QTL Mapping for Fiber Quality and Yield Traits Based on Introgression Lines Derived from Gossypium hirsutum × G. tomentosum


Abstract
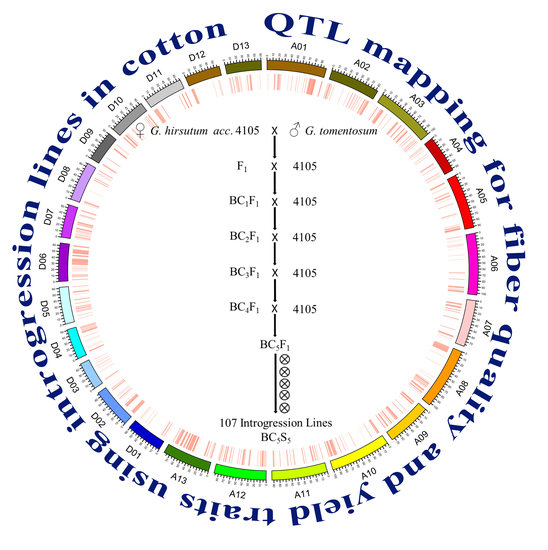
:
1. Introduction
2. Results
2.1. Phenotypic Performance of Yield and Fiber Quality Traits in ILs
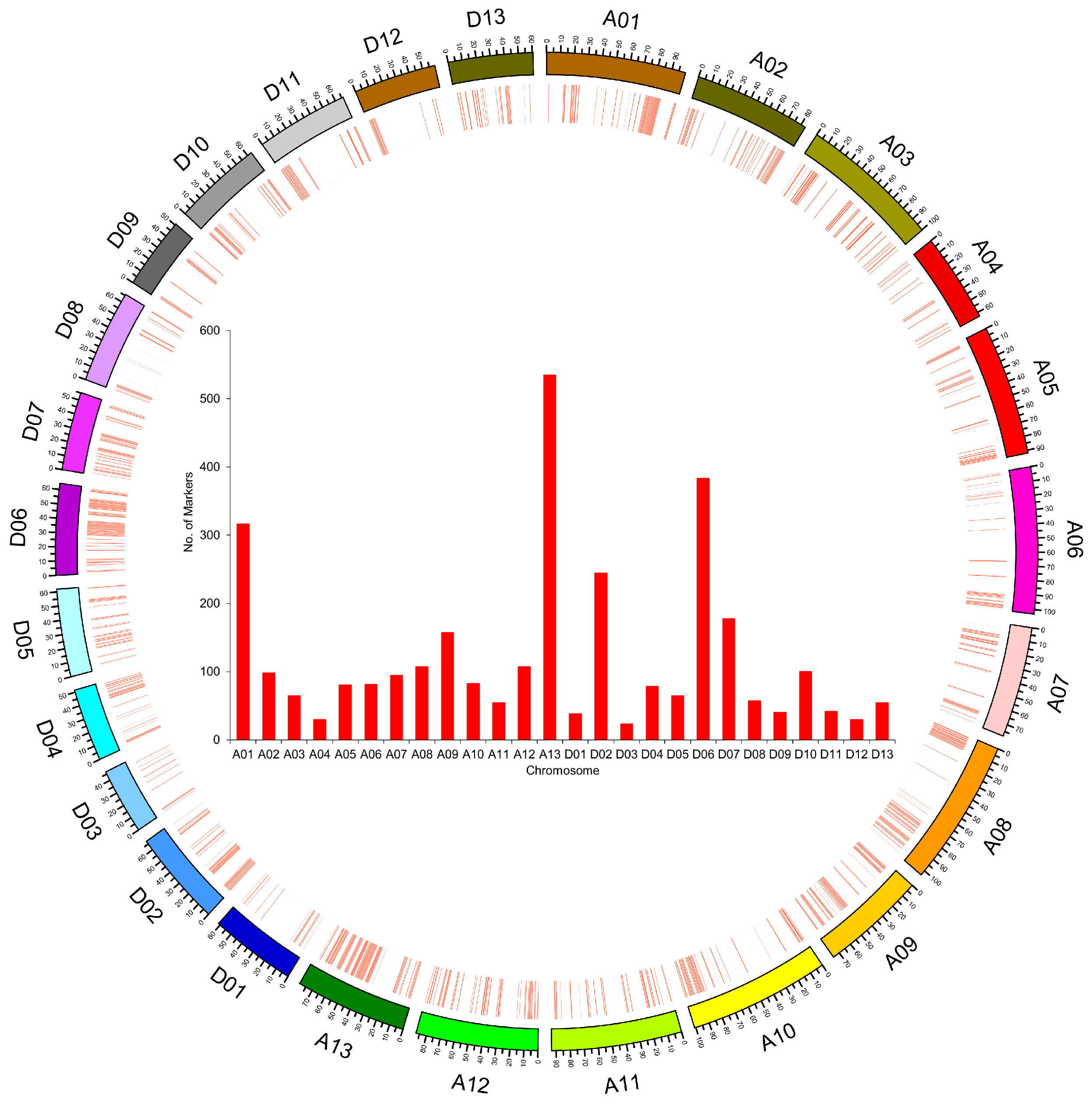
2.2. SLAF-seq and SNP Marker Development
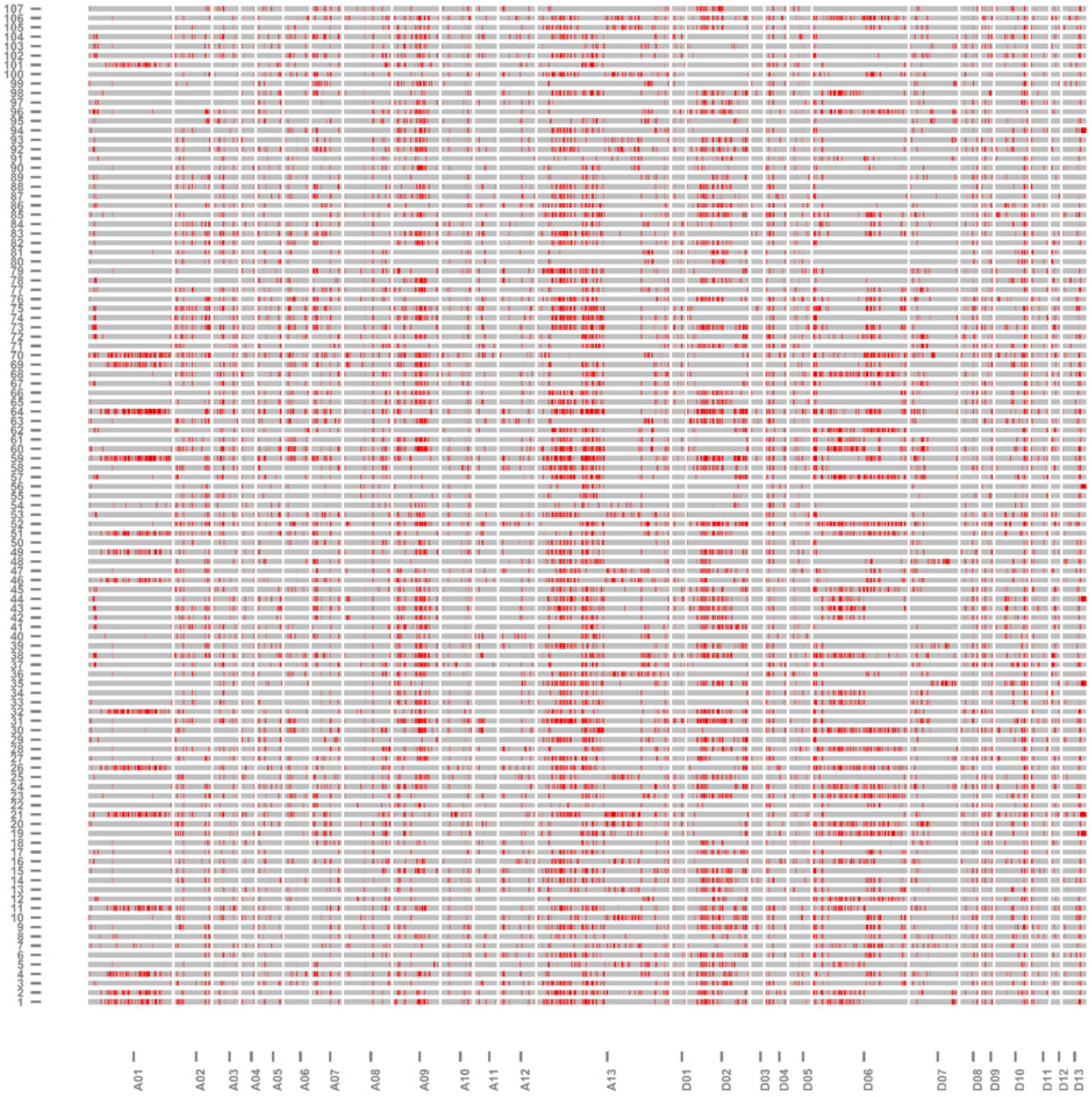
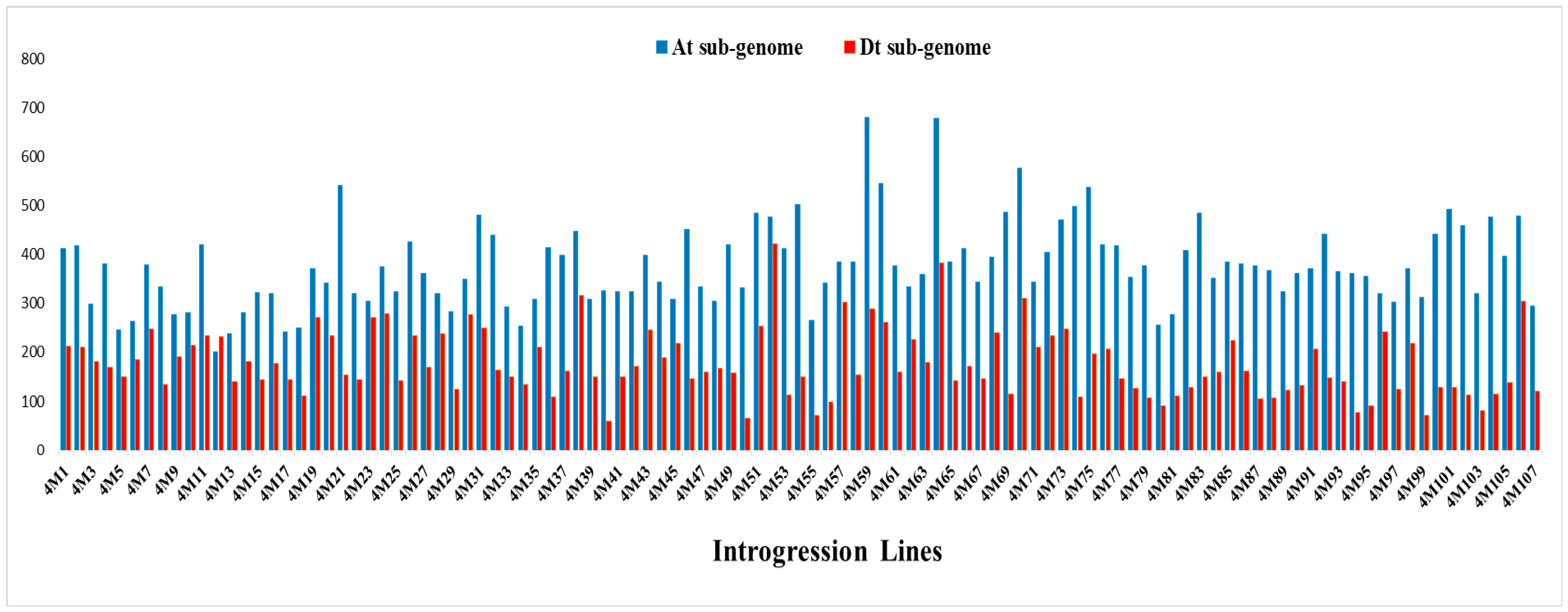
2.3. Genomic Component and Diversity of the ILs
2.4. QTL Mapping for Fiber Quality and Yield Traits
2.5. QTL Hotspot Analysis
3. Discussion
3.1. Advantage of the Permanent Population Derived from Wild Species
3.2. Characteristics of SLAF-Sequencing Strategy in Genotyping ILs
3.3. QTL Mapping Using SNP Markers in ILs
4. Materials and Methods
4.1. Plant Materials
4.2. Phenotypic Traits Collection and Analysis
4.3. DNA Extraction, SLAF-Library Construction, and High throughput Sequencing
4.4. SLAF-seq Data Grouping and Genotyping
4.5. QTL Analysis for Yield and Fiber Quality Traits
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| ILs | Introgression lines |
| SLAF | Specific locus amplified fragment |
| SNP | Single nucleotide polymorphism |
| QTL | Quantitative trait loci |
| At | A sub-genome |
| Dt | D sub-genome |
| PVE | Phenotypic variance explained |
| MIC | Micronaire value |
| FL | Fiber length |
| FU | Fiber uniformity |
| FS | Fiber strength |
| FE | Fiber elongation |
| BW | Boll weight |
| LP | Lint percentage |
| HSW | Hundred seed weight |
| PH | Plant height |
| BN | Boll number |
| FBN | Fruiting branches number |
| SI | Seed index |
| CV | Coefficient of variance |
| E1-3 | Environment 1-3 |
| LOD | Logarithm of odds |
References
- Fang, D.D.; Jenkins, J.N.; Deng, D.D.; McCarty, J.C.; Li, P.; Wu, J. Quantitative trait loci analysis of fiber quality traits using a random-mated recombinant inbred population in Upland cotton (Gossypium hirsutum L.). BMC Genom. 2014, 15, 397. [Google Scholar] [CrossRef] [PubMed]
- Hulse-Kemp, A.M.; Lemm, J.; Plieske, J.; Ashrafi, H.; Buyyarapu, R.; Fang, D.D.; Frelichowski, J.; Giband, M.; Hague, S.; Hinze, L.L.; et al. Development of a 63K SNP array for cotton and high-density mapping of intraspecific and interspecific populations of Gossypium spp. G3 (Bethesda) 2015, 5, 1187–1209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mei, H.; Zhu, X.; Zhang, T. Favorable QTL alleles for yield and its components identified by association mapping in Chinese Upland cotton cultivars. PLoS ONE 2013, 8, e82193. [Google Scholar] [CrossRef] [PubMed]
- Gore, M.A.; Fang, D.D.; Poland, J.A.; Zhang, J.; Percy, R.G.; Cantrell, R.G.; Thyssen, G.; Lipka, A.E. Linkage map construction and quantitative trait locus analysis of agronomic and fiber quality traits in cotton. Plant Genome 2014, 7, 1–10. [Google Scholar] [CrossRef]
- Said, J.I.; Lin, Z.; Zhang, X.; Song, M.; Zhang, J. A comprehensive meta QTL analysis for fiber quality, yield, yield related and morphological traits, drought tolerance, and disease resistance in tetraploid cotton. BMC Genom. 2013, 14, 776. [Google Scholar] [CrossRef] [PubMed]
- Qin, H.; Guo, W.; Zhang, Y.M.; Zhang, T. QTL mapping of yield and fiber traits based on a four-way cross population in Gossypium hirsutum L. Theor. Appl. Genet. 2008, 117, 883–894. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Zhang, K.; Li, S.; Yu, S.; Zhai, H.; Wu, M.; Li, X.; Fan, S.; Song, M.; Yang, D.; et al. Mapping quantitative trait loci for lint yield and fiber quality across environments in a Gossypium hirsutum × Gossypium barbadense backcross inbred line population. Theor. Appl. Genet. 2013, 126, 275–287. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Hu, Y.; Jiang, W.; Fang, L.; Guan, X.; Chen, J.; Zhang, J.; Saski, C.A.; Scheffler, B.E.; Stelly, D.M. Sequencing of allotetraploid cotton (Gossypium hirsutum L. acc. TM-1) provides a resource for fiber improvement. Nat. Biotechnol. 2015, 33, 531–537. [Google Scholar] [CrossRef] [PubMed]
- Cao, Z.; Zhu, X.; Chen, H.; Zhang, T. Fine mapping of clustered quantitative trait loci for fiber quality on chromosome 7 using a Gossypium barbadense introgressed line. Mol. Breed. 2015, 35, 215. [Google Scholar] [CrossRef]
- Shen, X.; Guo, W.; Zhu, X.; Yuan, Y.; Yu, J.Z.; Kohel, R.J.; Zhang, T. Molecular mapping of QTLs for fiber qualities in three diverse lines in Upland cotton using SSR markers. Mol. Breed. 2005, 15, 169–181. [Google Scholar] [CrossRef]
- Stewart, J. Potential for crop improvement with exotic germplasm and genetic engineering. In Proceedings of the World Cotton Research Conference-I; Comtable, G.A., Forrester, N.W., Eds.; CSIRO: Melbourne, Australia, 1995; pp. 313–327. [Google Scholar]
- Wang, B.; Nie, Y.; Lin, Z.; Zhang, X.; Liu, J.; Bai, J. Molecular diversity, genomic constitution, and QTL mapping of fiber quality by mapped SSRs in introgression lines derived from Gossypium hirsutum × G. darwinii Watt. Theor. Appl. Genet. 2012, 125, 1263–1274. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Draye, X.; Zhang, Z.; Zhuang, Z.; May, O.L.; Paterson, A.H.; Chee, P.W. Advanced backcross quantitative trait locus analysis of fiber elongation in a cross between Gossypium hirsutum and G. mustelinum. Crop Sci. 2016, 56, 1760–1768. [Google Scholar] [CrossRef]
- Grover, C.E.; Gallagher, J.P.; Jareczek, J.J.; Page, J.T.; Udall, J.A.; Gore, M.A.; Wendel, J.F. Re-evaluating the phylogeny of allopolyploid Gossypium L. Mol. Phylogenet. Evol. 2015, 92, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Rong, J.; Waghmare, V.N.; Chee, P.W.; May, O.L.; Wright, R.J.; Gannaway, J.R.; Paterson, A.H. QTL alleles for improved fiber quality from a wild Hawaiian cotton, Gossypium tomentosum. Theor. Appl. Genet. 2011, 123, 1075–1088. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.Y.; Oluoch, G.; Riaz Khan, M.K.; Wang, X.X.; Cai, X.Y.; Zhou, Z.L.; Wang, C.Y.; Wang, Y.H.; Li, X.Y.; Liu, F.; et al. Mapping QTLs for drought tolerance in an F2:3 population from an inter-specific cross between Gossypium tomentosum and Gossypium hirsutum. Genet. Mol. Res. 2016, 15. [Google Scholar] [CrossRef] [PubMed]
- Fonceka, D.; Tossim, H.-A.; Rivallan, R.; Vignes, H.; Lacut, E.; de Bellis, F.; Faye, I.; Ndoye, O.; Leal-Bertioli, S.C.M.; Valls, J.F.M.; et al. Construction of chromosome segment substitution lines in peanut (Arachis hypogaea L.) using a wild synthetic and qtl mapping for plant morphology. PLoS ONE 2012, 7, e48642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zamir, D. OPINION: Improving plant breeding with exotic genetic libraries. Nat. Rev. Genet. 2001, 2, 983–989. [Google Scholar] [CrossRef] [PubMed]
- Wan, X.Y.; Wan, J.M.; Su, C.C.; Wang, C.M.; Shen, W.B.; Li, J.M.; Wang, H.L.; Jiang, L.; Liu, S.J.; Chen, L.M.; et al. QTL detection for eating quality of cooked rice in a population of chromosome segment substitution lines. Theor. Appl. Genet. 2004, 110, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Yano, M. Genetic and molecular dissection of naturally occurring variation. Curr. Opin. Plant Biol. 2001, 4, 130–135. [Google Scholar] [CrossRef]
- Grandillo, S.; Tanksley, S.D.; Zamir, D. Exploitation of natural biodiversity through genomics. In Genomics-Assisted Crop Improvement; Varshney, R.K., Tuberosa, R., Eds.; Springer: Dordrecht, The Netherlands, 2007; pp. 121–150. [Google Scholar]
- Khan, M.K.; Chen, H.; Zhou, Z.; Ilyas, M.K.; Wang, X.; Cai, X.; Wang, C.; Liu, F.; Wang, K. Genome wide SSR high density genetic map construction from an interspecific cross of Gossypium hirsutum × Gossypium tomentosum. Front. Plant Sci. 2016, 7, 436. [Google Scholar] [CrossRef] [PubMed]
- Said, J.I.; Song, M.; Wang, H.; Lin, Z.; Zhang, X.; Fang, D.D.; Zhang, J. A comparative meta-analysis of QTL between intraspecific Gossypium hirsutum and interspecific G. hirsutum × G. barbadense populations. Mol. Genet. Genom. 2015, 290, 1003–1025. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Jin, X.; Wang, H.; Zhang, X.; Lin, Z. Structure, evolution, and comparative genomics of tetraploid cotton based on a high-density genetic linkage map. DNA Res. 2016, 23, 283–293. [Google Scholar] [CrossRef] [PubMed]
- Meredith, W. RFLP association with varietal origin and heterosis. In Proceedings of the Beltwide Cotton Production Conference, Nashville, TN, USA, 6–10 January 1992; pp. 6–10. [Google Scholar]
- Wang, B.; Draye, X.; Zhuang, Z.; Zhang, Z.; Liu, M.; Lubbers, E.L.; Jones, D.; May, O.L.; Paterson, A.H.; Chee, P.W. QTL analysis of cotton fiber length in advanced backcross populations derived from a cross between Gossypium hirsutum and G. mustelinum. Theor. Appl. Genet. 2017, 130, 1297–1308. [Google Scholar] [CrossRef] [PubMed]
- Jiang, N.; Shi, S.; Shi, H.; Khanzada, H.; Wassan, G.M.; Zhu, C.; Peng, X.; Yu, Q.; Chen, X.; He, X. Mapping QTL for seed germinability under low temperature using a new high-density genetic map of rice. Front. Plant Sci. 2017, 8, 1223. [Google Scholar] [CrossRef] [PubMed]
- Ariyadasa, R.; Mascher, M.; Nussbaumer, T.; Schulte, D.; Frenkel, Z.; Poursarebani, N.; Zhou, R.; Steuernagel, B.; Gundlach, H.; Taudien, S.; et al. A sequence-ready physical map of barley anchored genetically by two million single-nucleotide polymorphisms. Plant Physiol. 2014, 164, 412–423. [Google Scholar] [CrossRef] [PubMed]
- Ball, A.D.; Stapley, J.; Dawson, D.A.; Birkhead, T.R.; Burke, T.; Slate, J. A comparison of SNPs and microsatellites as linkage mapping markers: Lessons from the zebra finch (Taeniopygia guttata). BMC Genom. 2010, 11, 218. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Xie, W.; Wang, J.; Xing, Y.; Xu, C.; Li, X.; Xiao, J.; Zhang, Q. Gains in QTL detection using an ultra-high density SNP map based on population sequencing relative to traditional RFLP/SSR markers. PLoS ONE 2011, 6, e17595. [Google Scholar]
- Rout, K.; Sharma, M.; Gupta, V.; Mukhopadhyay, A.; Sodhi, Y.S.; Pental, D.; Pradhan, A.K. Deciphering allelic variations for seed glucosinolate traits in oilseed mustard (Brassica juncea) using two bi-parental mapping populations. Theor. Appl. Genet. 2015, 128, 657–666. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Liu, D.; Zhang, X.; Li, W.; Liu, H.; Hong, W.; Jiang, C.; Guan, N.; Ma, C.; Zeng, H.; et al. SLAF-seq: An efficient method of large-scale de novo SNP discovery and genotyping using high-throughput sequencing. PLoS ONE 2013, 8, e58700. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, L.; Xin, H.; Li, D.; Ma, C.; Ding, X.; Hong, W.; Zhang, X. Construction of a high-density genetic map for sesame based on large scale marker development by specific length amplified fragment (SLAF) sequencing. BMC Plant Biol. 2013, 13, 141. [Google Scholar] [CrossRef] [PubMed]
- Qi, Z.; Huang, L.; Zhu, R.; Xin, D.; Liu, C.; Han, X.; Jiang, H.; Hong, W.; Hu, G.; Zheng, H.; et al. A high-density genetic map for soybean based on specific length amplified fragment sequencing. PLoS ONE 2014, 9, e104871. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhang, Q.; Cheng, T.; Yang, W.; Pan, H.; Zhong, J.; Huang, L.; Liu, E. High-density genetic map construction and identification of a locus controlling weeping trait in an ornamental woody plant (Prunus mume Sieb. et Zucc). DNA Res. 2015, 22, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Shang, H.; Shi, Y.; Huang, L.; Li, J.; Ge, Q.; Gong, J.; Liu, A.; Chen, T.; Wang, D.; et al. Construction of a high-density genetic map by specific locus amplified fragment sequencing (SLAF-seq) and its application to Quantitative Trait Loci (QTL) analysis for boll weight in upland cotton (Gossypium hirsutum.). BMC Plant Biol. 2016, 16, 79. [Google Scholar] [CrossRef] [PubMed]
- Su, J.; Pang, C.; Wei, H.; Li, L.; Liang, B.; Wang, C.; Song, M.; Wang, H.; Zhao, S.; Jia, X. Identification of favorable SNP alleles and candidate genes for traits related to early maturity via GWAS in upland cotton. BMC Genom. 2016, 17, 687. [Google Scholar] [CrossRef] [PubMed]
- Shen, C.; Jin, X.; Zhu, D.; Lin, Z. Uncovering SNP and indel variations of tetraploid cottons by SLAF-seq. BMC Genom. 2017, 18, 247. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.L.; Sanchez, P.L.; Yu, S.-B.; Lorieux, M.; Eizenga, G.C. Chromosome segment substitution lines: A powerful tool for the introgression of valuable genes from oryza wild species into cultivated rice (O. sativa). Rice 2010, 3, 218–234. [Google Scholar] [CrossRef]
- Rong, J.; Feltus, F.A.; Waghmare, V.N.; Pierce, G.J.; Chee, P.W.; Draye, X.; Saranga, Y.; Wright, R.J.; Wilkins, T.A.; May, O.L.; et al. Meta-analysis of polyploid cotton QTL shows unequal contributions of subgenomes to a complex network of genes and gene clusters implicated in lint fiber development. Genetics 2007, 176, 2577–2588. [Google Scholar] [CrossRef] [PubMed]
- Lacape, J.-M.; Llewellyn, D.; Jacobs, J.; Arioli, T.; Becker, D.; Calhoun, S.; Al-Ghazi, Y.; Liu, S.; Palaï, O.; Georges, S. Meta-analysis of cotton fiber quality QTLs across diverse environments in a Gossypium hirsutum × G. barbadense RIL population. BMC Plant Biol. 2010, 10, 132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Si, Z.; Chen, H.; Zhu, X.; Cao, Z.; Zhang, T. Genetic dissection of lint yield and fiber quality traits of G. hirsutum in G. barbadense background. Mol. Breed. 2017, 37, 9. [Google Scholar] [CrossRef]
- Zhang, S.; Wang, T.; Liu, Q.; Gao, X.; Zhu, X.; Zhang, T.; Zhou, B. Quantitative trait locus analysis of boll-related traits in an intraspecific population of Gossypium hirsutum. Euphytica 2015, 203, 121–144. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- DePristo, M.A.; Banks, E.; Poplin, R.; Garimella, K.V.; Maguire, J.R.; Hartl, C.; Philippakis, A.A.; Del Angel, G.; Rivas, M.A.; Hanna, M. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011, 43, 491–498. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Li, H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 2011, 27, 2987–2993. [Google Scholar] [CrossRef] [PubMed]
- Krzywinski, M.; Schein, J.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An information aesthetic for comparative genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wan, X.; Li, H.; Pfeiffer, W.H.; Crouch, J.; Wan, J. Application of identified QTL-marker associations in rice quality improvement through a design-breeding approach. Theor. Appl. Genet. 2007, 115, 87–100. [Google Scholar] [CrossRef] [PubMed]
- McCouch, S. Report on QTL nomenclature. Rice Genet. Newsl. 1997, 14, 11–13. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Source of Variation | Replication (R) | ILs | Environment (E) | ILs × E | Error |
| D.F. = 1 | D.F. = 106 | D.F. = 2 | D.F. = 214 | D.F. = 321 | |
| MIC | 0.147 | 0.266 ** | 2.361 ** | 0.242 ** | 0.083 |
| FL | 0.054 | 3.948 ** | 451.793 ** | 3.003 ** | 1.141 |
| FU | 0.379 | 1.530 ** | 101.344 ** | 1.534 ** | 1.066 |
| FS | 14.395 | 12.410 ** | 650.072 ** | 10.213 ** | 5.096 |
| FE | 0.284 | 0.953 ** | 14.661 ** | 0.767 ** | 0.341 |
| BW | 0.812 | 0.410 ** | 47.524 ** | 0.321 ** | 0.174 |
| LW | 0.021 | 0.112 ** | 19.035 ** | 0.075 ** | 0.010 |
| LP | 54.740 | 15.260 ** | 1216.613 ** | 13.021 ** | 9.182 |
| Source of Variation | Replication (R) | ILs | Environment (E) | ILs × E | Error |
| D.F. = 1 | D.F. = 106 | D.F. = 1 | D.F. = 106 | D.F. = 213 | |
| HSW | 6.481 | 1.382 ** | 5243.981 ** | 1.679 ** | 0.921 |
| PH | 108.001 | 229.215 ** | 317,162.146 ** | 167.541 ** | 109.551 |
| BN | 444.164 | 123.641 ** | 17,213.992 ** | 121.418 ** | 92.814 |
| Source of Variation | Replication (R) | ILs | Error | ||
| D.F. = 1 | D.F. = 106 | D.F. = 105 | |||
| FBN | 86.580 | 76.332 ** | 29.093 | ||
| SI | 1.616 | 1.169 ** | 0.482 | ||
| Trait | Env. | 4105 Mean | ILs Mean | SD | CV (%) | Skewness | Kurtosis |
|---|---|---|---|---|---|---|---|
| MIC | E1 | 5.07 | 4.99 | 0.38 | 8.35 | −0.20 | −0.37 |
| E2 | - | 5.08 | 0.43 | 8.60 | −0.16 | −0.30 | |
| E3 | 4.81 | 4.85 | 0.33 | 7.80 | 0.04 | 0.16 | |
| FL | E1 | 26.99 | 26.29 | 1.26 | 5.34 | 0.25 | −0.48 |
| E2 | - | 29.09 | 1.52 | 5.53 | 0.39 | 0.30 | |
| E3 | 28.23 | 28.23 | 1.29 | 5.21 | 0.26 | −0.31 | |
| FU | E1 | 84.21 | 82.96 | 0.86 | 1.27 | −0.01 | −0.04 |
| E2 | - | 84.31 | 0.91 | 1.28 | −0.15 | 0.11 | |
| E3 | 83.69 | 83.31 | 0.95 | 1.52 | −0.60 | 0.65 | |
| FS | E1 | 30.46 | 28.42 | 2.76 | 11.11 | 0.50 | 0.53 |
| E2 | - | 28.77 | 2.16 | 8.51 | 0.35 | 0.01 | |
| E3 | 30.86 | 31.76 | 2.39 | 9.14 | −0.06 | 0.44 | |
| FE | E1 | 3.18 | 3.63 | 0.72 | 21.72 | 0.17 | 0.05 |
| E2 | - | 3.66 | 0.78 | 23.29 | 0.35 | 0.45 | |
| E3 | 2.82 | 3.20 | 0.56 | 21.57 | 0.29 | 0.48 | |
| BW | E1 | 4.47 | 4.81 | 0.49 | 10.39 | 0.12 | −0.04 |
| E2 | - | 5.31 | 0.58 | 10.93 | −0.83 | 2.70 | |
| E3 | 4.05 | 4.36 | 0.45 | 10.48 | 0.22 | 1.77 | |
| LW | E1 | 1.85 | 2.01 | 0.25 | 12.43 | −0.29 | 0.20 |
| E2 | - | 2.23 | 0.26 | 11.95 | −0.63 | 1.17 | |
| E3 | 1.5 | 1.64 | 0.24 | 14.66 | 1.14 | 0.68 | |
| LP | E1 | 42.82 | 41.80 | 2.76 | 6.60 | −1.01 | 1.97 |
| E2 | - | 42.02 | 2.70 | 6.42 | 0.82 | 2.93 | |
| E3 | 37.08 | 37.82 | 4.48 | 11.86 | -1.39 | −1.56 | |
| HSW | E2 | 10.85 | 9.97 | 0.81 | 8.18 | 0.055 | 0.39 |
| E3 | 17.2 | 16.93 | 1.35 | 8.00 | 0.07 | −0.53 | |
| PH | E2 | 142.20 | 139.68 | 13.99 | 10.021 | 0.06 | −0.48 |
| E3 | 80 | 84.82 | 10.24 | 12.08 | 0.43 | 0.73 | |
| BN | E2 | 39.25 | 35.11 | 8.93 | 25.43 | 0.98 | 1.61 |
| E3 | 59.5 | 47.36 | 11.79 | 24.91 | 0.42 | 0.33 | |
| FBN | E2 | 19.71 | 17.44 | 2.60 | 14.92 | −1.28 | 0.21 |
| SI | E3 | 10.6 | 10.44 | 0.90 | 8.70 | 0.11 | −0.05 |
| Chromosome | Size of Physical Distance (Mb) | Coverage of Introgressed Segments in Genome (Mb) | Percentage of Genome Coverage (%) |
|---|---|---|---|
| A1 | 99.88 | 38 | 38.04 |
| A2 | 83.45 | 34 | 40.74 |
| A3 | 100.26 | 22 | 21.94 |
| A4 | 62.91 | 13 | 20.66 |
| A5 | 82.05 | 29 | 35.35 |
| A6 | 103.17 | 24 | 23.26 |
| A7 | 78.25 | 24 | 30.67 |
| A8 | 103.63 | 39 | 37.64 |
| A9 | 75.00 | 30 | 40.00 |
| A10 | 100.87 | 29 | 28.75 |
| A11 | 93.32 | 19 | 20.36 |
| A12 | 87.48 | 40 | 45.72 |
| A13 | 79.96 | 62 | 77.54 |
| Average | 88.48 | 31.00 | 35.44 |
| D1 | 61.46 | 15 | 24.41 |
| D2 | 67.28 | 34 | 50.53 |
| D3 | 46.69 | 9 | 19.28 |
| D4 | 51.45 | 22 | 42.76 |
| D5 | 61.93 | 21 | 33.91 |
| D6 | 64.29 | 38 | 59.10 |
| D7 | 55.31 | 32 | 57.85 |
| D8 | 65.89 | 14 | 21.25 |
| D9 | 51.00 | 12 | 23.53 |
| D10 | 63.37 | 19 | 29.98 |
| D11 | 66.09 | 13 | 19.67 |
| D12 | 59.11 | 12 | 20.30 |
| D13 | 60.53 | 21 | 34.69 |
| Average | 59.57 | 20.15 | 33.64 |
| Cluster Name | Approximate Position on Chromosome (Mb) | Number of QTLs | Name of QTLs |
|---|---|---|---|
| A01-cluster | 77.52–86.96 | 5 | qFS-A01-1 |
| qFS-A01-2 | |||
| qLP-A01-1 | |||
| qLP-A01-2 | |||
| qBN-A01-1 | |||
| A09-cluster | 45.81–46.41 | 3 | qFS-A09-1 |
| qFS-A09-2 | |||
| qBW-A09-1 | |||
| A13-cluster | 48.09–58.85 | 5 | qLP-A13-1 |
| qHSW-A13-1 | |||
| qHSW-A13-2 | |||
| qPH-A13-1 | |||
| qSI-A13-1 | |||
| D02-cluster | 18.22–22.44 | 4 | qFS-D02-1 |
| qFE-D02-1 | |||
| qLP-D02-1 | |||
| qFBN-D02-1 | |||
| D10-cluster | 56.48–61.85 | 3 | qFL-D10-2 |
| qBW-D10-1 | |||
| qLW-D10-1 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Keerio, A.A.; Shen, C.; Nie, Y.; Ahmed, M.M.; Zhang, X.; Lin, Z. QTL Mapping for Fiber Quality and Yield Traits Based on Introgression Lines Derived from Gossypium hirsutum × G. tomentosum. Int. J. Mol. Sci. 2018, 19, 243. https://doi.org/10.3390/ijms19010243
Keerio AA, Shen C, Nie Y, Ahmed MM, Zhang X, Lin Z. QTL Mapping for Fiber Quality and Yield Traits Based on Introgression Lines Derived from Gossypium hirsutum × G. tomentosum. International Journal of Molecular Sciences. 2018; 19(1):243. https://doi.org/10.3390/ijms19010243
Chicago/Turabian StyleKeerio, Ayaz Ali, Chao Shen, Yichun Nie, Muhammad Mahmood Ahmed, Xianlong Zhang, and Zhongxu Lin. 2018. "QTL Mapping for Fiber Quality and Yield Traits Based on Introgression Lines Derived from Gossypium hirsutum × G. tomentosum" International Journal of Molecular Sciences 19, no. 1: 243. https://doi.org/10.3390/ijms19010243