A Different Microbiome Gene Repertoire in the Airways of Cystic Fibrosis Patients with Severe Lung Disease

 , , ,
, , , 
Abstract
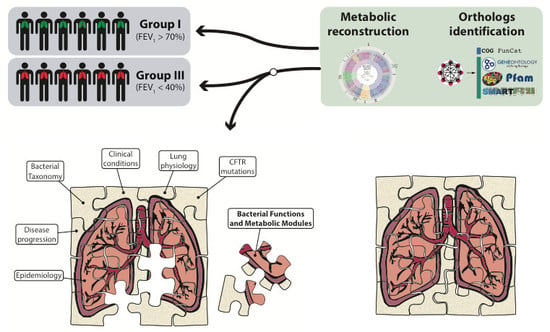
:
1. Introduction
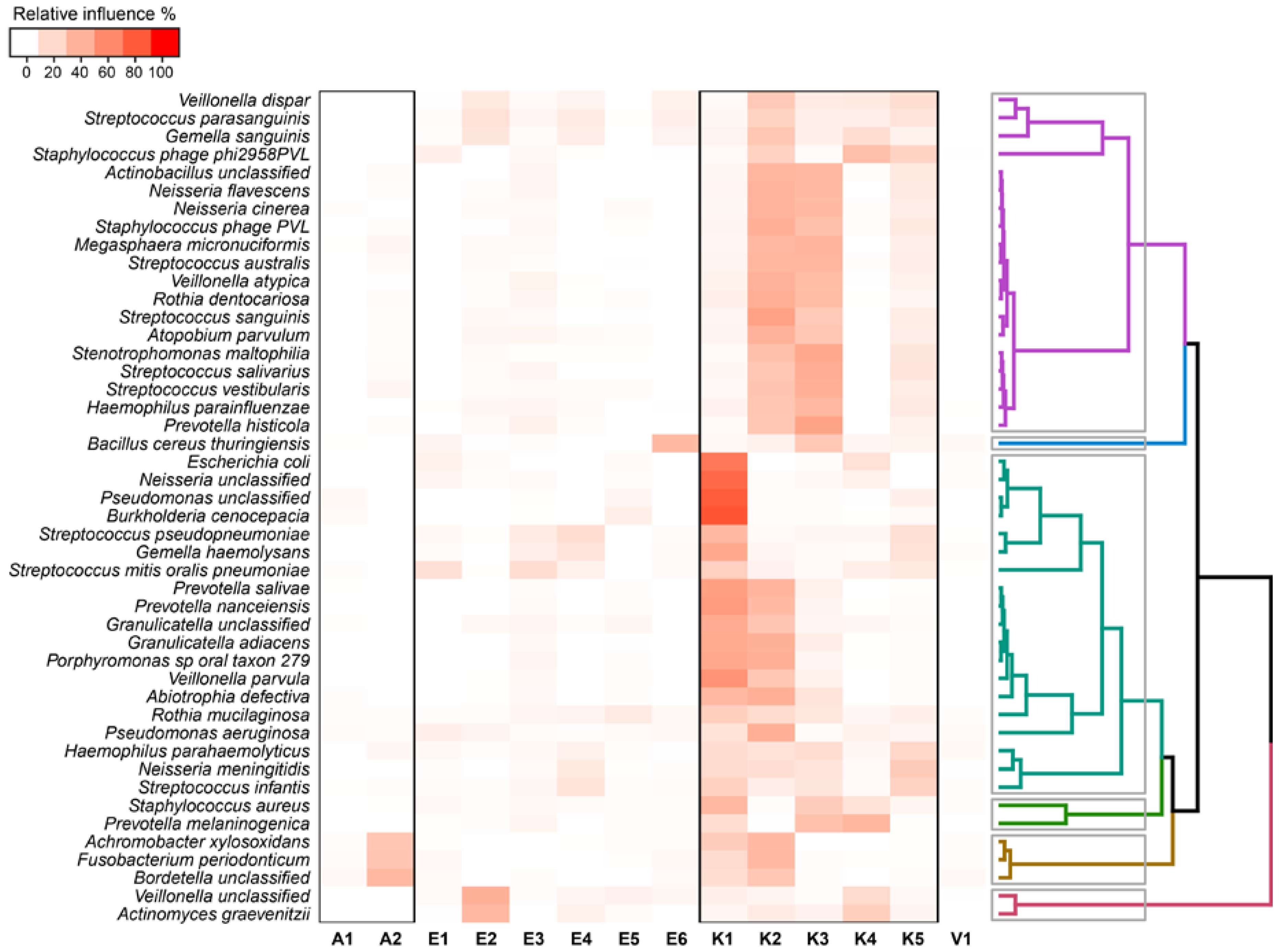
2. Results
2.1. Clinical Characteristics of Enrolled Patients and Culture-Based Diagnostic Microbiology
2.2. Metabolic Community Structure between Patient Groups
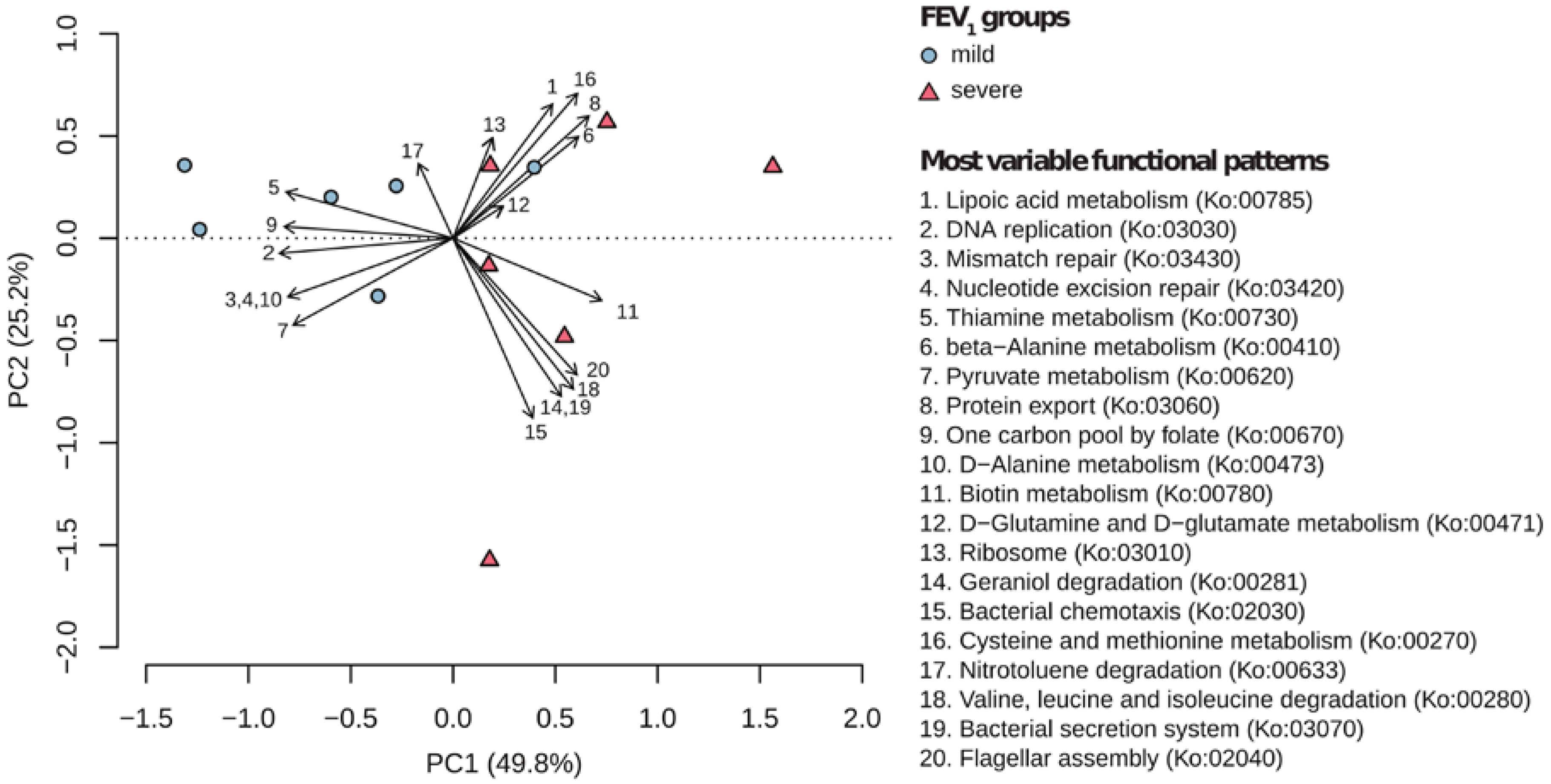
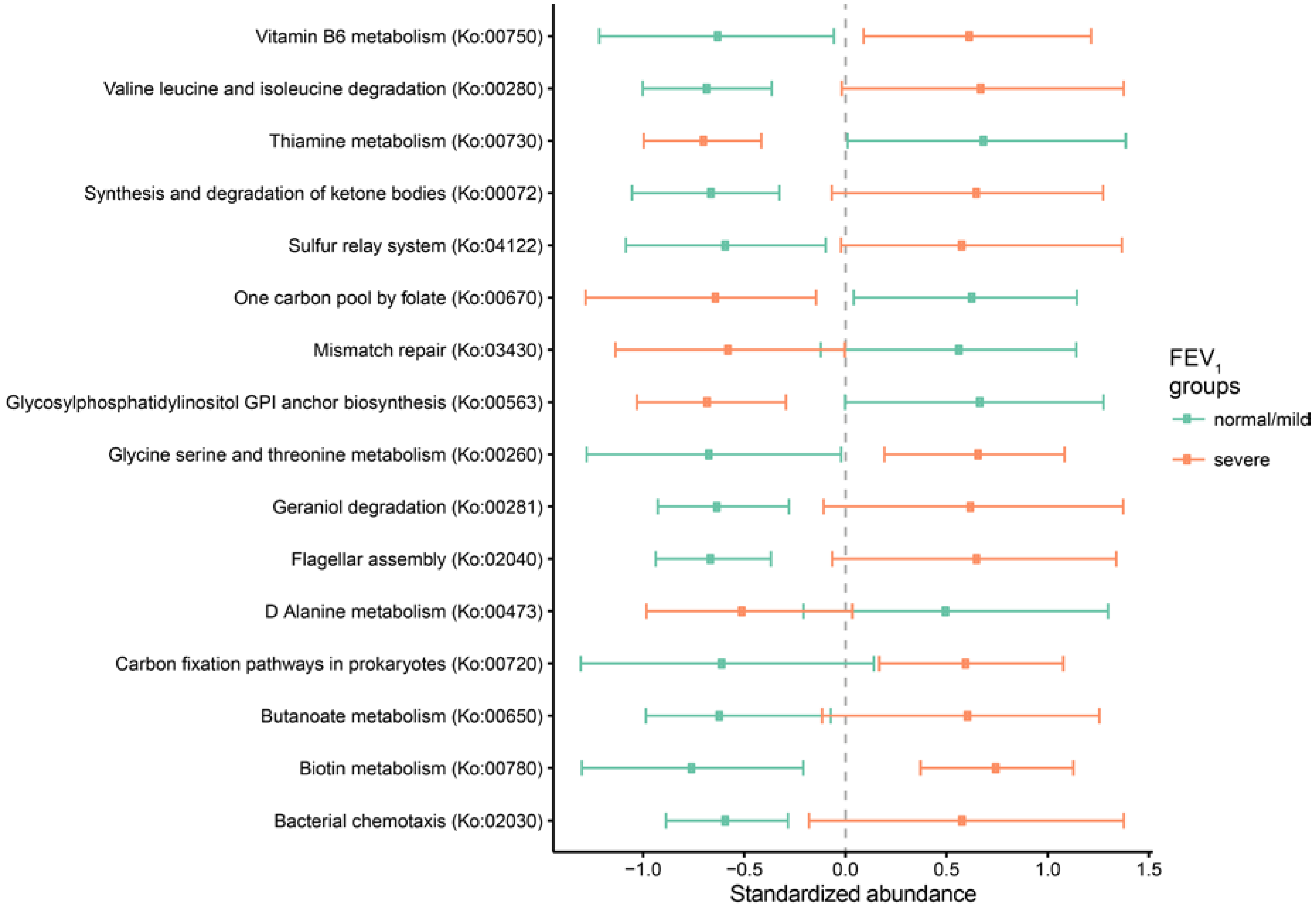
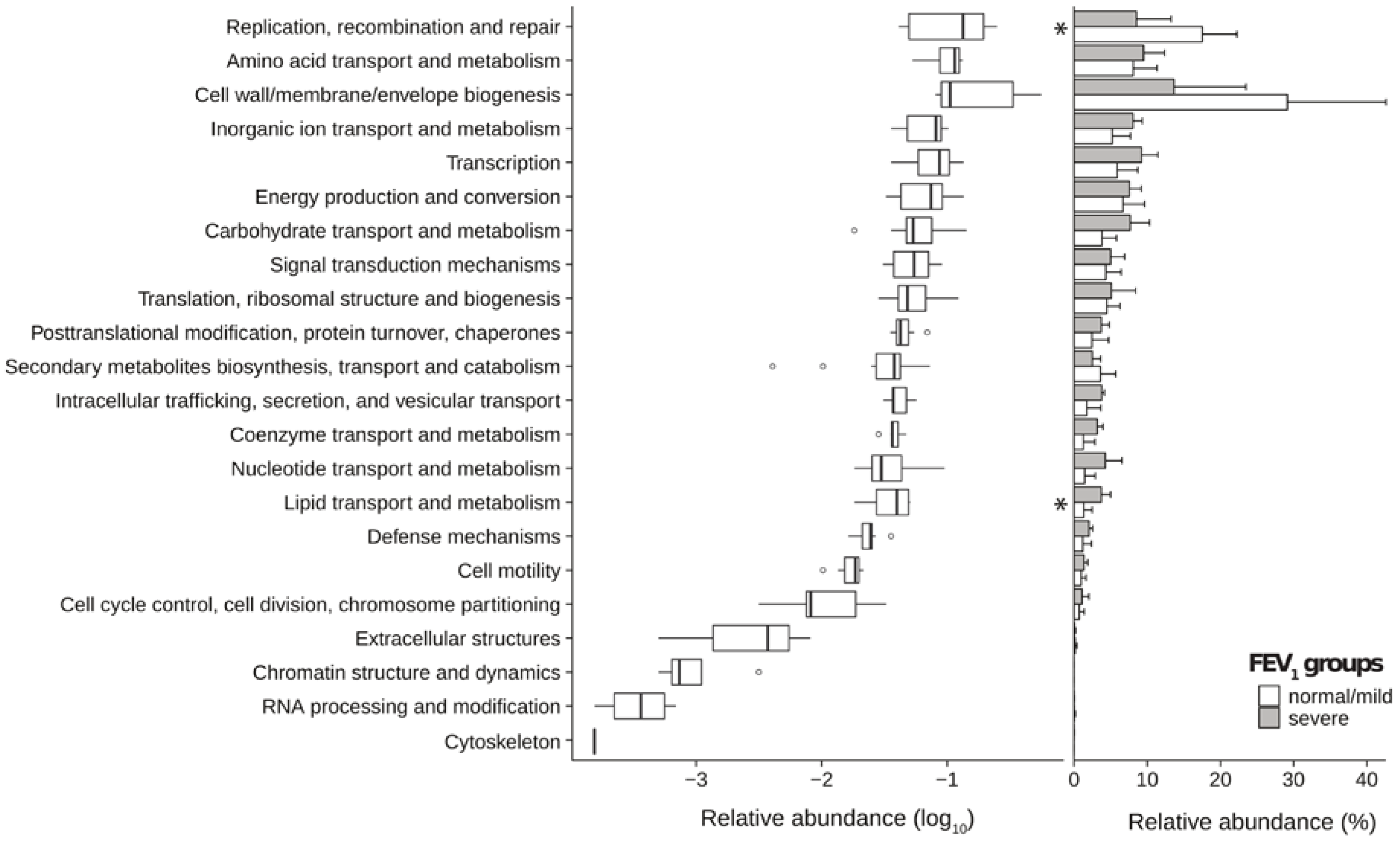
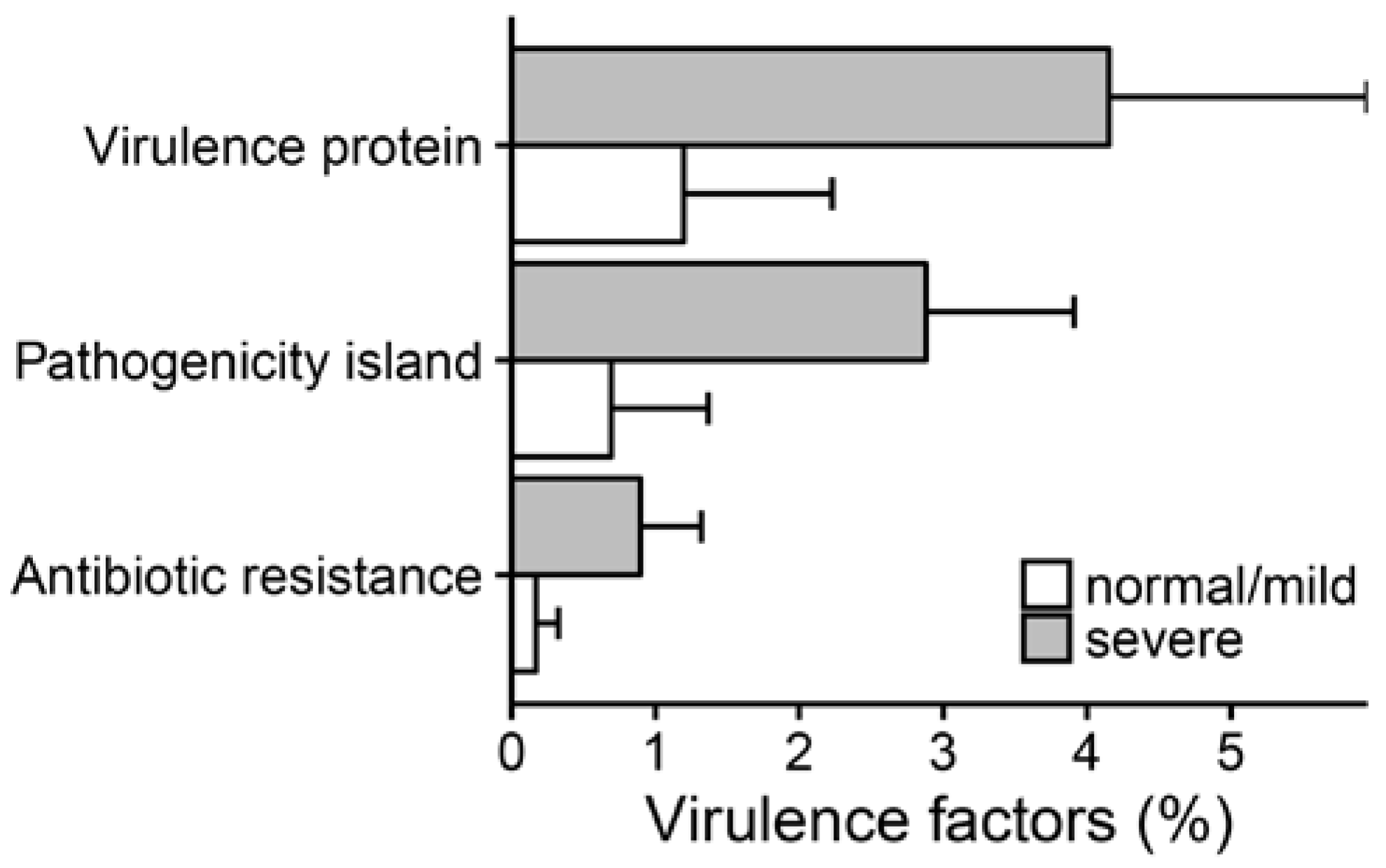
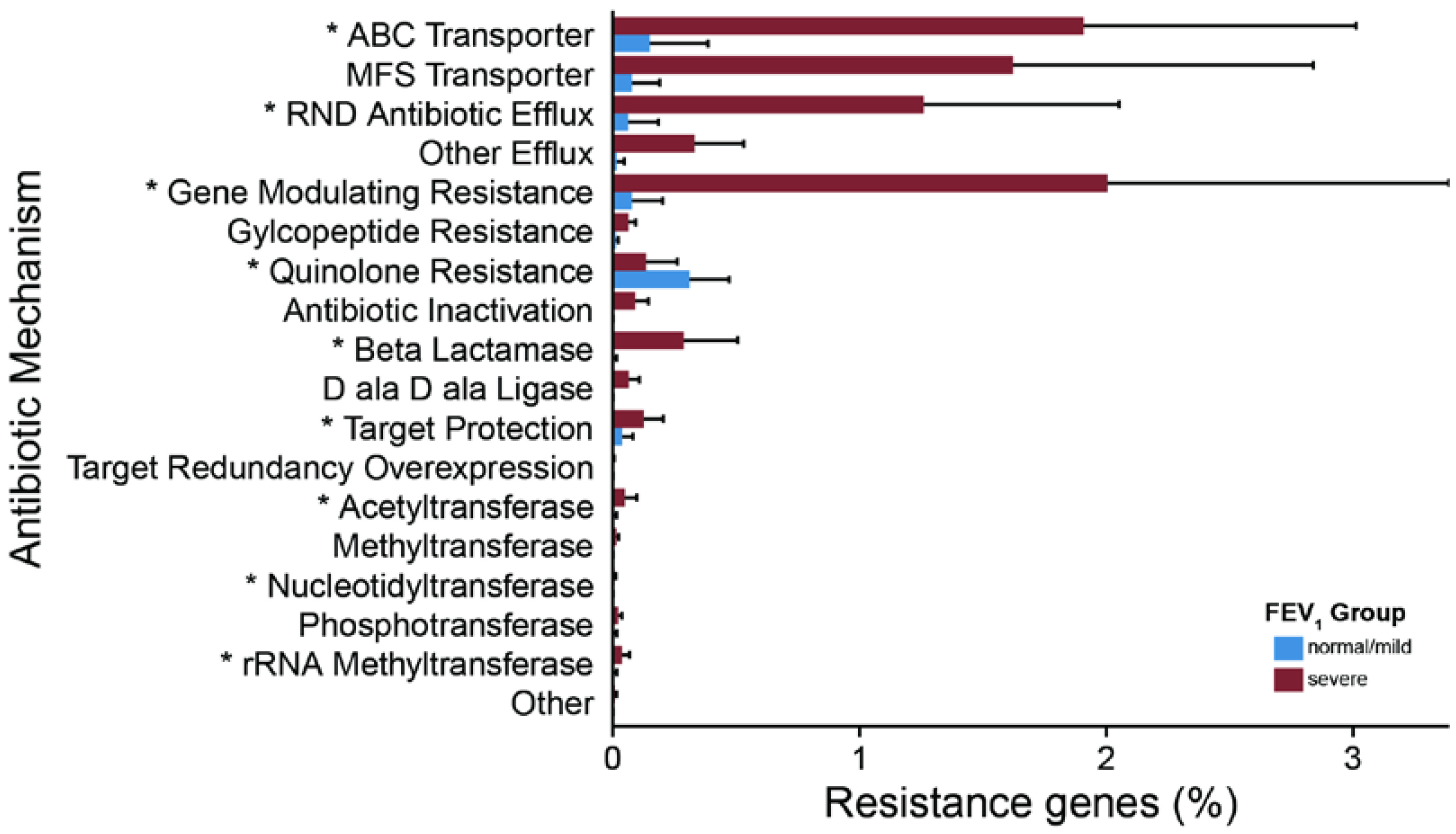
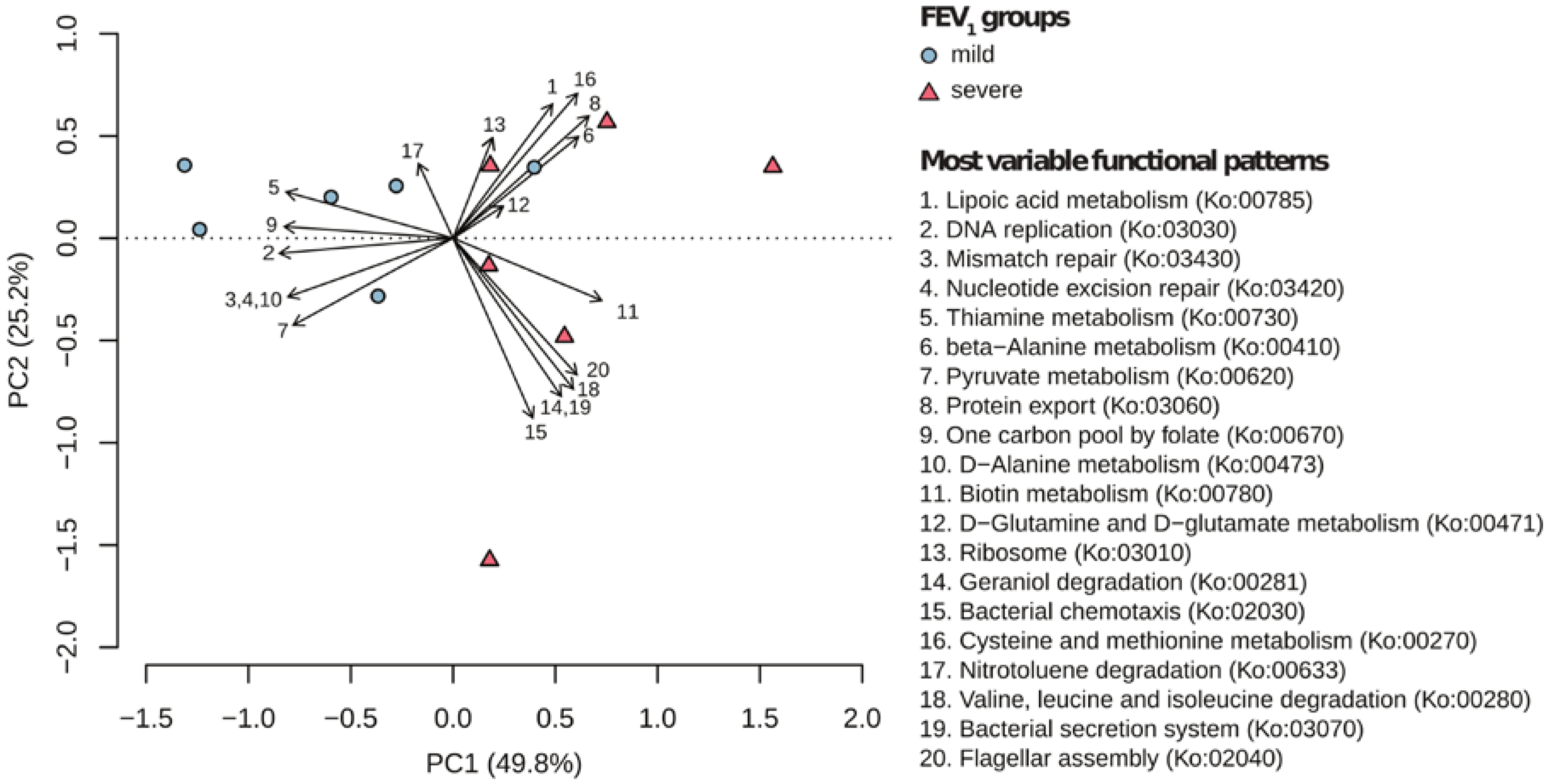
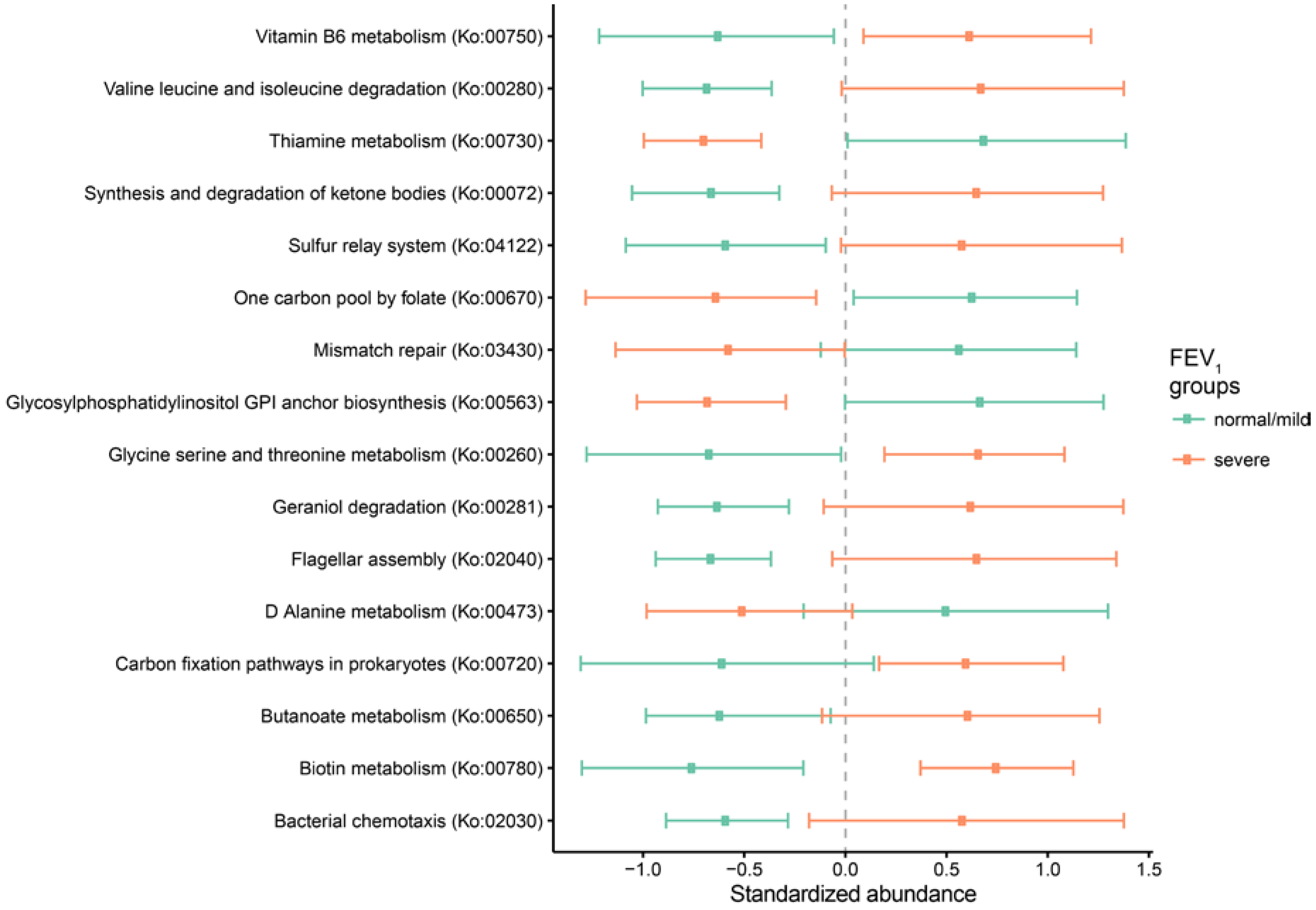
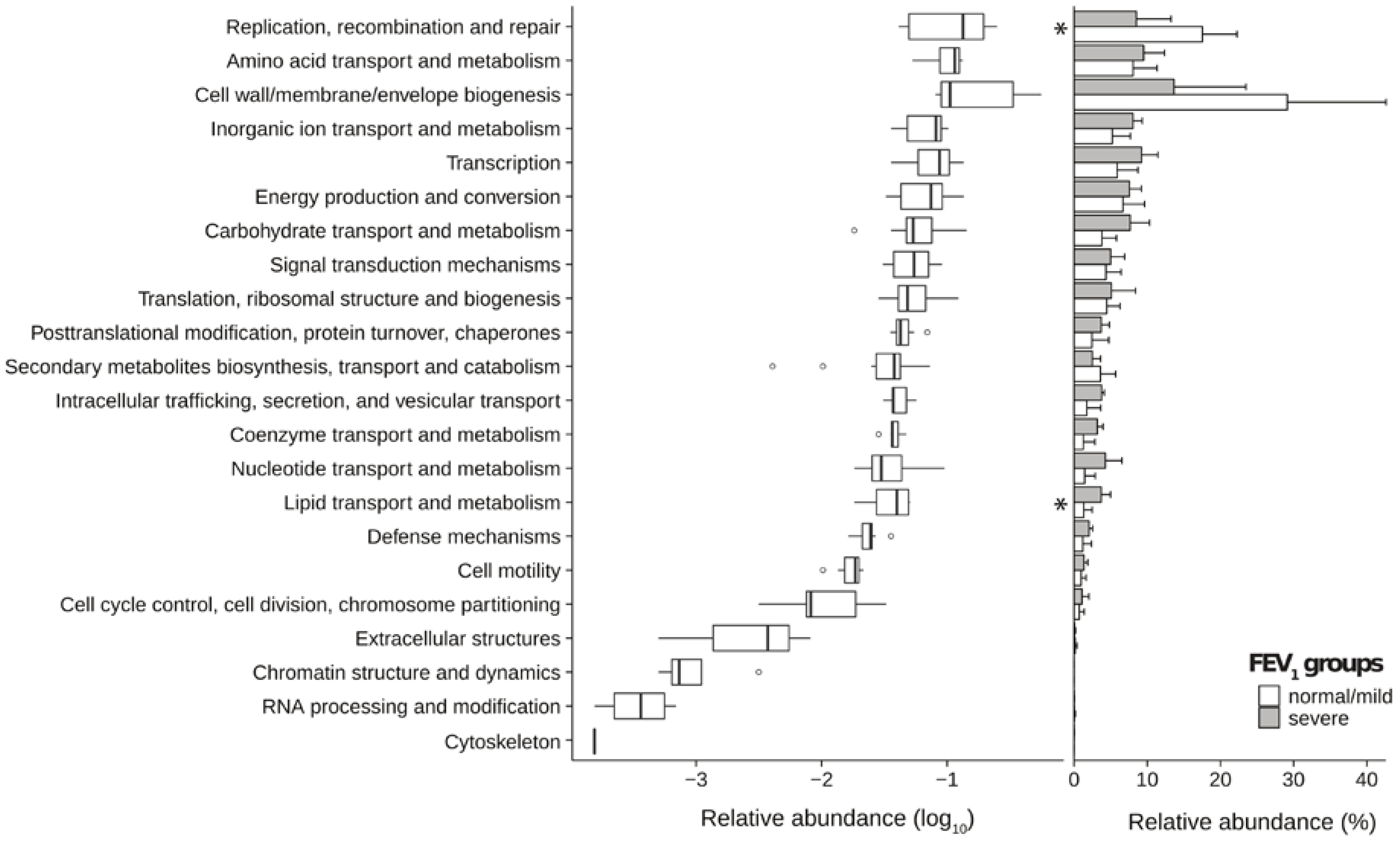
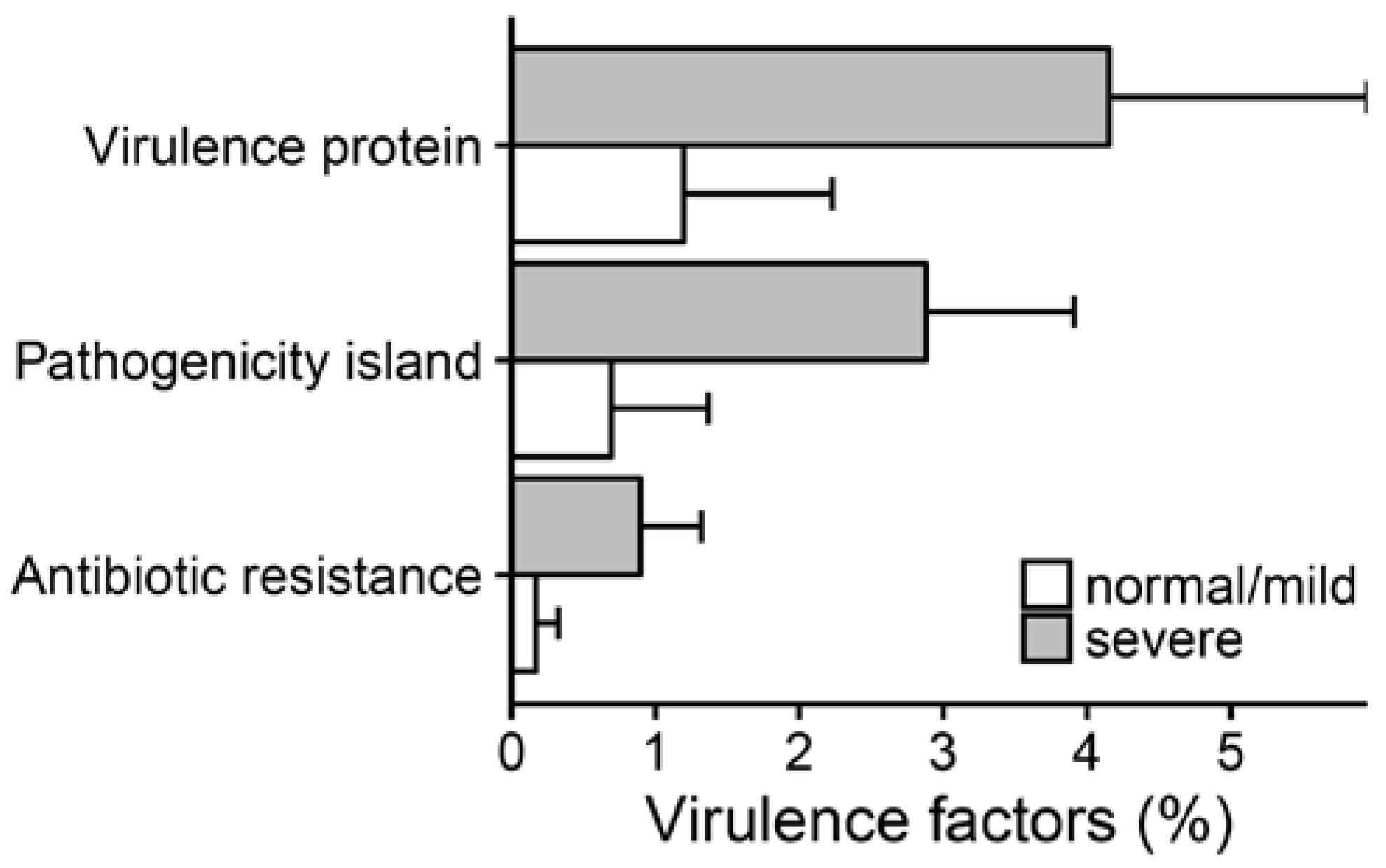
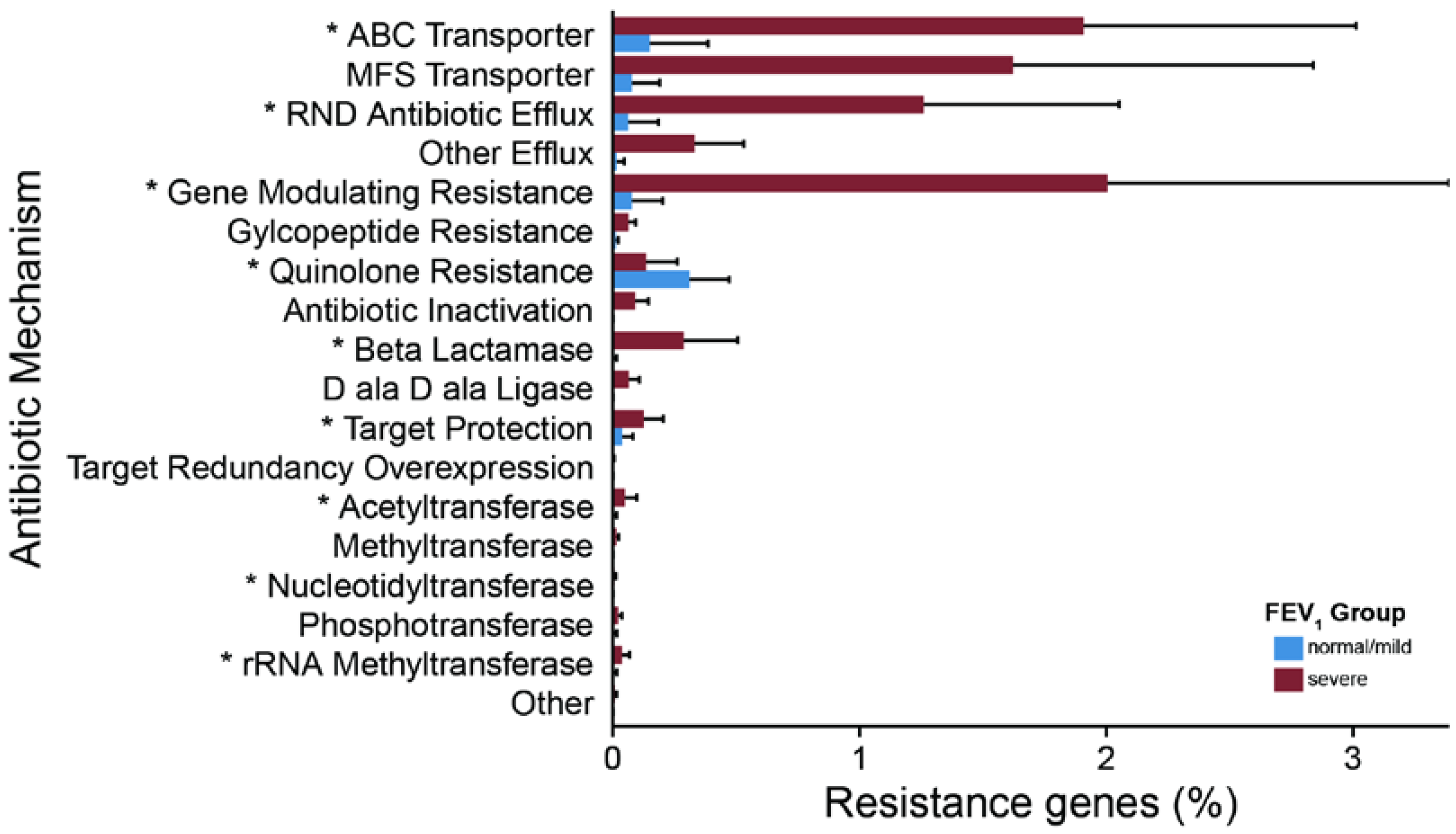
2.3. Metagenomic Functional Differences Suggest Distinct Ecological Roles within the Cystic Fibrosis Microbiome
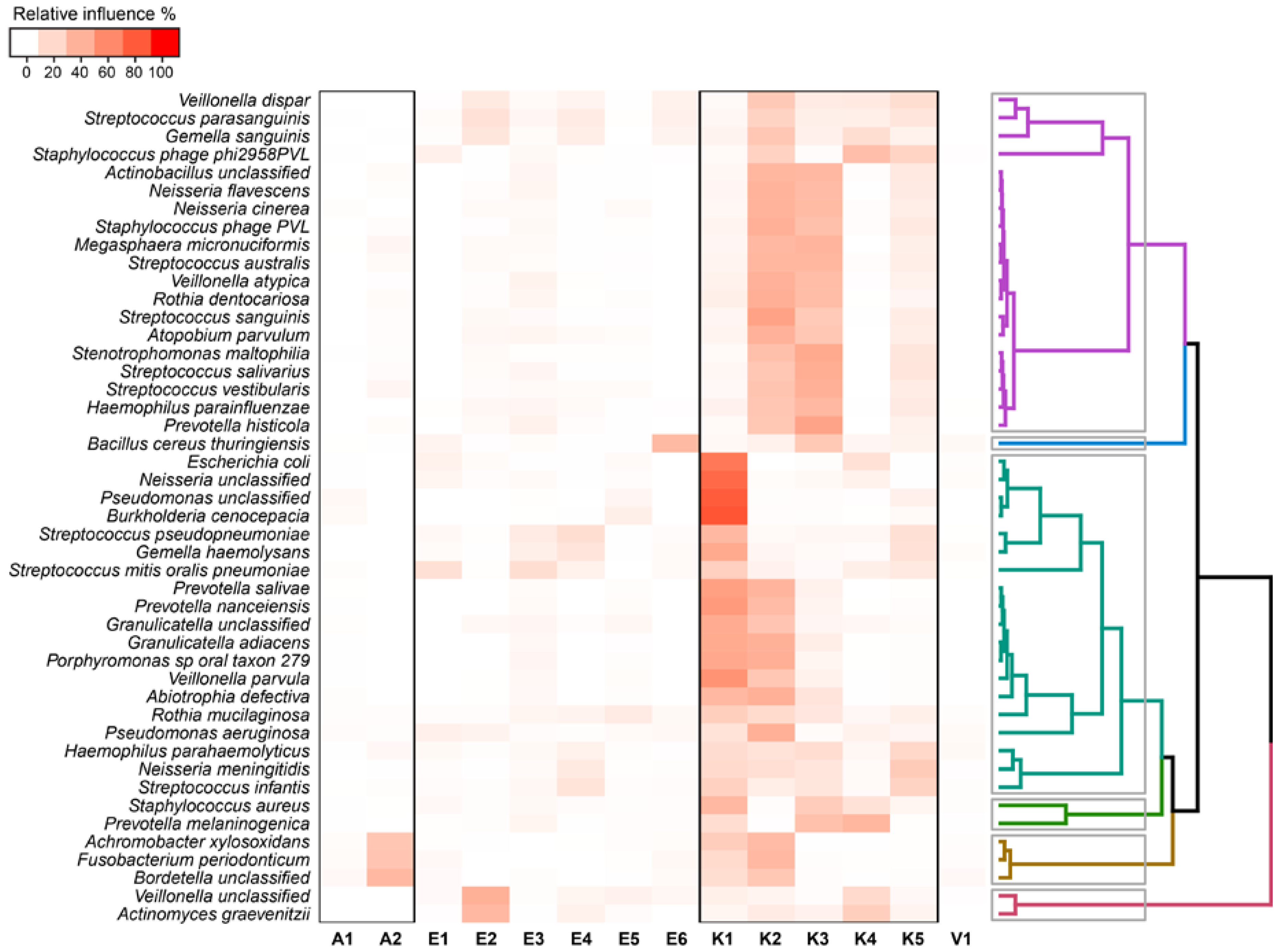
2.4. Relationship between Functional Potential and Taxonomic Microbial Composition
3. Discussion
4. Materials and Methods
4.1. Ethics Statement
4.2. Patients
4.3. Sample Processing
4.4. DNA Extraction Procedures and Sequencing
4.5. Bioinformatic Analyses
4.6. Statistical Analyses
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Cystic Fibrosis Foundation. Patient Registry Annual Data Report 2015; Cystic Fibrosis Foundation: Bethesda, MD, USA, 2016. [Google Scholar]
- Gibson, R.L.; Burns, J.L.; Ramsey, B.W. Pathophysiology and Management of Pulmonary Infections in Cystic Fibrosis. Am. J. Respir. Crit. Care Med. 2003, 168, 918–951. [Google Scholar] [CrossRef] [PubMed]
- Lipuma, J.J. The changing microbial epidemiology in cystic fibrosis. Clin. Microbiol. Rev. 2010, 23, 299–323. [Google Scholar] [CrossRef] [PubMed]
- Carmody, L.A.; Zhao, J.; Schloss, P.D.; Petrosino, J.F.; Murray, S.; Young, V.B.; Li, J.Z.; LiPuma, J.J. Changes in cystic fibrosis airway microbiota at pulmonary exacerbation. Ann. Am. Thorac. Soc. 2013, 10, 179–187. [Google Scholar] [CrossRef] [PubMed]
- Coburn, B.; Wang, P.W.; Diaz Caballero, J.; Clark, S.T.; Brahma, V.; Donaldson, S.; Zhang, Y.; Surendra, A.; Gong, Y.; Elizabeth Tullis, D.; et al. Lung microbiota across age and disease stage in cystic fibrosis. Sci. Rep. 2015, 5, 10241. [Google Scholar] [CrossRef] [PubMed]
- Hogan, D.A.; Willger, S.D.; Dolben, E.L.; Hampton, T.H.; Stanton, B.A.; Morrison, H.G.; Sogin, M.L.; Czum, J.; Ashare, A. Analysis of lung microbiota in bronchoalveolar lavage, protected brush and sputum samples from subjects with mild-to-moderate cystic fibrosis lung disease. PLoS ONE 2016, 11, e0149998. [Google Scholar] [CrossRef] [PubMed]
- Rogers, G.B.; Shaw, D.; Marsh, R.L.; Carroll, M.P.; Serisier, D.J.; Bruce, K.D. Respiratory microbiota: addressing clinical questions, informing clinical practice. Thorax 2015, 70, 74–81. [Google Scholar] [CrossRef] [PubMed]
- Cuthbertson, L.; Rogers, G.B.; Walker, A.W.; Oliver, A.; Green, L.E.; Daniels, T.W.V.; Carroll, M.P.; Parkhill, J.; Bruce, K.D.; van der Gast, C.J. Respiratory microbiota resistance and resilience to pulmonary exacerbation and subsequent antimicrobial intervention. ISME J. 2015, 10, 1081–1091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maughan, H.; Wang, P.W.; Diaz Caballero, J.; Fung, P.; Gong, Y.; Donaldson, S.L.; Yuan, L.; Keshavjee, S.; Zhang, Y.; Yau, Y.C.W.; et al. Analysis of the cystic fibrosis lung microbiota via serial Illumina sequencing of bacterial 16S rRNA hypervariable regions. PLoS ONE 2012, 7, e45791. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.J.; LiPuma, J.J. The Microbiome in Cystic Fibrosis. Clin. Chest Med. 2016, 37, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Surette, M.G. The cystic fibrosis lung microbiome. Ann. Am. Thorac. Soc. 2014, 11, 61–65. [Google Scholar] [CrossRef] [PubMed]
- Carmody, L.A.; Zhao, J.; Kalikin, L.M.; LeBar, W.; Simon, R.H.; Venkataraman, A.; Schmidt, T.M.; Abdo, Z.; Schloss, P.D.; LiPuma, J.J. The daily dynamics of cystic fibrosis airway microbiota during clinical stability and at exacerbation. Microbiome 2015, 3, 12. [Google Scholar] [CrossRef] [PubMed]
- Paganin, P.; Fiscarelli, E.V.; Tuccio, V.; Chiancianesi, M.; Bacci, G.; Morelli, P.; Dolce, D.; Dalmastri, C.; de Alessandri, A.; Lucidi, V.; et al. Changes in cystic fibrosis airway microbial community associated with a severe decline in lung function. PLoS ONE 2015, 10, e0124348. [Google Scholar] [CrossRef] [PubMed]
- Bacci, G.; Paganin, P.; Lopez, L.; Vanni, C.; Dalmastri, C.; Cantale, C.; Daddiego, L.; Perrotta, G.; Dolce, D.; Morelli, P.; et al. Pyrosequencing unveils cystic fibrosis lung microbiome differences associated with a severe lung function decline. PLoS ONE 2016, 11, e0156807. [Google Scholar] [CrossRef]
- Narayanamurthy, V.; Sweetnam, J.M.; Denner, D.R.; Chen, L.W.; Naureckas, E.T.; Laxman, B.; White, S.R. The metabolic footprint of the airway bacterial community in cystic fibrosis. Microbiome 2017, 5, 67. [Google Scholar] [CrossRef] [PubMed]
- Sharon, G.; Garg, N.; Debelius, J.; Knight, R.; Dorrestein, P.C.; Mazmanian, S.K. Specialized metabolites from the microbiome in health and disease. Cell Metab. 2014, 20, 719–730. [Google Scholar] [CrossRef] [PubMed]
- Lim, Y.W.; Evangelista, J.S.; Schmieder, R.; Bailey, B.; Haynes, M.; Furlan, M.; Maughan, H.; Edwards, R.; Rohwer, F.; Conrad, D. Clinical insights from metagenomic analysis of sputum samples from patients with cystic fibrosis. J. Clin. Microbiol. 2014, 52, 425–437. [Google Scholar] [CrossRef] [PubMed]
- Willner, D.; Haynes, M.R.; Furlan, M.; Hanson, N.; Kirby, B.; Lim, Y.W.; Rainey, P.B.; Schmieder, R.; Youle, M.; Conrad, D.; et al. Case studies of the spatial heterogeneity of DNA viruses in the cystic fibrosis lung. Am. J. Respir. Cell Mol. Biol. 2012, 46, 127–131. [Google Scholar] [CrossRef] [PubMed]
- Whiteson, K.L.; Meinardi, S.; Lim, Y.W.; Schmieder, R.; Maughan, H.; Quinn, R.; Blake, D.R.; Conrad, D.; Rohwer, F. Breath gas metabolites and bacterial metagenomes from cystic fibrosis airways indicate active pH neutral 2,3-butanedione fermentation. ISME J. 2014, 8, 1247–1258. [Google Scholar] [CrossRef] [PubMed]
- Losada, P.M.; Chouvarine, P.; Dorda, M.; Hedtfeld, S.; Mielke, S.; Schulz, A.; Wiehlmann, L.; Tümmler, B. The cystic fibrosis lower airways microbial metagenome. ERJ Open Res. 2016, 2, 00096-2015. [Google Scholar] [CrossRef]
- Dhooghe, B.; Noël, S.; Huaux, F.; Leal, T.; Diaz Caballero, J.; Clark, S.T.; Coburn, B.; Zhang, Y.; Wang, P.W.; Donaldson, S.L.; et al. Sputum DNA sequencing in cystic fibrosis: Non-invasive access to the lung microbiome and to pathogen details. J. Cyst. Fibros. 2014, 5, 20. [Google Scholar] [CrossRef]
- Gibson, M.K.; Forsberg, K.J.; Dantas, G. Improved annotation of antibiotic resistance determinants reveals microbial resistomes cluster by ecology. ISME J. 2014, 9, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Elith, J.; Leathwick, J.R.; Hastie, T. A working guide to boosted regression trees. J. Anim. Ecol. 2008, 77, 802–813. [Google Scholar] [CrossRef] [PubMed]
- Lambiase, A.; Catania, M.R.; Del Pezzo, M.; Rossano, F.; Terlizzi, V.; Sepe, A.; Raia, V. Achromobacter xylosoxidans respiratory tract infection in cystic fibrosis patients. Eur. J. Clin. Microbiol. Infect. Dis. 2011, 30, 973–980. [Google Scholar] [CrossRef] [PubMed]
- De Baets, F.; Schelstraete, P.; van Daele, S.; Haerynck, F.; Vaneechoutte, M. Achromobacter xylosoxidans in cystic fibrosis: Prevalence and clinical relevance. J. Cyst. Fibros. 2007, 6, 75–78. [Google Scholar] [CrossRef] [PubMed]
- Worlitzsch, D.; Rintelen, C.; Böhm, K.; Wollschläger, B.; Merkel, N.; Borneff-Lipp, M.; Döring, G. Antibiotic-resistant obligate anaerobes during exacerbations of cystic fibrosis patients. Clin. Microbiol. Infect. 2009, 15, 454–460. [Google Scholar] [CrossRef] [PubMed]
- Cox, M.J.; Allgaier, M.; Taylor, B.; Baek, M.S.; Huang, Y.J.; Daly, R.A.; Karaoz, U.; Andersen, G.L.; Brown, R.; Fujimura, K.E.; et al. Airway microbiota and pathogen abundance in age-stratified cystic fibrosis patients. PLoS ONE 2010, 5, e11044. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Murray, S.; Lipuma, J.J. Modeling the impact of antibiotic exposure on human microbiota. Sci. Rep. 2014, 4, 4345. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Schloss, P.D.; Kalikin, L.M.; Carmody, L.A.; Foster, B.K.; Petrosino, J.F.; Cavalcoli, J.D.; VanDevanter, D.R.; Murray, S.; Li, J.Z.; et al. Decade-long bacterial community dynamics in cystic fibrosis airways. Proc. Natl. Acad. Sci. USA 2012, 109, 5809–5814. [Google Scholar] [CrossRef] [PubMed]
- Rogers, G.B.; van der Gast, C.J.; Cuthbertson, L.; Thomson, S.K.; Bruce, K.D.; Martin, M.L.; Serisier, D.J. Clinical measures of disease in adult non-CF bronchiectasis correlate with airway microbiota composition. Thorax 2013, 68, 731–737. [Google Scholar] [CrossRef] [PubMed]
- Dickson, R.P.; Erb-Downward, J.R.; Martinez, F.J.; Huffnagle, G.B. The Microbiome and the respiratory tract. Annu. Rev. Physiol. 2016, 78, 481–504. [Google Scholar] [CrossRef] [PubMed]
- Quinn, R.A.; Lim, Y.W.; Maughan, H.; Conrad, D.; Rohwer, F.; Whiteson, K.L. Biogeochemical forces shape the composition and physiology of polymicrobial communities in the cystic fibrosis lung. MBio 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Willner, D.; Furlan, M.; Haynes, M.; Schmieder, R.; Angly, F.E.; Silva, J.; Tammadoni, S.; Nosrat, B.; Conrad, D.; Rohwer, F. Metagenomic analysis of respiratory tract DNA viral communities in cystic fibrosis and non-cystic fibrosis individuals. PLoS ONE 2009, 4, e7370. [Google Scholar] [CrossRef] [PubMed]
- Lim, Y.W.; Schmieder, R.; Haynes, M.; Willner, D.; Furlan, M.; Youle, M.; Abbott, K.; Edwards, R.; Evangelista, J.; Conrad, D.; et al. Metagenomics and metatranscriptomics: Windows on CF-associated viral and microbial communities. J. Cyst. Fibros. 2013, 12, 154–164. [Google Scholar] [CrossRef] [PubMed]
- Nelson, J.W.; Tredgett, M.W.; Sheehan, J.K.; Thornton, D.J.; Notman, D.; Govan, J.R. Mucinophilic and chemotactic properties of Pseudomonas aeruginosa in relation to pulmonary colonization in cystic fibrosis. Infect. Immun. 1990, 58, 1489–1495. [Google Scholar] [PubMed]
- Feldman, M.; Bryan, R.; Rajan, S.; Scheffler, L.; Brunnert, S.; Tang, H.; Prince, A. Role of flagella in pathogenesis of Pseudomonas aeruginosa pulmonary infection. Infect. Immun. 1998, 66, 43–51. [Google Scholar] [PubMed]
- Wolfgang, M.C.; Jyot, J.; Goodman, A.L.; Ramphal, R.; Lory, S. Pseudomonas aeruginosa regulates flagellin expression as part of a global response to airway fluid from cystic fibrosis patients. Proc. Natl. Acad. Sci. USA 2004, 101, 6664–6668. [Google Scholar] [CrossRef] [PubMed]
- Luzar, M.A.; Thomassen, M.J.; Montie, T.C. Flagella and motility alterations in Pseudomonas aeruginosa strains from patients with cystic fibrosis: Relationship to patient clinical condition. Infect. Immun. 1985, 50, 577–582. [Google Scholar] [PubMed]
- Deretic, V.; Govan, J.R.W.; Konyecsni, W.M.; Martin, D.W. Mucoid Pseudomonas aeruginosa in cystic fibrosis: Mutations in the muc loci affect transcription of the algR and algD genes in response to environmental stimuli. Mol. Microbiol. 1990, 4, 189–196. [Google Scholar] [CrossRef] [PubMed]
- Chawley, P.; Samal, H.B.; Prava, J.; Suar, M.; Mahapatra, R.K. Comparative genomics study for identification of drug and vaccine targets in Vibrio cholerae: MurA ligase as a case study. Genomics 2014, 103, 83–93. [Google Scholar] [CrossRef] [PubMed]
- Rubin, B.K. Mucus structure and properties in cystic fibrosis. Paediatr. Respir. Rev. 2007, 8, 4–7. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil extracellular traps kill bacteria. Science 2004, 303, 1532–1535. [Google Scholar] [CrossRef] [PubMed]
- Whitchurch, C.B.; Tolker-Nielsen, T.; Ragas, P.C.; Mattick, J.S. Extracellular DNA required for bacterial biofilm formation. Science 2002, 295, 1487. [Google Scholar] [CrossRef] [PubMed]
- Voynow, J.A.; Fischer, B.M.; Zheng, S. Proteases and cystic fibrosis. Int. J. Biochem. Cell Biol. 2008, 40, 1238–1245. [Google Scholar] [CrossRef] [PubMed]
- Doring, G.; Flume, P.; Heijerman, H.; Elborn, J.S.; Consensus Study Group. Treatment of lung infection in patients with cystic fibrosis: Current and future strategies. J. Cyst. Fibros. 2012, 11, 461–479. [Google Scholar] [CrossRef] [PubMed]
- Doring, G.; Hoiby, N.; Consensus Study Group. Early intervention and prevention of lung disease in cystic fibrosis: A European consensus. J. Cyst. Fibros. 2004, 3, 67–91. [Google Scholar] [CrossRef] [PubMed]
- Chavez, A.; Mian, A.; Scurlock, A.M.; Blackall, D.; Com, G. Antibiotic hypersensitivity in CF: Drug-induced life-threatening hemolytic anemia in a pediatric patient. J. Cyst. Fibros. 2010, 9, 433–438. [Google Scholar] [CrossRef] [PubMed]
- Pehrsson, E.C.; Forsberg, K.J.; Gibson, M.K.; Ahmadi, S.; Dantas, G. Novel resistance functions uncovered using functional metagenomic investigations of resistance reservoirs. Front. Microbiol. 2013, 4. [Google Scholar] [CrossRef] [PubMed]
- King, P.; Pham, L.K.; Waltz, S.; Sphar, D.; Yamamoto, R.T.; Conrad, D.; Taplitz, R.; Torriani, F.; Forsyth, R.A. Longitudinal metagenomic analysis of hospital air identifies clinically relevant microbes. PLoS ONE 2016, 11. [Google Scholar] [CrossRef]
- Blaser, M.J. The microbiome revolution. J. Clin. Invest. 2014, 124, 4162–4165. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, H.J.; Borowitz, D.S.; Christiansen, D.H.; Morris, E.M.; Nash, M.L.; Ramsey, B.W.; Rosenstein, B.J.; Smith, A.L.; Wohl, M.E. Effect of aerosolized recombinant human DNase on exacerbations of respiratory symptoms and on pulmonary function in patients with cystic fibrosis. The Pulmozyme Study Group. N. Engl. J. Med. 1994, 331, 637–642. [Google Scholar] [CrossRef] [PubMed]
- Ramsey, B.W.; Pepe, M.S.; Quan, J.M.; Otto, K.L.; Montgomery, A.B.; Williams-Warren, J.; Vasiljev-K, M.; Borowitz, D.; Bowman, C.M.; Marshall, B.C.; et al. Intermittent administration of inhaled tobramycin in patients with cystic fibrosis. Cystic Fibrosis Inhaled Tobramycin Study Group. N. Engl. J. Med. 1999, 340, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Kerem, E.; Conway, S.; Elborn, S.; Heijerman, H.; Committee, C. others Standards of care for patients with cystic fibrosis: A European consensus. J. Cyst. Fibros. 2005, 4, 7–26. [Google Scholar] [CrossRef] [PubMed]
- Flume, P.A.; Mogayzel, P.J.; Robinson, K.A.; Goss, C.H.; Rosenblatt, R.L.; Kuhn, R.J.; Marshall, B.C.; Bujan, J.; Downs, A.; Finder, J.; et al. Cystic fibrosis pulmonary guidelines: Treatment of pulmonary exacerbations. Am. J. Respir. Crit. Care Med. 2009, 180, 802–808. [Google Scholar] [CrossRef] [PubMed]
- Smyth, A.R.; Bell, S.C.; Bojcin, S.; Bryon, M.; Duff, A.; Flume, P.; Kashirskaya, N.; Munck, A.; Ratjen, F.; Schwarzenberg, S.J.; et al. European cystic fibrosis society standards of care: Best practice guidelines. J. Cyst. Fibros. 2014, 13, S23–S42. [Google Scholar] [CrossRef] [PubMed]
- Taylor-Robinson, D.; Whitehead, M.; Diderichsen, F.; Olesen, H.V.; Pressler, T.; Smyth, R.L.; Diggle, P. Understanding the natural progression in %FEV1 decline in patients with cystic fibrosis: A longitudinal study. Thorax 2012, 67, 860–866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flume, P.A.; O’Sullivan, B.P.; Robinson, K.A.; Goss, C.H.; Mogayzel, P., Jr.; Willey-Courand, D.B.; Bujan, J.; Finder, J.; Lester, M. Cystic Fibrosis Pulmonary Guidelines: Chronic Medications for Maintenance of Lung Health. Am. J. Respir. Crit. Care Med. 2007, 176, 957–969. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.R.; Hankinson, J.; Brusasco, V.; Burgos, F.; Casaburi, R.; Coates, A.; Crapo, R.; Enright, P.; van der Grinten, C.P.M.; Gustafsson, P.; et al. Standardisation of spirometry. Eur. Respir. J. 2005, 26, 319–338. [Google Scholar] [CrossRef] [PubMed]
- Willner, D.; Daly, J.; Whiley, D.; Grimwood, K.; Wainwright, C.E.; Hugenholtz, P. Comparison of DNA extraction methods for microbial community profiling with an application to pediatric bronchoalveolar lavage samples. PLoS ONE 2012, 7, e34605. [Google Scholar] [CrossRef] [PubMed]
- Bacci, G.; Bazzicalupo, M.; Benedetti, A.; Mengoni, A. StreamingTrim 1.0: A Java software for dynamic trimming of 16S rRNA sequence data from metagenetic studies. Mol. Ecol. Resour. 2014, 14, 426–434. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Abubucker, S.; Segata, N.; Goll, J.; Schubert, A.M.; Izard, J.; Cantarel, B.L.; Rodriguez-Mueller, B.; Zucker, J.; Thiagarajan, M.; Henrissat, B.; et al. Metabolic reconstruction for metagenomic data and its application to the human microbiome. PLoS Comput. Biol. 2012, 8, e1002358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Truong, D.T.; Franzosa, E.A.; Tickle, T.L.; Scholz, M.; Weingart, G.; Pasolli, E.; Tett, A.; Huttenhower, C.; Segata, N. MetaPhlAn2 for enhanced metagenomic taxonomic profiling. Nat. Methods 2015, 12, 902–903. [Google Scholar] [CrossRef] [PubMed]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Li, R.; Raes, J.; Arumugam, M.; Burgdorf, K.S.; Manichanh, C.; Nielsen, T.; Pons, N.; Levenez, F.; Yamada, T.; et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010, 464, 59–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, W.; Lomsadze, A.; Borodovsky, M. Ab initio gene identification in metagenomic sequences. Nucleic Acids Res. 2010, 38, e132. [Google Scholar] [CrossRef] [PubMed]
- Eddy, S.R. Accelerated Profile HMM Searches. PLoS Comput. Biol. 2011, 7, e1002195. [Google Scholar] [CrossRef] [PubMed]
- Powell, S.; Szklarczyk, D.; Trachana, K.; Roth, A.; Kuhn, M.; Muller, J.; Arnold, R.; Rattei, T.; Letunic, I.; Doerks, T.; et al. eggNOG v3.0: Orthologous groups covering 1133 organisms at 41 different taxonomic ranges. Nucleic Acids Res. 2012, 40, D284–D289. [Google Scholar] [CrossRef] [PubMed]
- Pearson, W.R. An introduction to sequence similarity (“homology”) searching. Curr. Protoc. Bioinform. 2013. [Google Scholar] [CrossRef]
- Saier, M.H.; Reddy, V.S.; Tamang, D.G.; Västermark, A. The transporter classification database. Nucleic Acids Res. 2014, 42, D251–D258. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.E.; Smith, J.; Lam, M.; Zemla, A.; Dyer, M.D.; Slezak, T. MvirDB—A microbial database of protein toxins, virulence factors and antibiotic resistance genes for bio-defence applications. Nucleic Acids Res. 2007, 35, D391–D394. [Google Scholar] [CrossRef] [PubMed]
- Oksanen, J.; Blanchet, F.; Kindt, R.; Legendre, P.; Minchin, P.; O’Hara, R.; Simpson, G.; Solymos, P.; Stevens, M.H.H.; Wagner, H. Vegan: Community ecology package. R package version 2.0–10. 2013. Available online: http://CRAN.R-project.org/package=vegan.
- Kuang, J.; Huang, L.; He, Z.; Chen, L.; Hua, Z.; Jia, P.; Li, S.; Liu, J.; Li, J.; Zhou, J.; et al. Predicting taxonomic and functional structure of microbial communities in acid mine drainage. ISME J. 2016, 10, 1527–1539. [Google Scholar] [CrossRef] [PubMed]
- Turnbaugh, P.J.; Hamady, M.; Yatsunenko, T.; Cantarel, B.L.; Duncan, A.; Ley, R.E.; Sogin, M.L.; Jones, W.J.; Roe, B.A.; Affourtit, J.P.; et al. A core gut microbiome in obese and lean twins. Nature 2009, 457, 480–484. [Google Scholar] [CrossRef] [PubMed]
- John, R.; Dalling, J.W.; Harms, K.E.; Yavitt, J.B.; Stallard, R.F.; Mirabello, M.; Hubbell, S.P.; Valencia, R.; Navarrete, H.; Vallejo, M.; et al. Soil nutrients influence spatial distributions of tropical tree species. Proc. Natl. Acad. Sci. USA 2007, 104, 864–869. [Google Scholar] [CrossRef] [PubMed]
- Wickham, H. Ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2009; ISBN 978-0-387-98140-6. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Study ID | Age | Gender | CFTR Genotype | BMI | Average Annual FEV1% Value | Lung Disease Status | Number of Exacerbations in the Last 5 Years | Maintenance Antimicrobial Therapy 1 |
|---|---|---|---|---|---|---|---|---|
| BS29 | 25 | F | F508del/L1077P | 23.1 | 72 | normal/mild | 20 (2–7) | AT |
| BS47 | 33 | M | F508del/N1303K | 23.8 | 94 | normal/mild | 5 (1–2) | AA, AC |
| MS1 | 30 | F | G1244 E/G1244 E | 22.9 | 83 | normal/mild | 5 (0–4) | AC |
| GNR19 | 18 | M | F508del/F508del | 21.4 | 80 | normal/mild | 9 (1–3) | None |
| GNR5 | 24 | M | F508del/12491G>A | 22.7 | 72 | normal/mild | 9 (0–3) | None |
| BNR22 | 22 | F | F508del/G85E | 22.1 | 81 | normal/mild | 23 (2–7) | AT, AZ |
| BS19 | 36 | M | F508del/W1282X | 24.9 | 37 | severe | 18 (3–4) | AC, AZ |
| BS51 | 36 | M | F508del/2789+5G>A | 21 | 38 | severe | 16 (2–6) | AC, AZ |
| BS85 | 34 | M | F508del/1259insA | 18.8 | 21 | severe | 15 (2–5) | AC, AZ |
| BNR15 | 46 | F | F508del/F508del | 19.7 | 37 | severe | 16 (2–4) | AC, AZ |
| BNR20 | 25 | M | F508del/F508del | 23.3 | 36 | severe | 36 (4–11) | AA, AC |
| BNR49 | 26 | M | N1303K/G85E | 19.9 | 29 | severe | 10 (1–3) | AC |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bacci, G.; Mengoni, A.; Fiscarelli, E.; Segata, N.; Taccetti, G.; Dolce, D.; Paganin, P.; Morelli, P.; Tuccio, V.; De Alessandri, A.; et al. A Different Microbiome Gene Repertoire in the Airways of Cystic Fibrosis Patients with Severe Lung Disease. Int. J. Mol. Sci. 2017, 18, 1654. https://doi.org/10.3390/ijms18081654
Bacci G, Mengoni A, Fiscarelli E, Segata N, Taccetti G, Dolce D, Paganin P, Morelli P, Tuccio V, De Alessandri A, et al. A Different Microbiome Gene Repertoire in the Airways of Cystic Fibrosis Patients with Severe Lung Disease. International Journal of Molecular Sciences. 2017; 18(8):1654. https://doi.org/10.3390/ijms18081654
Chicago/Turabian StyleBacci, Giovanni, Alessio Mengoni, Ersilia Fiscarelli, Nicola Segata, Giovanni Taccetti, Daniela Dolce, Patrizia Paganin, Patrizia Morelli, Vanessa Tuccio, Alessandra De Alessandri, and et al. 2017. "A Different Microbiome Gene Repertoire in the Airways of Cystic Fibrosis Patients with Severe Lung Disease" International Journal of Molecular Sciences 18, no. 8: 1654. https://doi.org/10.3390/ijms18081654