Functional Testing of SLC26A4 Variants—Clinical and Molecular Analysis of a Cohort with Enlarged Vestibular Aqueduct from Austria

 , ,
, , 
Abstract
:1. Introduction
2. Results
2.1. Clinical Features
2.2. Detection of Sequence Variations in the GJB2 and SLC26A4 Genes
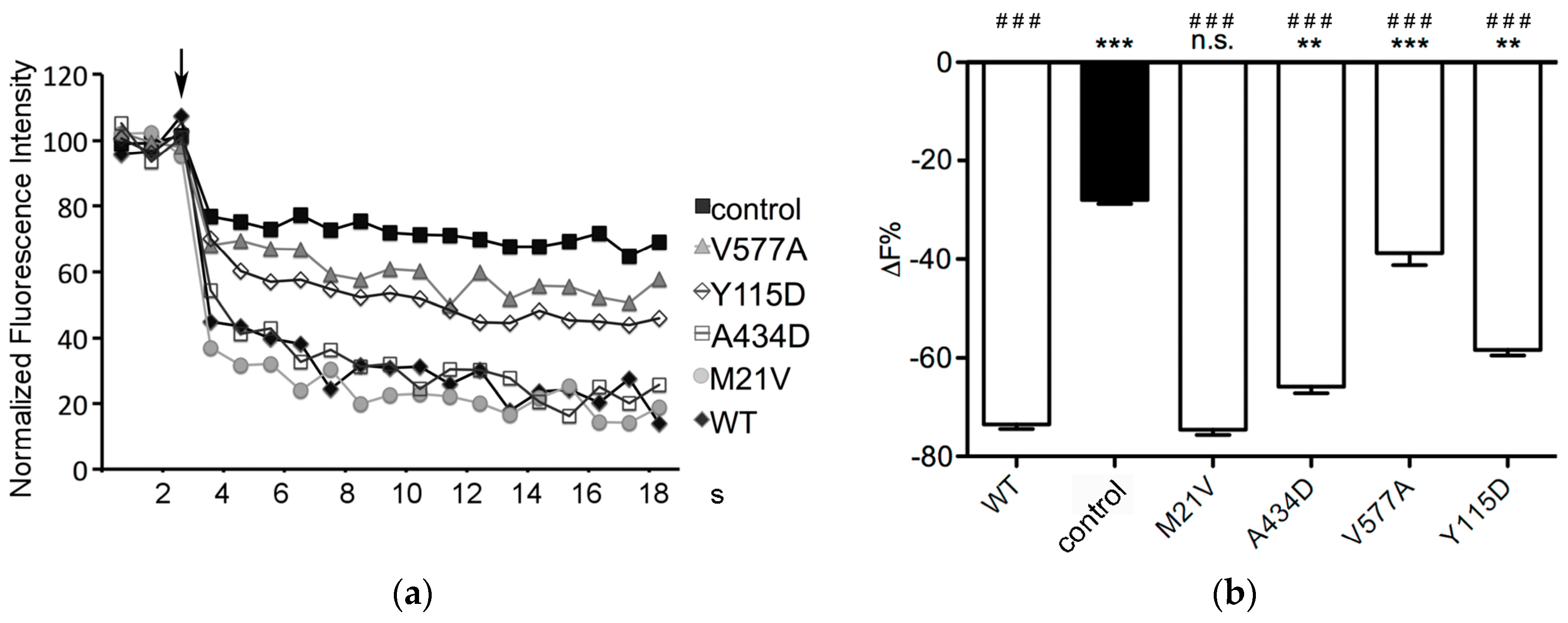
2.3. Function of SLC26A4 Protein Variants
2.4. Subcellular Localization of Pendrin Variants
2.5. Total and Plasma Membrane Expression Levels of Pendrin Variants
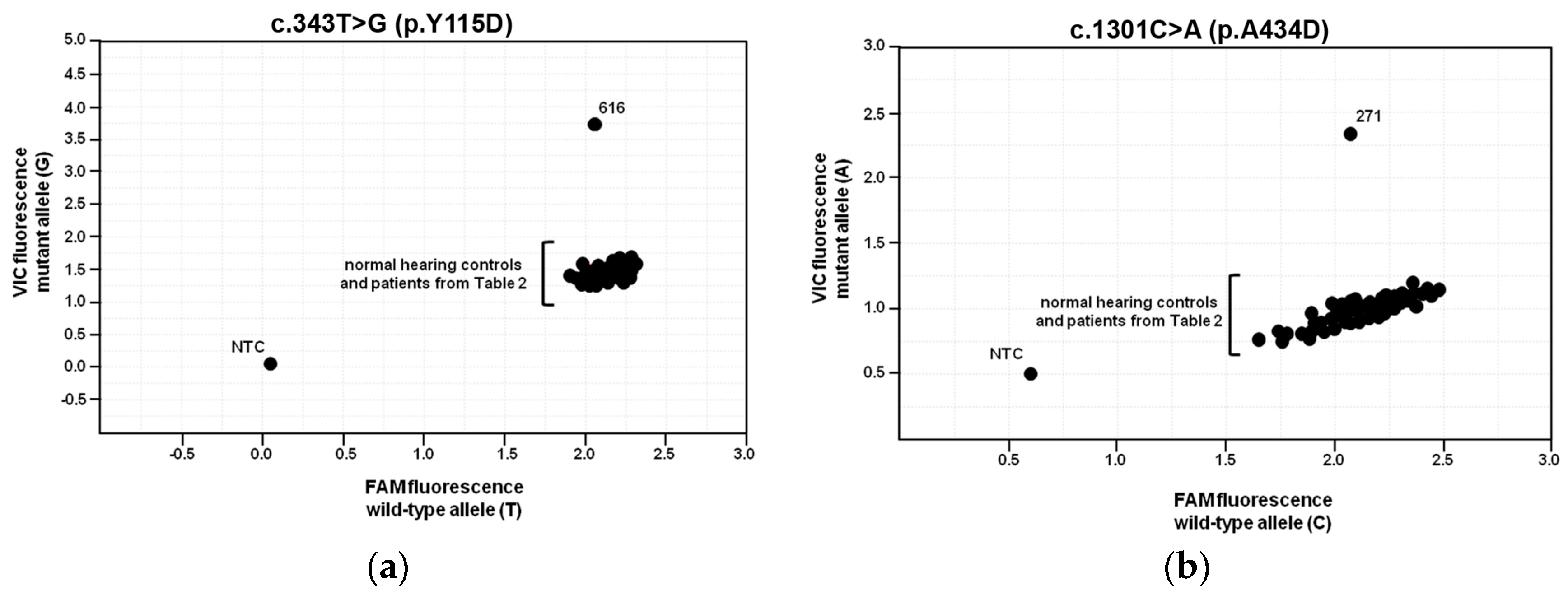
2.6. Population Frequencies of c.343T>G and c.1301C>A
3. Discussion
4. Materials and Methods
4.1. Patients
4.2. Genomic DNA Samples
4.3. Genomic DNA Analysis
4.4. Plasmid Constructs
4.5. Cell Lines
4.6. Pendrin Functional Test
4.7. Co-Localization Experiments
4.8. Determination of Wild Type and Mutant Pendrin Total Expression Levels by Imaging
4.9. Determination of Wild Type and Mutant Pendrin Expression Levels in the Plasma Membrane Region
4.10. Sequence Variation Genotyping Assays
4.11. Salts and Chemicals
4.12. Statistical Analysis
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| ARNSHL | Autosomal Recessive Non-Syndromic Hearing Loss |
| ATP6V1B1 | ATPase H+ Transporting V1 Subunit B1 |
| CT | Computed Tomography |
| DFNB4 | Deafness B4 |
| ECFP | Enhanced Cyan Fluorescent Protein |
| ER | Endoplasmic Reticulum |
| EVA | Enlarged Vestibular Aqueduct |
| EYFP | Enhanced Yellow Fluorescent Protein |
| FOXI1 | Forkhead Box I1 |
| GJB2 | Gap Junction Protein β2 |
| HBSS | Hank’s Balanced Salt Solution |
| IP2 | Incomplete Partition 2 |
| KCNJ10 | Potassium Channel, Inwardly Rectifying, Subfamily J, Member 10 |
| MAF | Minor Allele Frequency |
| PS | Pendred Syndrome |
| RTA | Renal Tubular Acidosis |
| SLC26A4 | Solute Carrier Family 26 Member A4 |
| SNP | Single Nucleotide Polymorphism |
References
- Da Silva Costa, S.M.; Ramos, P.Z.; Arrojo Martins, F.T.; Sartorato, E.L. Genetic Diagnosis of Deafness. In The Role of Pendrin in Health and Disease; Dossena, S., Paulmichl, M., Eds.; Springer International Publishing: Basel, Switzerland, 2017; pp. 61–83. [Google Scholar]
- Rehm, H.L. Disease-targeted sequencing: A cornerstone in the clinic. Nat. Rev. Genet. 2013, 14, 295–300. [Google Scholar] [CrossRef] [PubMed]
- Kenneson, A.; Van Naarden Braun, K.; Boyle, C. GJB2 (connexin 26) variants and nonsyndromic sensorineural hearing loss: A HuGE review. Genet. Med. 2002, 4, 258–274. [Google Scholar] [CrossRef] [PubMed]
- Everett, L.A.; Glaser, B.; Beck, J.C.; Idol, J.R.; Buchs, A.; Heyman, M.; Adawi, F.; Hazani, E.; Nassir, E.; Baxevanis, A.D.; et al. Pendred syndrome is caused by mutations in a putative sulphate transporter gene (PDS). Nat. Genet. 1997, 17, 411–422. [Google Scholar] [CrossRef] [PubMed]
- Alasti, F.; Van Camp, G.; Smith, R.J.H. Pendred Syndrome/DFNB4. In GeneReviews(R); Pagon, R.A., Adam, M.P., Ardinger, H.H., Wallace, S.E., Amemiya, A., Bean, L.J.H., Bird, T.D., Ledbetter, N., Mefford, H.C., Smith, R.J.H., et al., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Ito, T.; Muskett, J.; Chattaraj, P.; Choi, B.Y.; Lee, K.Y.; Zalewski, C.K.; King, K.A.; Li, X.; Wangemann, P.; Shawker, T.; et al. SLC26A4 mutation testing for hearing loss associated with enlargement of the vestibular aqueduct. World J. Otorhinolaryngol. 2013, 3, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Roesch, S.; Tóth, M.; Rasp, G. Pendrin-Linked Deafness in Humans. In The Role of Pendrin in Health and Disease; Dossena, S., Paulmichl, M., Eds.; Springer International Publishing: Basel, Switzerland, 2017; pp. 37–60. [Google Scholar]
- Hilgert, N.; Smith, R.J.; Van Camp, G. Forty-six genes causing nonsyndromic hearing impairment: Which ones should be analyzed in DNA diagnostics? Mutat. Res. 2009, 681, 189–196. [Google Scholar] [CrossRef] [PubMed]
- Dossena, S.; Rodighiero, S.; Vezzoli, V.; Bazzini, C.; Sironi, C.; Meyer, G.; Furst, J.; Ritter, M.; Garavaglia, M.L.; Fugazzola, L.; et al. Fast fluorometric method for measuring pendrin (SLC26A4) Cl−/I− transport activity. Cell. Physiol. Biochem. 2006, 18, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, A.; Hisatome, I.; Taniguchi, S.; Sasaki, N.; Yamamoto, Y.; Miake, J.; Fukui, H.; Shimizu, H.; Okamura, T.; Okura, T.; et al. Mechanism of iodide/chloride exchange by pendrin. Endocrinology 2004, 145, 4301–4308. [Google Scholar] [CrossRef] [PubMed]
- Shcheynikov, N.; Yang, D.; Wang, Y.; Zeng, W.; Karniski, L.P.; Therefore, I.; Wall, S.M.; Muallem, S. The Slc26a4 transporter functions as an electroneutral Cl−/I−/HCO3− exchanger: Role of Slc26a4 and Slc26a6 in I− and HCO3− secretion and in regulation of CFTR in the parotid duct. J. Physiol. 2008, 586, 3813–3824. [Google Scholar] [CrossRef] [PubMed]
- Everett, L.A.; Morsli, H.; Wu, D.K.; Green, E.D. Expression pattern of the mouse ortholog of the Pendred’s syndrome gene (Pds) suggests a key role for pendrin in the inner ear. Proc. Natl. Acad. Sci. USA 1999, 96, 9727–9732. [Google Scholar] [CrossRef] [PubMed]
- Royaux, I.E.; Suzuki, K.; Mori, A.; Katoh, R.; Everett, L.A.; Kohn, L.D.; Green, E.D. Pendrin, the protein encoded by the Pendred syndrome gene (PDS), is an apical porter of iodide in the thyroid and is regulated by thyroglobulin in FRTL-5 cells. Endocrinology 2000, 141, 839–845. [Google Scholar] [CrossRef] [PubMed]
- Shearer, A.E.; Hildebrand, M.S.; Smith, R.J.H. Hereditary hearing loss and deafness overview. In GeneReviews((R)); Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Mefford, H.C., Stephens, K., Amemiya, A., Ledbetter, N., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Fraser, G.R. Association of congenital deafness with goitre (Pendred’s syndrome) a study of 207 families. Ann. Hum. Genet. 1965, 28, 201–249. [Google Scholar] [CrossRef] [PubMed]
- Phelps, P.D.; Coffey, R.A.; Trembath, R.C.; Luxon, L.M.; Grossman, A.B.; Britton, K.E.; Kendall-Taylor, P.; Graham, J.M.; Cadge, B.C.; Stephens, S.G.; et al. Radiological malformations of the ear in Pendred syndrome. Clin. Radiol. 1998, 53, 268–273. [Google Scholar] [CrossRef]
- King, K.A.; Choi, B.Y.; Zalewski, C.; Madeo, A.C.; Manichaikul, A.; Pryor, S.P.; Ferruggiaro, A.; Eisenman, D.; Kim, H.J.; Niparko, J.; et al. SLC26A4 genotype, but not cochlear radiologic structure, is correlated with hearing loss in ears with an enlarged vestibular aqueduct. Laryngoscope 2010, 120, 384–389. [Google Scholar] [CrossRef] [PubMed]
- Rah, Y.C.; Kim, A.R.; Koo, J.W.; Lee, J.H.; Oh, S.H.; Choi, B.Y. Audiologic presentation of enlargement of the vestibular aqueduct according to the SLC26A4 genotypes. Laryngoscope 2015, 125, E216–E222. [Google Scholar] [CrossRef] [PubMed]
- Greinwald, J.; DeAlarcon, A.; Cohen, A.; Uwiera, T.; Zhang, K.; Benton, C.; Halstead, M.; Meinzen-Derr, J. Significance of unilateral enlarged vestibular aqueduct. Laryngoscope 2013, 123, 1537–1546. [Google Scholar] [CrossRef] [PubMed]
- Soh, L.M.; Druce, M.; Grossman, A.B.; Differ, A.M.; Rajput, L.; Bitner-Glindzicz, M.; Korbonits, M. Evaluation of genotype-phenotype relationships in patients referred for endocrine assessment in suspected Pendred syndrome. Eur. J. Endocrinol. 2015, 172, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Muskett, J.A.; Chattaraj, P.; Heneghan, J.F.; Reimold, F.R.; Shmukler, B.E.; Brewer, C.C.; King, K.A.; Zalewski, C.K.; Shawker, T.H.; Butman, J.A.; et al. Atypical patterns of segregation of familial enlargement of the vestibular aqueduct. Laryngoscope 2016, 126, E240–E247. [Google Scholar] [CrossRef] [PubMed]
- Pryor, S.P.; Madeo, A.C.; Reynolds, J.C.; Sarlis, N.J.; Arnos, K.S.; Nance, W.E.; Yang, Y.; Zalewski, C.K.; Brewer, C.C.; Butman, J.A.; et al. SLC26A4/PDS genotype-phenotype correlation in hearing loss with enlargement of the vestibular aqueduct (EVA): Evidence that Pendred syndrome and non-syndromic EVA are distinct clinical and genetic entities. J. Med. Genet. 2005, 42, 159–165. [Google Scholar] [CrossRef] [PubMed]
- Chattaraj, P.; Reimold, F.R.; Muskett, J.A.; Shmukler, B.E.; Chien, W.W.; Madeo, A.C.; Pryor, S.P.; Zalewski, C.K.; Butman, J.A.; Brewer, C.C.; et al. Use of SLC26A4 mutation testing for unilateral enlargement of the vestibular aqueduct. JAMA Otolaryngol. Head Neck Surg. 2013, 139, 907–913. [Google Scholar] [CrossRef] [PubMed]
- Tsukada, K.; Nishio, S.Y.; Hattori, M.; Usami, S. Ethnic-specific spectrum of GJB2 and SLC26A4 mutations: Their origin and a literature review. Ann. Otol. Rhinol. Laryngol. 2015, 124, 61S–76S. [Google Scholar] [CrossRef] [PubMed]
- Wolf, A.; Frohne, A.; Allen, M.; Parzefall, T.; Koenighofer, M.; Schreiner, M.M.; Schoefer, C.; Frei, K.; Lucas, T. A novel mutation in SLC26A4 causes nonsyndromic autosomal recessive hearing impairment. Otol. Neurotol. 2017, 38, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- De Moraes, V.C.; Bernardinelli, E.; Zocal, N.; Fernandez, J.A.; Nofziger, C.; Castilho, A.M.; Sartorato, E.L.; Paulmichl, M.; Dossena, S. Reduction of cellular expression levels is a common feature of functionally affected pendrin (SLC26A4) protein variants. Mol. Med. 2016. [Google Scholar] [CrossRef] [PubMed]
- Noordman, B.J.; van Beeck Calkoen, E.; Witte, B.; Goverts, T.; Hensen, E.; Merkus, P. Prognostic factors for sudden drops in hearing level after minor head injury in patients with an enlarged vestibular aqueduct: A meta-analysis. Otol. Neurotol. 2015, 36, 4–11. [Google Scholar] [CrossRef] [PubMed]
- Zalewski, C.K.; Chien, W.W.; King, K.A.; Muskett, J.A.; Baron, R.E.; Butman, J.A.; Griffith, A.J.; Brewer, C.C. Vestibular dysfunction in patients with enlarged vestibular aqueduct. Otolaryngol. Head Neck Surg. 2015, 153, 257–262. [Google Scholar] [CrossRef] [PubMed]
- Noguchi, Y.; Fukuda, S.; Fukushima, K.; Gyo, K.; Hara, A.; Nakashima, T.; Ogawa, K.; Okamoto, M.; Sato, H.; Usami, S.I.; et al. A nationwide study on enlargement of the vestibular aqueduct in Japan. Auris Nasus Larynx 2017, 44, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.G.; Sim, N.S.; Kim, S.H.; Kim, U.K.; Kim, S.; Choi, J.Y. Enlarged cochlear aqueducts: A potential route for CSF gushers in patients with enlarged vestibular aqueducts. Otol. Neurotol. 2013, 34, 1660–1665. [Google Scholar] [CrossRef] [PubMed]
- Manzoor, N.F.; Wick, C.C.; Wahba, M.; Gupta, A.; Piper, R.; Murray, G.S.; Otteson, T.; Megerian, C.A.; Semaan, M.T. Bilateral sequential cochlear implantation in patients with enlarged vestibular aqueduct (EVA) syndrome. Otol. Neurotol. 2016, 37, e96–e103. [Google Scholar] [CrossRef] [PubMed]
- Grimmer, J.F.; Hedlund, G. Vestibular symptoms in children with enlarged vestibular aqueduct anomaly. Int. J. Pediatr. Otorhinolaryngol. 2007, 71, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Escamez, J.A.; Carey, J.; Chung, W.H.; Goebel, J.A.; Magnusson, M.; Mandala, M.; Newman-Toker, D.E.; Strupp, M.; Suzuki, M.; Trabalzini, F.; et al. Diagnostic criteria for Meniere’s disease. J. Vestib. Res. 2015, 25, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Lempert, T.; Olesen, J.; Furman, J.; Waterston, J.; Seemungal, B.; Carey, J.; Bisdorff, A.; Versino, M.; Evers, S.; Newman-Toker, D. Vestibular migraine: Diagnostic criteria. J. Vestib. Res. 2012, 22, 167–172. [Google Scholar] [CrossRef] [PubMed]
- Zelante, L.; Gasparini, P.; Estivill, X.; Melchionda, S.; D’Agruma, L.; Govea, N.; Mila, M.; Monica, M.D.; Lutfi, J.; Shohat, M.; et al. Connexin26 mutations associated with the most common form of non-syndromic neurosensory autosomal recessive deafness (DFNB1) in Mediterraneans. Hum. Mol. Genet. 1997, 6, 1605–1609. [Google Scholar] [CrossRef] [PubMed]
- Azaiez, H.; Chamberlin, G.P.; Fischer, S.M.; Welp, C.L.; Prasad, S.D.; Taggart, R.T.; del Castillo, I.; Van Camp, G.; Smith, R.J. GJB2: The spectrum of deafness-causing allele variants and their phenotype. Hum. Mutat. 2004, 24, 305–311. [Google Scholar] [CrossRef] [PubMed]
- Hwa, H.L.; Ko, T.M.; Hsu, C.J.; Huang, C.H.; Chiang, Y.L.; Oong, J.L.; Chen, C.C.; Hsu, C.K. Mutation spectrum of the connexin 26 (GJB2) gene in Taiwanese patients with prelingual deafness. Genet. Med. 2003, 5, 161–165. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Ying, Z.; Cai, Z.; Sun, D.; He, Z.; Gao, Y.; Zhang, T.; Zhu, Y.; Chen, Y.; Guan, M.X. GJB2 Mutation Spectrum and Genotype-Phenotype Correlation in 1067 Han chinese subjects with non-syndromic hearing loss. PLoS ONE 2015, 10, e0128691. [Google Scholar] [CrossRef] [PubMed]
- Wilch, E.; Azaiez, H.; Fisher, R.A.; Elfenbein, J.; Murgia, A.; Birkenhager, R.; Bolz, H.; Da Silva-Costa, S.M.; Del Castillo, I.; Haaf, T.; et al. A novel DFNB1 deletion allele supports the existence of a distant cis-regulatory region that controls GJB2 and GJB6 expression. Clin. Genet. 2010, 78, 267–274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yashima, T.; Noguchi, Y.; Kawashima, Y.; Rai, T.; Ito, T.; Kitamura, K. Novel ATP6V1B1 mutations in distal renal tubular acidosis and hearing loss. Acta Otolaryngol. 2010, 130, 1002–1008. [Google Scholar] [CrossRef] [PubMed]
- Propst, E.J.; Blaser, S.; Stockley, T.L.; Harrison, R.V.; Gordon, K.A.; Papsin, B.C. Temporal bone imaging in GJB2 deafness. Laryngoscope 2006, 116, 2178–2186. [Google Scholar] [CrossRef] [PubMed]
- Kenna, M.A.; Rehm, H.L.; Frangulov, A.; Feldman, H.A.; Robson, C.D. Temporal bone abnormalities in children with GJB2 mutations. Laryngoscope 2011, 121, 630–635. [Google Scholar] [CrossRef] [PubMed]
- Dewan, K.; Wippold, F.J., 2nd; Lieu, J.E. Enlarged vestibular aqueduct in pediatric sensorineural hearing loss. Otolaryngol. Head Neck Surg. 2009, 140, 552–558. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.H.; Larson, D.A.; Shott, G.; Rasmussen, B.; Cohen, A.P.; Benton, C.; Halsted, M.; Choo, D.; Meinzen-Derr, J.; Greinwald, J.H., Jr. Audiologic and temporal bone imaging findings in patients with sensorineural hearing loss and GJB2 mutations. Laryngoscope 2009, 119, 554–558. [Google Scholar] [CrossRef] [PubMed]
- Madeo, A.C.; Manichaikul, A.; Reynolds, J.C.; Sarlis, N.J.; Pryor, S.P.; Shawker, T.H.; Griffith, A.J. Evaluation of the thyroid in patients with hearing loss and enlarged vestibular aqueducts. Arch. Otolaryngol. Head Neck Surg. 2009, 135, 670–676. [Google Scholar] [CrossRef] [PubMed]
- Birkeland, A.C.; Lesperance, M.M. Clinical challenges in diagnosing the genetic etiology of hearing loss. In Genetics of Deafness. Monographs in Human Genetics; Karger Publishers: Basel, Switzerland, 2016; Volume 20, pp. 40–55. [Google Scholar]
- Azaiez, H.; Yang, T.; Prasad, S.; Sorensen, J.L.; Nishimura, C.J.; Kimberling, W.J.; Smith, R.J. Genotype-phenotype correlations for SLC26A4-related deafness. Hum. Genet. 2007, 122, 451–457. [Google Scholar] [CrossRef] [PubMed]
- Choi, B.Y.; Madeo, A.C.; King, K.A.; Zalewski, C.K.; Pryor, S.P.; Muskett, J.A.; Nance, W.E.; Butman, J.A.; Brewer, C.C.; Griffith, A.J. Segregation of enlarged vestibular aqueducts in families with non-diagnostic SLC26A4 genotypes. J. Med. Genet. 2009, 46, 856–861. [Google Scholar] [CrossRef] [PubMed]
- Chattaraj, P.; Munjal, T.; Honda, K.; Rendtorff, N.D.; Ratay, J.S.; Muskett, J.A.; Risso, D.S.; Roux, I.; Gertz, E.M.; Schaffer, A.A.; et al. A common SLC26A4-linked haplotype underlying non-syndromic hearing loss with enlargement of the vestibular aqueduct. J. Med. Genet. 2017, 54, 665–673. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Gurrola, J.G.; Wu, H.; Chiu, S.M.; Wangemann, P.; Snyder, P.M.; Smith, R.J. Mutations of KCNJ10 together with mutations of SLC26A4 cause digenic nonsyndromic hearing loss associated with enlarged vestibular aqueduct syndrome. Am. J. Hum. Genet. 2009, 84, 651–657. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Vidarsson, H.; Rodrigo-Blomqvist, S.; Rosengren, S.S.; Enerback, S.; Smith, R.J. Transcriptional control of SLC26A4 is involved in Pendred syndrome and nonsyndromic enlargement of vestibular aqueduct (DFNB4). Am. J. Hum. Genet. 2007, 80, 1055–1063. [Google Scholar] [CrossRef] [PubMed]
- Hulander, M.; Kiernan, A.E.; Blomqvist, S.R.; Carlsson, P.; Samuelsson, E.J.; Johansson, B.R.; Steel, K.P.; Enerback, S. Lack of pendrin expression leads to deafness and expansion of the endolymphatic compartment in inner ears of Foxi1 null mutant mice. Development 2003, 130, 2013–2025. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Wangemann, P. Free radical stress-mediated loss of Kcnj10 protein expression in stria vascularis contributes to deafness in Pendred syndrome mouse model. Am. J. Physiol. Ren. Physiol. 2008, 294, F139–F148. [Google Scholar] [CrossRef] [PubMed]
- Pique, L.M.; Brennan, M.L.; Davidson, C.J.; Schaefer, F.; Greinwald, J., Jr.; Schrijver, I. Mutation analysis of the SLC26A4, FOXI1 and KCNJ10 genes in individuals with congenital hearing loss. PeerJ 2014, 2, e384. [Google Scholar] [CrossRef] [PubMed]
- Landa, P.; Differ, A.M.; Rajput, K.; Jenkins, L.; Bitner-Glindzicz, M. Lack of significant association between mutations of KCNJ10 or FOXI1 and SLC26A4 mutations in Pendred syndrome/enlarged vestibular aqueducts. BMC Med. Genet. 2013, 14, 85. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Wang, X.; Sun, L.; Jiang, H. Screening of SLC26A4, FOXI1, KCNJ10, and GJB2 in bilateral deafness patients with inner ear malformation. Otolaryngol. Head Neck Surg. 2012, 146, 972–978. [Google Scholar] [CrossRef] [PubMed]
- Cirello, V.; Bazzini, C.; Vezzoli, V.; Muzza, M.; Rodighiero, S.; Castorina, P.; Maffini, A.; Botta, G.; Persani, L.; Beck-Peccoz, P.; et al. Molecular and functional studies of 4 candidate loci in Pendred syndrome and nonsyndromic hearing loss. Mol. Cell. Endocrinol. 2012, 351, 342–350. [Google Scholar] [CrossRef] [PubMed]
- Pera, A.; Dossena, S.; Rodighiero, S.; Gandia, M.; Botta, G.; Meyer, G.; Moreno, F.; Nofziger, C.; Hernandez-Chico, C.; Paulmichl, M. Functional assessment of allelic variants in the SLC26A4 gene involved in Pendred syndrome and nonsyndromic EVA. Proc. Natl. Acad. Sci. USA 2008, 105, 18608–18613. [Google Scholar] [CrossRef] [PubMed]
- Dossena, S.; Bizhanova, A.; Nofziger, C.; Bernardinelli, E.; Ramsauer, J.; Kopp, P.; Paulmichl, M. Identification of allelic variants of pendrin (SLC26A4) with loss and gain of function. Cell. Physiol. Biochem. 2011, 28, 467–476. [Google Scholar] [CrossRef] [PubMed]
- Taylor, J.P.; Metcalfe, R.A.; Watson, P.F.; Weetman, A.P.; Trembath, R.C. Mutations of the PDS gene, encoding pendrin, are associated with protein mislocalization and loss of iodide efflux: Implications for thyroid dysfunction in Pendred syndrome. J. Clin. Endocrinol. Metab. 2002, 87, 1778–1784. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.S.; Park, H.J.; Yoo, S.Y.; Namkung, W.; Jo, M.J.; Koo, S.K.; Park, H.Y.; Lee, W.S.; Kim, K.H.; Lee, M.G. Heterogeneity in the processing defect of SLC26A4 mutants. J. Med. Genet. 2008, 45, 411–419. [Google Scholar] [CrossRef] [PubMed]
- Choi, B.Y.; Stewart, A.K.; Nishimura, K.K.; Cha, W.J.; Seong, M.W.; Park, S.S.; Kim, S.W.; Chun, Y.S.; Chung, J.W.; Park, S.N.; et al. Efficient molecular genetic diagnosis of enlarged vestibular aqueducts in East Asians. Genet. Test. Mol. Biomark. 2009, 13, 679–687. [Google Scholar] [CrossRef] [PubMed]
- Ishihara, K.; Okuyama, S.; Kumano, S.; Iida, K.; Hamana, H.; Murakoshi, M.; Kobayashi, T.; Usami, S.; Ikeda, K.; Haga, Y.; et al. Salicylate restores transport function and anion exchanger activity of missense pendrin mutations. Hear. Res. 2010, 270, 110–118. [Google Scholar] [CrossRef] [PubMed]
- Choi, B.Y.; Stewart, A.K.; Madeo, A.C.; Pryor, S.P.; Lenhard, S.; Kittles, R.; Eisenman, D.; Kim, H.J.; Niparko, J.; Thomsen, J.; et al. Hypo-functional SLC26A4 variants associated with nonsyndromic hearing loss and enlargement of the vestibular aqueduct: Genotype-phenotype correlation or coincidental polymorphisms? Hum. Mutat. 2009, 30, 599–608. [Google Scholar] [CrossRef] [PubMed]
- Rose, J.; Muskett, J.A.; King, K.A.; Zalewski, C.K.; Chattaraj, P.; Butman, J.A.; Kenna, M.A.; Chien, W.W.; Brewer, C.C.; Griffith, A.J. Hearing loss associated with enlarged vestibular aqueduct and zero or one mutant allele of SLC26A4. Laryngoscope 2017, 127, E238–E243. [Google Scholar] [CrossRef] [PubMed]
- Albert, S.; Blons, H.; Jonard, L.; Feldmann, D.; Chauvin, P.; Loundon, N.; Sergent-Allaoui, A.; Houang, M.; Joannard, A.; Schmerber, S.; et al. SLC26A4 gene is frequently involved in nonsyndromic hearing impairment with enlarged vestibular aqueduct in Caucasian populations. Eur. J. Hum. Genet. 2006, 14, 773–779. [Google Scholar] [CrossRef] [PubMed]
- Shearer, A.E.; Eppsteiner, R.W.; Booth, K.T.; Ephraim, S.S.; Gurrola, J., 2nd; Simpson, A.; Black-Ziegelbein, E.A.; Joshi, S.; Ravi, H.; Giuffre, A.C.; et al. Utilizing ethnic-specific differences in minor allele frequency to recategorize reported pathogenic deafness variants. Am. J. Hum. Genet. 2014, 95, 445–453. [Google Scholar] [CrossRef] [PubMed]
- Leung, K.J.; Quesnel, A.M.; Juliano, A.F.; Curtin, H.D. Correlation of CT, MR, and histopathology in incomplete partition-ii cochlear anomaly. Otol. Neurotol. 2016, 37, 434–437. [Google Scholar] [CrossRef] [PubMed]
- Sennaroglu, L.; Saatci, I. A new classification for cochleovestibular malformations. Laryngoscope 2002, 112, 2230–2241. [Google Scholar] [CrossRef] [PubMed]
- DiCiommo, D.P.; Duckett, A.; Burcescu, I.; Bremner, R.; Gallie, B.L. Retinoblastoma protein purification and transduction of retina and retinoblastoma cells using improved alphavirus vectors. Investig. Ophthalmol. Vis. Sci. 2004, 45, 3320–3329. [Google Scholar] [CrossRef] [PubMed]
- Fugazzola, L.; Cirello, V.; Dossena, S.; Rodighiero, S.; Muzza, M.; Castorina, P.; Lalatta, F.; Ambrosetti, U.; Beck-Peccoz, P.; Botta, G.; et al. High phenotypic intrafamilial variability in patients with Pendred syndrome and a novel duplication in the SLC26A4 gene: Clinical characterization and functional studies of the mutated SLC26A4 protein. Eur. J. Endocrinol. 2007, 157, 331–338. [Google Scholar] [CrossRef] [PubMed]
- Dror, A.A.; Politi, Y.; Shahin, H.; Lenz, D.R.; Dossena, S.; Nofziger, C.; Fuchs, H.; Hrabe de Angelis, M.; Paulmichl, M.; Weiner, S.; et al. Calcium oxalate stone formation in the inner ear as a result of an Slc26a4 mutation. J. Biol. Chem. 2010, 285, 21724–21735. [Google Scholar] [CrossRef] [PubMed]
- Dossena, S.; Nofziger, C.; Brownstein, Z.; Kanaan, M.; Avraham, K.B.; Paulmichl, M. Functional characterization of pendrin mutations found in the Israeli and Palestinian populations. Cell. Physiol. Biochem. 2011, 28, 477–484. [Google Scholar] [CrossRef] [PubMed]
- Procino, G.; Milano, S.; Tamma, G.; Dossena, S.; Barbieri, C.; Nicoletti, M.C.; Ranieri, M.; Di Mise, A.; Nofziger, C.; Svelto, M.; et al. Co-regulated pendrin and aquaporin 5 expression and trafficking in Type-B intercalated cells under potassium depletion. Cell. Physiol. Biochem. 2013, 32, 184–199. [Google Scholar] [CrossRef] [PubMed]
- Bernardinelli, E.; Costa, R.; Nofziger, C.; Paulmichl, M.; Dossena, S. Effect of known inhibitors of ion transport on pendrin (SLC26A4) activity in a human kidney cell line. Cell. Physiol. Biochem. 2016, 38, 1984–1998. [Google Scholar] [CrossRef] [PubMed]
- Galietta, L.J.; Haggie, P.M.; Verkman, A.S. Green fluorescent protein-based halide indicators with improved chloride and iodide affinities. FEBS Lett. 2001, 499, 220–224. [Google Scholar] [CrossRef]
- Adler, J.; Parmryd, I. Quantifying colocalization by correlation: The Pearson correlation coefficient is superior to the Mander’s overlap coefficient. Cytom. A 2010, 77, 733–742. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient ID | Ethnicity | Sex | Age (year) | EVA, Side | Mondini Malformation, Side | Side Affected by HL | Type of HL | Frequencies Affected by HL, Side |
|---|---|---|---|---|---|---|---|---|
| 119 | Caucasian | Male | 33 | B | No | B | Sensorineural | All, B |
| 267 | Caucasian (Arab) | Female | 23 | B | No | B | Sensorineural | All, B |
| 271 | Caucasian | Female | 8 | B | Yes, B | B | Sensorineural | All, B |
| 272 | Caucasian | Male | 37 | R | No | R | Conductive | Low and Middle, R |
| 278 | Caucasian | Female | 42 | L | No | B | Sensorineural | All, B |
| 305 | Caucasian | Male | 16 | B | Yes, B | B | Sensorineural | Middle and High, R; All, L |
| 307 | Caucasian | Male | 18 | B | No | B | Sensorineural | All, B |
| 308 | Caucasian (Arab) | Male | 12 | B | No | B | Sensorineural | All, B |
| 358 | Caucasian | Female | 32 | B | Yes, B | B | Sensorineural | All, B |
| 359 | Caucasian | Female | 46 | B | No | B | Sensorineural | All, R; Middle and High, L |
| 365 | Caucasian | Male | 76 | A | No | B | Sensorineural | All, B |
| 395 | Caucasian + African | Female | 17 | L | No | B | Sensorineural | Middle, R; All, L |
| 421 | Caucasian | Male | 21 | B | No | B | Sensorineural | All, B |
| 568 | Caucasian | Female | 19 | B | No | B | Sensorineural | All, B |
| 569 | Caucasian | Male | 28 | B | Yes, B | B | Sensorineural | All, B |
| 610 | Caucasian | Female | 51 | B | No | B | Sensorineural | All, B |
| 616 | Caucasian | Female | 32 | B | No | B | Mixed | Middle and High, R; Low and Middle, L |
| 622 | Caucasian | Male | 10 | B | Yes, B | B | Sensorineural | All, B |
| 632 | Caucasian | Female | 50 | B | No | B | Sensorineural | All, B |
| Patient ID | GJB2 | SLC26A4 | Causative Gene | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Nucleotide Change | Amino Acid Change | Nucleotide Change | Amino Acid Change | ||||||
| Allele 1 | Allele 2 | Allele 1 | Allele 2 | Allele 1 | Allele 2 | Allele 1 | Allele 2 | ||
| 119 | WT | WT | WT | WT | WT | WT | WT | WT | undetermined |
| 267 | c.88A>G | WT | p.I30V | WT | WT | WT | WT | WT | undetermined |
| 271 | WT | WT | WT | WT | c.1301C>A | c.1730T>C | p.A434D | p.V577A | SLC26A4 |
| 272 | WT | WT | WT | WT | WT | WT | WT | WT | undetermined |
| 278 | WT | WT | WT | WT | WT | WT | WT | WT | undetermined |
| 305 | WT | WT | WT | WT | WT | WT | WT | WT | undetermined |
| 307 | c.35delG | c.35delG | p.G12VfsX13 | p.G12VfsX13 | WT | WT | WT | WT | GJB2 |
| 308 | c.88A>G | WT | p.I30V | WT | WT | WT | WT | WT | undetermined |
| 358 | WT | WT | WT | WT | c.61A>G | WT | p.M21V | WT | not SLC26A4 |
| 359 | WT | WT | WT | WT | WT | WT | WT | WT | undetermined |
| 365 | WT | WT | WT | WT | WT | WT | WT | WT | undetermined |
| 395 | WT | WT | WT | WT | WT | WT | WT | WT | undetermined |
| 421 | c.35delG | c.35delG | p.G12VfsX13 | p.G12VfsX13 | WT | WT | WT | WT | GJB2 |
| 568 | WT | WT | WT | WT | WT | WT | WT | WT | undetermined |
| 569 | WT | WT | WT | WT | WT | WT | WT | WT | undetermined |
| 610 | WT | WT | WT | WT | WT | WT | WT | WT | undetermined |
| 616 | WT | WT | WT | WT | c.343T>G | WT | p.Y115D | WT | undetermined |
| 622 | WT | WT | WT | WT | WT | WT | WT | WT | undetermined |
| 632 | WT | WT | WT | WT | WT | WT | WT | WT | undetermined |
| SLC26A4 Variant | Function | Subcellular Localization | Expression Levels | Classification |
|---|---|---|---|---|
| p.M21V | not affected | plasma membrane | not affected | benign |
| p.Y115D | 33% reduction | ER | reduced | pathogenic |
| p.A434D | 16% reduction | plasma membrane and ER | reduced | pathogenic |
| p.V577A | 76% reduction | ER | reduced | pathogenic |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roesch, S.; Bernardinelli, E.; Nofziger, C.; Tóth, M.; Patsch, W.; Rasp, G.; Paulmichl, M.; Dossena, S. Functional Testing of SLC26A4 Variants—Clinical and Molecular Analysis of a Cohort with Enlarged Vestibular Aqueduct from Austria. Int. J. Mol. Sci. 2018, 19, 209. https://doi.org/10.3390/ijms19010209
Roesch S, Bernardinelli E, Nofziger C, Tóth M, Patsch W, Rasp G, Paulmichl M, Dossena S. Functional Testing of SLC26A4 Variants—Clinical and Molecular Analysis of a Cohort with Enlarged Vestibular Aqueduct from Austria. International Journal of Molecular Sciences. 2018; 19(1):209. https://doi.org/10.3390/ijms19010209
Chicago/Turabian StyleRoesch, Sebastian, Emanuele Bernardinelli, Charity Nofziger, Miklós Tóth, Wolfgang Patsch, Gerd Rasp, Markus Paulmichl, and Silvia Dossena. 2018. "Functional Testing of SLC26A4 Variants—Clinical and Molecular Analysis of a Cohort with Enlarged Vestibular Aqueduct from Austria" International Journal of Molecular Sciences 19, no. 1: 209. https://doi.org/10.3390/ijms19010209