-light-chain-enhancer of activated B cells |
| OPG | Osteoprotegerin |
| BMD | Bone Mineral Density |
| Ch-GNPs | Chitosan Gold Nanoparticles |
| miRNAs | MicroRNAs |
| BMI | Body Mass Index |
| MUFA | Monounsaturated Fatty Acids |
| PUFA | Polyunsaturated Fatty Acids |
| TNF | Tumor Necrosis Factor |
References
- Armitage, G.C. Diagnosis of periodontal diseases. J. Periodontol. 2003, 74, 1237–1247. [Google Scholar] [CrossRef] [PubMed]
- Buset, S.L.; Walter, C.; Friedmann, A.; Weiger, R.; Borgnakke, W.S.; Zitzmann, N.U. Are periodontal diseases really silent? A systematic review of their effect on quality of life. J. Clin. Periodontol. 2016, 43, 333–344. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, W.M.M.; Papapanou, P.N. Epidemiology of periodontal disease in children and adolescents. Periodontol. 2000 2001, 26, 16–32. [Google Scholar] [CrossRef] [PubMed]
- König, J.; Holtfreter, B.; Kocher, T. Periodontal health in Europe: Future trends based on treatment needs and the provision of periodontal services—Position paper 1. Eur. J. Dent. Educ. 2010, 14, 4–24. [Google Scholar] [CrossRef] [PubMed]
- Madianos, P.; Papaioannou, W.; Herrera, D.; Sanz, M.; Baeumer, A.; Bogren, A.; Bouchard, P.; Chomyszyn-Gajewska, M.; Demirel, K.; Gaspersic, R.; et al. EFP Delphi study on the trends in periodontology and periodontics in Europe for the year 2025. J. Clin. Periodontol. 2016, 43, 472–481. [Google Scholar] [CrossRef] [PubMed]
- Armitage, G.C. Periodontal diagnoses and classification of periodontal diseases. Periodontol. 2000 2004, 34, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Mendes, L.; Azevedo, N.F.; Felino, A.; Pinto, M.G. Relationship between invasion of the periodontium by periodontal pathogens and periodontal disease: A systematic review. Virulence 2015, 6, 208–215. [Google Scholar] [CrossRef] [PubMed]
- Ferretti, F. Unhealthy Behaviours: An International Comparison. PLoS ONE 2015, 10, e0141834. [Google Scholar] [CrossRef] [PubMed]
- Santoni, G.; Cardinali, C.; Morelli, M.B.; Santoni, M.; Nabissi, M.; Amantini, C. Danger- and pathogen-associated molecular patterns recognition by pattern-recognition receptors and ion channels of the transient receptor potential family triggers the inflammasome activation in immune cells and sensory neurons. J. Neuroinflamm. 2015, 12, 21. [Google Scholar] [CrossRef] [PubMed]
- Schroder, K.; Tschopp, J. The inflammasomes. Cell 2010, 140, 821–832. [Google Scholar] [CrossRef] [PubMed]
- Pedra, J.H.F.; Cassel, S.L.; Sutterwala, F.S. Sensing pathogens and danger signals by the inflammasome. Curr. Opin. Immunol. 2009, 21, 10–16. [Google Scholar] [CrossRef] [PubMed]
- Olsen, I.; Yilmaz, Ö. Modulation of inflammasome activity by Porphyromonas gingivalis in periodontitis and associated systemic diseases. J. Oral Microbiol. 2016, 8, 30385. [Google Scholar] [CrossRef] [PubMed]
- Dagenais, M.; Skeldon, A.; Saleh, M. The inflammasome: In memory of Dr. Jurg Tschopp. Cell Death Differ. 2012, 19, 5–12. [Google Scholar] [CrossRef] [PubMed]
- Varela-López, A.; Quiles, J.L.; Cordero, M.; Giampieri, F.; Bullón, P. Oxidative stress and dietary fat type in relation to periodontal disease. Antioxidants 2015, 4, 322–344. [Google Scholar] [CrossRef] [PubMed]
- Sies, H. Biochemistry of oxidative stress. Angew. Chem. Int. Ed. Engl. 1986, 25, 1058–1071. [Google Scholar] [CrossRef]
- Valko, M.; Leibfritz, D.; Moncol, J.; Cronin, M.T.D.; Mazur, M.; Telser, J. Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell Biol. 2007, 39, 44–84. [Google Scholar] [CrossRef] [PubMed]
- Battino, M.; Bullon, P.; Wilson, M.; Newman, H. Oxidative Injury and inflammatory periodontal diseases : The challenge of anti-oxidants to free radicals and reactive oxygen species. Crit. Rev. Oral Biol. Med. 1999, 10, 458–476. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; Mariño, G.; Levine, B. Autophagy and the integrated stress response. Mol. Cell 2010, 40, 280–293. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Kroemer, G. Autophagy in the pathogenesis of disease. Cell 2008, 132, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Bullon, P.; Cordero, M.D.; Quiles, J.L.; Ramirez-Tortosa, M.D.C.; Gonzalez-Alonso, A.; Alfonsi, S.; García-Marín, R.; de Miguel, M.; Battino, M. Autophagy in periodontitis patients and gingival fibroblasts: Unraveling the link between chronic diseases and inflammation. BMC Med. 2012, 10, 122. [Google Scholar] [CrossRef] [PubMed]
- Scherz-Shouval, R.; Elazar, Z. ROS, mitochondria and the regulation of autophagy. Trends Cell Biol. 2007, 17, 422–427. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011, 469, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Jung, C.H.; Ro, S.-H.; Cao, J.; Otto, N.M.; Kim, D.-H. mTOR regulation of autophagy. FEBS Lett. 2010, 584, 1287–1295. [Google Scholar] [CrossRef] [PubMed]
- Stafford, P.; Higham, J.; Pinnock, A.; Murdoch, C.; Douglas, C.W.I.; Stafford, G.P.; Lambert, D.W. Gingipain-dependent degradation of mammalian target of rapamycin pathway proteins by the periodontal pathogen Porphyromonas gingivalis during invasion. Mol. Oral Microbiol. 2013, 28, 366–378. [Google Scholar] [CrossRef] [PubMed]
- Blanchard, P.-G.; Festuccia, W.T.; Houde, V.P.; St-Pierre, P.; Brûlé, S.; Turcotte, V.; Côté, M.; Bellmann, K.; Marette, A.; Deshaies, Y. Major involvement of mTOR in the PPARγ-induced stimulation of adipose tissue lipid uptake and fat accretion. J. Lipid Res. 2012, 53, 1117–1125. [Google Scholar] [CrossRef] [PubMed]
- Tain, Y.-L.; Hsu, C.-N.; Chan, J. PPARs link early life nutritional insults to later programmed hypertension and metabolic syndrome. Int. J. Mol. Sci. 2015, 17, 20. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Yeon, J.E.; Ko, E.J.; Yoon, E.L.; Suh, S.J.; Kang, K.; Kim, H.R.; Kang, S.H.; Yoo, Y.J.; Je, J.; et al. Peroxisome proliferator-activated receptor-delta agonist ameliorated inflammasome activation in nonalcoholic fatty liver disease. World J. Gastroenterol. 2015, 21, 12787–12799. [Google Scholar] [CrossRef] [PubMed]
- Baltacioğlu, E.; Akalin, F. A.; Alver, A.; Değer, O.; Karabulut, E. Protein carbonyl levels in serum and gingival crevicular fluid in patients with chronic periodontitis. Arch. Oral Biol. 2008, 53, 716–722. [Google Scholar] [CrossRef] [PubMed]
- Bullón, P.; Román-Malo, L.; Marín-Aguilar, F.; Alvarez-Suarez, J.M.; Giampieri, F.; Battino, M.; Cordero, M.D. Lipophilic antioxidants prevent lipopolysaccharide-induced mitochondrial dysfunction through mitochondrial biogenesis improvement. Pharmacol. Res. 2015, 91, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Varela-Lopez, A.; Bullon, P.; Battino, M.; Ramirez-Tortosa, M.C.; Ochoa, J.J.; Cordero, M.D.; Ramirez-Tortosa, C.L.; Rubini, C.; Zizzi, A.; Quiles, J.L. Coenzyme Q Protects against age-related alveolar bone loss associated to n-6 polyunsaturated fatty acid rich-diets by modulating mitochondrial mechanisms. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2016, 71, 593–600. [Google Scholar] [CrossRef] [PubMed]
- Michalik, L.; Auwerx, J.; Berger, J.P.; Chatterjee, V.K.; Glass, C.K.; Gonzalez, F.J.; Grimaldi, P.A.; Kadowaki, T.; Lazar, M.A.; Rahilly, S.O.; et al. International Union of Pharmacology. LXI. Peroxisome proliferator-activated receptors. Pharmacol. Rev. 2006, 58, 726–741. [Google Scholar] [CrossRef] [PubMed]
- Chung, J.H.; Seo, A.Y.; Chung, S.W.; Kim, M.K.; Leeuwenburgh, C.; Yu, B.P.; Chung, H.Y. Molecular mechanism of PPAR in the regulation of age-related inflammation. Ageing Res. Rev. 2008, 7, 126–136. [Google Scholar] [CrossRef] [PubMed]
- Croasdell, A.; Duffney, P.F.; Kim, N.; Lacy, S.H.; Sime, P.J.; Phipps, R.P. PPARγ and the innate immune system mediate the resolution of inflammation. PPAR Res. 2015, 2015, 549691. [Google Scholar] [CrossRef] [PubMed]
- Michalik, L.; Feige, J.N.; Gelman, L.; Pedrazzini, T.; Keller, H.; Desvergne, B.; Wahli, W. Selective expression of a dominant-negative form of peroxisome proliferator-activated receptor in keratinocytes leads to impaired epidermal healing. Mol. Endocrinol. 2005, 19, 2335–2348. [Google Scholar] [CrossRef] [PubMed]
- Tan, N.S. Critical roles of PPARβ/δ in keratinocyte response to inflammation. Genes Dev. 2001, 15, 3263–3277. [Google Scholar] [CrossRef] [PubMed]
- Iglesias, J.; Morales, L.; Barreto, G.E. Metabolic and inflammatory adaptation of reactive astrocytes: Role of PPARs. Mol. Neurobiol. 2017, 54, 2518–2538. [Google Scholar] [CrossRef] [PubMed]
- Desreumaux, P.; Dubuquoy, L.; Nutten, S.; Peuchmaur, M.; Englaro, W.; Schoonjans, K.; Derijard, B.; Desvergne, B.; Wahli, W.; Chambon, P.; et al. Attenuation of colon inflammation through activators of the retinoid X receptor (RXR)/peroxisome Proliferator-activated receptor γ (PPARγ) heterodimer. A basis for new therapeutic strategies. J. Exp. Med. 2001, 193, 827–838. [Google Scholar] [CrossRef] [PubMed]
- Evans, R.M. The steroid and thyroid hormone receptor superfamily. Science 1988, 240, 889–895. [Google Scholar] [CrossRef] [PubMed]
- Delerive, P.; de Bosscher, K.; Besnard, S.; Vanden Berghe, W.; Peters, J.M.; Gonzalez, F.J.; Fruchart, J.C.; Tedgui, A.; Haegeman, G.; Staels, B. Peroxisome proliferator-activated receptor α negatively regulates the vascular inflammatory gene response by negative cross-talk with transcription factors NF-κB and AP-1. J. Biol. Chem. 1999, 274, 32048–32054. [Google Scholar] [CrossRef] [PubMed]
- Di Paola, R.; Briguglio, F.; Paterniti, I.; Mazzon, E.; Oteri, G.; Militi, D.; Cordasco, G.; Cuzzocrea, S. Emerging role of PPARβ/δ in inflammatory process associated to experimental periodontitis. Mediat. Inflamm. 2011, 2011, 787159. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Yi, T.; Yun, J.-H. Maintained stemness of human periodontal ligament stem cells isolated following prolonged storage of extracted teeth. J. Periodontol. 2016, 87, e148–e158. [Google Scholar] [CrossRef] [PubMed]
- Andrukhov, O.; Ertlschweiger, S.; Moritz, A.; Bantleon, H.-P.; Rausch-Fan, X. Different effects of P. gingivalis LPS and E. coli LPS on the expression of interleukin-6 in human gingival fibroblasts. Acta Odontol. Scand. 2014, 72, 337–345. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Zhou, Q.; Shi, Y.; Liu, J.; Zhong, F.; Hao, X.; Li, C.; Chen, N.; Wang, W. SUMOylation of PPARγ by rosiglitazone prevents LPS-induced NCoR degradation mediating down regulation of chemokines expression in renal proximal tubular cells. PLoS ONE 2013, 8, e79815. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhou, J.; Fu, S.; Wang, C.; Zhou, B. Preventive effects of protocatechuic acid on LPS-induced inflammatory response in human gingival fibroblasts via activating PPARγ. Inflammation 2015, 38, 1080–1084. [Google Scholar] [CrossRef] [PubMed]
- Di Paola, R.; Mazzon, E.; Maiere, D.; Zito, D.; Britti, D.; de Majo, M.; Genovese, T.; Cuzzocrea, S. Rosiglitazone reduces the evolution of experimental periodontitis in the rat. J. Dent. Res. 2006, 85, 156–161. [Google Scholar] [CrossRef] [PubMed]
- Folwaczny, M.; Manolis, V.; Markus, C.; Glas, J. Variants of the human PPARG locus and the susceptibility to chronic periodontitis. Innate Immun. 2011, 17, 541–547. [Google Scholar] [CrossRef] [PubMed]
- Hirano, E.; Sugita, N.; Kikuchi, A.; Shimada, Y.; Sasahara, J.; Iwanaga, R.; Tanaka, K.; Yoshie, H. Peroxisome proliferator-activated receptor gamma polymorphism and periodontitis in pregnant Japanese women. J. Periodontol. 2010, 81, 897–906. [Google Scholar] [CrossRef] [PubMed]
- Yoshihara, A.; Sugita, N.; Iwasaki, M.; Wang, Y.; Miyazaki, H.; Yoshie, H.; Nakamura, K. The interaction between β-3 adrenergic receptor and peroxisome proliferator-activated receptor gamma gene polymorphism to periodontal disease in community-dwelling elderly japanese. J. Periodontol. 2015, 86, 955–963. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Sugita, N.; Yoshihara, A.; Iwasaki, M.; Miyazaki, H.; Nakamura, K.; Yoshie, H. PPARγ gene polymorphism, C-reactive protein level, BMI and periodontitis in post-menopausal Japanese women. Gerodontology 2014, 33, 44–51. [Google Scholar] [CrossRef] [PubMed]
- Briguglio, E.; di Paola, R.; Paterniti, I.; Mazzon, E.; Oteri, G.; Cordasco, G.; Cuzzocrea, S. WY-14643, a potent peroxisome proliferator activator receptor-α PPARα agonist ameliorates the inflammatory process associated to experimental periodontitis. PPAR Res. 2010, 2010, 193019. [Google Scholar] [CrossRef] [PubMed]
- Harsløf, T.; Tofteng, C.L.; Husted, L.B.; Nyegaard, M.; Børglum, A.; Carstens, M.; Stenkjær, L.; Brixen, K.; Eiken, P.; Jensen, J.-E.B.; et al. Polymorphisms of the peroxisome proliferator-activated receptor γ (PPARγ) gene are associated with osteoporosis. Osteoporos. Int. 2011, 22, 2655–2666. [Google Scholar] [CrossRef] [PubMed]
- Wan, Y.; Chong, L.-W.; Evans, R.M. PPARγ regulates osteoclastogenesis in mice. Nat. Med. 2007, 13, 1496–1503. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.-J.; Yoon, K.-A.; Yoon, H.-J.; Hong, J.M.; Lee, M.-J.; Lee, I.-K.; Kim, S.-Y. Liver X receptor activation inhibits osteoclastogenesis by suppressing NF-κB activity and c-Fos induction and prevents inflammatory bone loss in mice. J. Leukoc. Biol. 2013, 94, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Siddhivarn, C.; Banes, A.; Champagne, C.; Riche, E.L.; Weerapradist, W.; Offenbacher, S. Prostaglandin D2 pathway and peroxisome proliferator-activated receptor γ-1 expression are induced by mechanical loading in an osteoblastic cell line. J. Periodontal Res. 2006, 41, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Siddhivarn, C.; Banes, A.; Champagne, C.; Riché, E.L.; Weerapradist, W.; Offenbacher, S. Mechanical loading and Δ12prostaglandin J2 induce bone morphogenetic protein-2, peroxisome proliferator-activated receptor γ-1, and bone nodule formation in an osteoblastic cell line. J. Periodontal Res. 2007, 42, 383–392. [Google Scholar] [CrossRef] [PubMed]
- Jung, W.-K.; Park, I.-S.; Park, S.-J.; Yea, S.S.; Choi, Y.H.; Oh, S.; Park, S.-G.; Choi, I.-W. The 15-deoxy-Δ12,14-prostaglandin J2 inhibits LPS-stimulated AKT and NF-κB activation and suppresses interleukin-6 in osteoblast-like cells MC3T3E-1. Life Sci. 2009, 85, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Brennan-Calanan, R.M.; Genco, R.J.; Wilding, G.E.; Hovey, K.M.; Trevisan, M.; Wactawski-Wende, J. Osteoporosis and oral infection: Independent risk factors for oral bone loss. J. Dent. Res. 2008, 87, 323–327. [Google Scholar] [CrossRef] [PubMed]
- Al Habashneh, R.; Alchalabi, H.; Khader, Y.S.; Hazza’a, A.M.; Odat, Z.; Johnson, G.K. Association between periodontal disease and osteoporosis in postmenopausal women in Jordan. J. Periodontol. 2010, 81, 1613–1621. [Google Scholar] [CrossRef] [PubMed]
- Pepelassi, E.; Nicopoulou-Karayianni, K.; Archontopoulou, A.D.; Mitsea, A.; Kavadella, A.; Tsiklakis, K.; Vrotsos, I.; Devlin, H.; Horner, K. The relationship between osteoporosis and periodontitis in women aged 45–70 years. Oral Dis. 2012, 18, 353–359. [Google Scholar] [CrossRef] [PubMed]
- Yoshihara, A.; Seida, Y.; Hanada, N.; Miyazaki, H. A longitudinal study of the relationship between periodontal disease and bone mineral density in community-dwelling older adults. J. Clin. Periodontol. 2004, 31, 680–684. [Google Scholar] [CrossRef] [PubMed]
- Hassumi, M.Y.; Silva-Filho, V.J.; Campos-Júnior, J.C.; Vieira, S.M.; Cunha, F.Q.; Alves, P.M.; Alves, J.B.; Kawai, T.; Gonçalves, R.B.; Napimoga, M.H. PPARγ agonist rosiglitazone prevents inflammatory periodontal bone loss by inhibiting osteoclastogenesis. Int. Immunopharmacol. 2009, 9, 1150–1158. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Sugita, N.; Yoshihara, A.; Iwasaki, M.; Miyazaki, H.; Nakamura, K.; Yoshie, H. Peroxisome proliferator-activated receptor (PPAR)γ polymorphism, vitamin d, bone mineral density and periodontitis in postmenopausal women. Oral Dis. 2013, 19, 501–506. [Google Scholar] [CrossRef] [PubMed]
- Bhattarai, G.; Lee, Y.H.; Lee, N.H.; Park, I.S.; Lee, M.H.; Yi, H.K. PPARγ delivered by Ch-GNPs onto titanium surfaces inhibits implant-induced inflammation and induces bone mineralization of MC-3T3E1 osteoblast-like cells. Clin. Oral Implant. Res. 2013, 24, 1101–1109. [Google Scholar] [CrossRef] [PubMed]
- Bullon, P.; Newman, H.N.; Battino, M. Obesity, diabetes mellitus, atherosclerosis and chronic periodontitis: A shared pathology via oxidative stress and mitochondrial dysfunction? Periodontol. 2000 2014, 64, 139–153. [Google Scholar] [CrossRef] [PubMed]
- Dandona, P.; Aljada, A.; Bandyopadhyay, A. Inflammation: The link between insulin resistance, obesity and diabetes. Trends Immunol. 2004, 25, 4–7. [Google Scholar] [CrossRef] [PubMed]
- Rinaudo, P.; Wang, E. Fetal programming and metabolic syndrome. Annu. Rev. Physiol. 2012, 74, 107–130. [Google Scholar] [CrossRef] [PubMed]
- Lendvai, A.; Deutsch, M.J.; Plosch, T.; Ensenauer, R. The peroxisome proliferator-activated receptors under epigenetic control in placental metabolism and fetal development. Am. J. Physiol. Endocrinol. Metab. 2016, 310, E797–E810. [Google Scholar] [CrossRef] [PubMed]
- Monsalve, F.A.; Pyarasani, R.D.; Delgado-Lopez, F.; Moore-Carrasco, R. Peroxisome proliferator-activated receptor targets for the treatment of metabolic diseases. Mediat. Inflamm. 2013, 2013, 549627. [Google Scholar] [CrossRef] [PubMed]
- Yessoufou, A.; Wahli, W. Multifaceted roles of peroxisome proliferator-activated receptors (PPARs) at the cellular and whole organism levels. Swiss Med. Wkly. 2010, 140, w13071. [Google Scholar] [CrossRef] [PubMed]
- Emilsson, V.; Thorleifsson, G.; Zhang, B.; Leonardson, A.S.; Zink, F.; Zhu, J.; Carlson, S.; Helgason, A.; Walters, G.B.; Gunnarsdottir, S.; et al. Genetics of gene expression and its effect on disease. Nature 2008, 452, 423–428. [Google Scholar] [CrossRef] [PubMed]
- Lin, Q.; Gao, Z.; Alarcon, R.M.; Ye, J.; Yun, Z. A role of miR-27 in the regulation of adipogenesis. FEBS J. 2009, 276, 2348–2358. [Google Scholar] [CrossRef] [PubMed]
- Takanabe, R.; Ono, K.; Abe, Y.; Takaya, T.; Horie, T.; Wada, H.; Kita, T.; Satoh, N.; Shimatsu, A.; Hasegawa, K. Up-regulated expression of microRNA-143 in association with obesity in adipose tissue of mice fed high-fat diet. Biochem. Biophys. Res. Commun. 2008, 376, 728–732. [Google Scholar] [CrossRef] [PubMed]
- Maeda, A.; Gohda, T.; Funabiki, K.; Horikoshi, S.; Tomino, Y. Peroxisome proliferator-activated receptor γ gene polymorphism is associated with serum triglyceride levels and body mass index in Japanese type 2 diabetic patients. J. Clin. Lab. Anal. 2004, 18, 317–321. [Google Scholar] [CrossRef] [PubMed]
- Garaulet, M.; Smith, C.E.; Hernández-González, T.; Lee, Y.-C.; Ordovás, J.M. PPARγ Pro12Ala interacts with fat intake for obesity and weight loss in a behavioural treatment based on the Mediterranean diet. Mol. Nutr. Food Res. 2011, 55, 1771–1779. [Google Scholar] [CrossRef] [PubMed]
- Rühl, R.; Landrier, J.F. Dietary regulation of adiponectin by direct and indirect lipid activators of nuclear hormone receptors. Mol. Nutr. Food Res. 2016, 60, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Ding, G.; Cheng, L.; Qin, Q.; Frontin, S.; Yang, Q. PPARδ modulates lipopolysaccharide-induced TNF-α inflammation signaling in cultured cardiomyocytes. J. Mol. Cell. Cardiol. 2006, 40, 821–828. [Google Scholar] [CrossRef] [PubMed]
- Palomer, X.; Barroso, E.; Zarei, M.; Botteri, G.; Vázquez-Carrera, M. PPARβ/δ and lipid metabolism in the heart. Biochim. Biophys. Acta 2016, 1861, 1569–1578. [Google Scholar] [CrossRef] [PubMed]
- Andriankaja, O.M.; Galicia, J.; Dong, G.; Xiao, W.; Alawi, F.; Graves, D.T. Gene expression dynamics during diabetic periodontitis. J. Dent. Res. 2012, 91, 1160–1165. [Google Scholar] [CrossRef] [PubMed]
- Salvi, G.E.; Beck, J.D.; Offenbacher, S. PGE2, IL-1β, and TNF-α responses in diabetics as modifiers of periodontal disease expression. Ann. Periodontol. 1998, 3, 40–50. [Google Scholar] [CrossRef] [PubMed]
- Nareika, A.; Maldonado, A.; He, L.; Game, B.A.; Slate, E.H.; Sanders, J.J.; London, S.D.; Lopes-Virella, M.F.; Huang, Y. High glucose-boosted inflammatory responses to lipopolysaccharide are suppressed by statin. J. Periodontal Res. 2007, 42, 31–38. [Google Scholar] [CrossRef] [PubMed]


© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Román-Malo, L.; Bullon, P. Influence of the Periodontal Disease, the Most Prevalent Inflammatory Event, in Peroxisome Proliferator-Activated Receptors Linking Nutrition and Energy Metabolism. Int. J. Mol. Sci. 2017, 18, 1438. https://doi.org/10.3390/ijms18071438
Román-Malo L, Bullon P. Influence of the Periodontal Disease, the Most Prevalent Inflammatory Event, in Peroxisome Proliferator-Activated Receptors Linking Nutrition and Energy Metabolism. International Journal of Molecular Sciences. 2017; 18(7):1438. https://doi.org/10.3390/ijms18071438
Chicago/Turabian StyleRomán-Malo, Lourdes, and Pedro Bullon. 2017. "Influence of the Periodontal Disease, the Most Prevalent Inflammatory Event, in Peroxisome Proliferator-Activated Receptors Linking Nutrition and Energy Metabolism" International Journal of Molecular Sciences 18, no. 7: 1438. https://doi.org/10.3390/ijms18071438