1. Introduction
Satellite cells as a primary source of muscle progenitors were initially identified in the frog skeletal muscle by Mauro in 1961 [
1]. Satellite cells locating a niche between the basal lamina and plasma membrane of mature myofibers play an indispensable role in supporting muscle growth, maintenance and regeneration of postnatal skeletal muscle [
2,
3,
4,
5]. Numerous studies have shown that satellite cells are usually quiescent in adult muscles [
6,
7,
8]. In response to muscle injury, satellite cells become activated and re-enter the cell cycle to proliferate and differentiate. Notch signaling played an important role in the regeneration of injured muscle [
9].
The Notch signaling pathway is one of the major regulatory pathways in myogenesis [
9,
10,
11,
12]. There are four Notch receptors (Notch1–Notch4), which are activated after binding to its ligands, such as Jagged 1, Jagged 2, Delta-like 1, Delta-like 3 and Delta-like 4 in mammals [
13]. Once bound, Notch undergoes protease cleavage to free Notch intracellular domain (NICD) and translocates into nucleus, where it interacts directly with the transcription factor RBPJ (also known as CBF1, recombination signal binding protein for immunoglobulin kappa J region) to activate its target genes, such as hes related family bHLH transcription factor with YRPW motif (HEY) and HES family [
14]. Notch1–3, HES1, HES2, HES5, HEY1 and hes related family bHLH transcription factor with YRPW motif-like (HEYL) were highly expressed in the quiescent satellite cells, which implies that Notch signaling is essential for maintenance of satellite cell quiescence. Notch is downregulated when satellite cells are activated [
15,
16,
17]. Furthermore, the expression of Notch signaling is decreased in the quiescent mice satellite cells with RBPJ defect, which facilitated the spontaneous activation of satellite cells and allowed them to undergo terminal differentiation, thereby resulting in a severe depletion of satellite cells’ pool [
15,
16]. Knocking down Notch1 reduces PSCs (porcine satellite cells) proliferation, and overexpressing Notch1 with recombinant human NF-κB (nuclear factor kappa B) protein promotes PSCs proliferation though influencing cell cycle progression [
18]. Furthermore, activation of Notch signaling promotes proliferation in mice satellite cells [
10,
19,
20]. There exist two waves of Notch signaling during the activation of satellite cells. The first wave of Notch signaling maintains the quiescent state of the satellite cells. The second wave of Notch signaling rises after satellite cells are activated [
4]. This explanation is supported by Notch expression being decreased at first and then increasing during muscle injury [
9,
16]. Furthermore, Notch signaling is also related to satellite cell differentiation. Due to the crosstalk between Notch and Wnt cascade via GSK3β, the inhibition of Notch signaling would be suitable for a rapid temporal switch to suppress Notch signaling and activate Wnt signaling, which facilitated satellite cell differentiation and fusion of myoblasts to myotubes [
20]. Still, how the Notch signaling pathway regulates skeletal muscle myogenesis is not clear.
Small noncoding RNAs about 22 nucleotides long in the animal are classified as microRNAs (miRNAs), which regulate the expression of posttranscriptional protein-coding genes through interacting with the 3′-untranslated region (3′-UTR) of their target mRNAs [
21]. Currently, 382 hairpin precursor miRNAs and 411 mature miRNAs have been identified in the miRBase database (Version 21), which is still far less than by comparing with human 2588 miRNA (
http://mirbase.org/index.shtml). Numerous studies demonstrated that miRNA play an indispensable role in regulating muscle development [
22]. In skeletal muscle, highly expressed miRNAs are called myomiRs, which include miR-1, miR-133a, miR-133b, miR-206, miR-208a, miR-208b, miR-486 and miR-499 [
23]. Besides the myomiRs, there are also many miRNAs regulating satellite cell myogenesis, for example miR-31 [
24]; miR-27a [
25,
26], miR128 [
27], miR-221/miR-222 [
28], miR-682 [
29] and miR-181 [
30]; miR-186 [
31], miR-24 [
32], miR-214 [
33,
34], miR-26a [
35], miR-29b/c [
36], miR-322/424 [
37] and miR-503 [
37] have been identified to regulate the quiescence, proliferation and differentiation stage of satellite cells respectively. Both Notch signaling and miRNA have been identified to play critical roles in regulating myogenesis, and four miRNAs, miR-1, miR-206, miR-146a and 146b, have been reported to regulate skeletal muscle myogenesis via Notch signaling [
38,
39,
40,
41]. However, none of these four miRNAs were studied in porcine skeletal muscle.
In this study, we hypothesized that the Notch signaling pathway might contribute to PSCs’ proliferation and differentiation by regulating specific genes and miRNAs. We used next generation sequencing technology (NGST) to characterize mRNA-seq and miRNA-seq of control and stably transfected with N1ICD in PSCs. We have identified many differentially-expressed genes (DEGs) and differentially-expressed microRNAs (DEMs) by using bioinformatics analysis. We have also established relations between these DEGs and DEMS to identify the miRNA-mRNA pairs that may be connected to the Notch signaling in PSCs’ development. These findings could provide a reference for further research of molecular mechanisms underlying Notch signaling in PSCs.
3. Discussion
Our study, we believe, is the first comprehensive report about the miRNA and mRNA signatures in PSCs with overexpressed NI1CD, and the overall findings of this study will facilitate our understanding of the molecular events underlying the roles of microRNAs and genes in the Notch signaling pathway in regulating PSCs’ development.
Notch signaling has been identified to be critical in modulating satellite cell function, and it can be used as a therapeutic application to overcome skeletal muscle atrophy [
39,
44]. However, all previous research used only methods of transient transfection of NICD, which usually has the problems of low transfection efficiency and short duration. In our study, we constructed a stably-transfected N1ICD model to avoid the problems mentioned above. In
Figure 1A, Notch1 was mainly located in the nucleus of PSCs. In
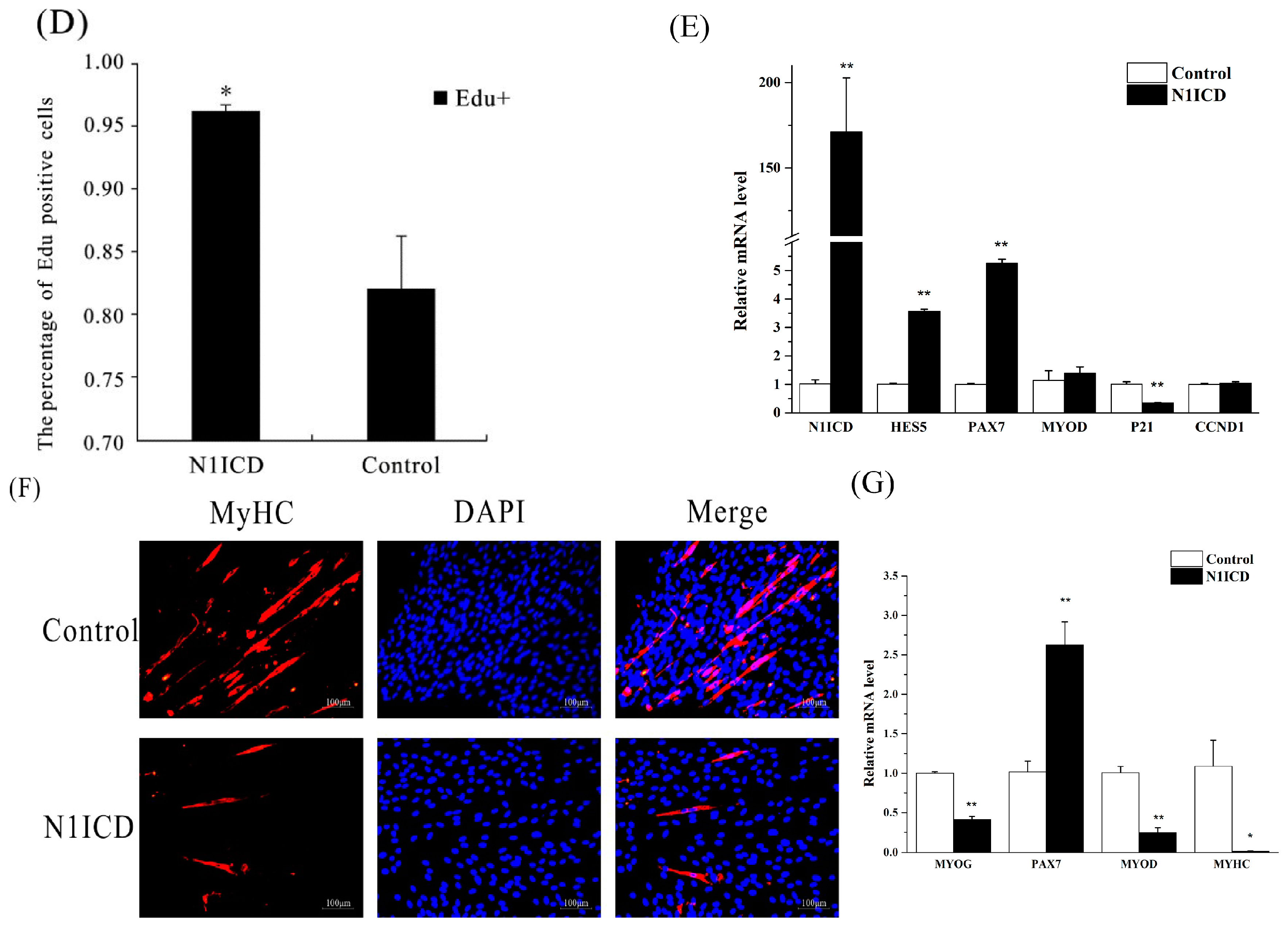
Figure 1B, the green fluorescence in the pEGFP-N1ICD group was higher than the control group, which suggests that N1ICD overexpression was successful in PSCs. However, the degree of green fluorescence of the pEGFP-N1ICD group was lower. A possible explanation is the transfecting N1ICD fragment changed the skeleton of the vector, which affects the ability of the protein expression of the vector.
In 2016, Shan et al. reviewed that activation of Notch1 signaling promotes the muscle satellite cells’ quiescence and proliferation [
45]. To date, several studies have confirmed several specific genes and miRNAs regulating the proliferation of satellite cells through the Notch signaling pathway [
39,
40,
46]. However, the regulated network of the Notch signaling pathway has still not been explored in proliferating satellite cells. In this study, our Edu experiment indicated that overexpression of N1ICD promotes PSCs’ proliferation. Meanwhile, the Q-PCR result showed that PAX7 dramatically increases while P21, also known as cyclin-dependent kinase inhibitor 1, significantly decreases PSCs proliferation with overexpressed N1ICD. To further uncover the regulating network in PSCs proliferation, we used NGST to assess the expression change in both miRNAs and mRNAs. A total of 349 miRNAs and 10,735 genes was identified in both proliferation and differentiation of PSCs. Furthermore, 44 miRNAs and 295 genes were significantly different in satellite cell proliferation by using integrative methods. After analyzing the sequencing results using NGST, we found the porcine N1ICD was approximately 1.6-fold higher after N1ICD overexpression. The Notch downstream regulated genes HEYL (about 41.6-fold), HEY1 (about 6.36-fold) and HES4 (about 2.73-fold) were also upregulated during PSCs proliferation. HEYL is the most prominent gene among all DEGs that were significantly changed. Interestingly, JAG1 (about 3.48-fold) was also increased. Moreover, HEYL, HEY1 and JAG1 are known for regulating skeletal muscle development [
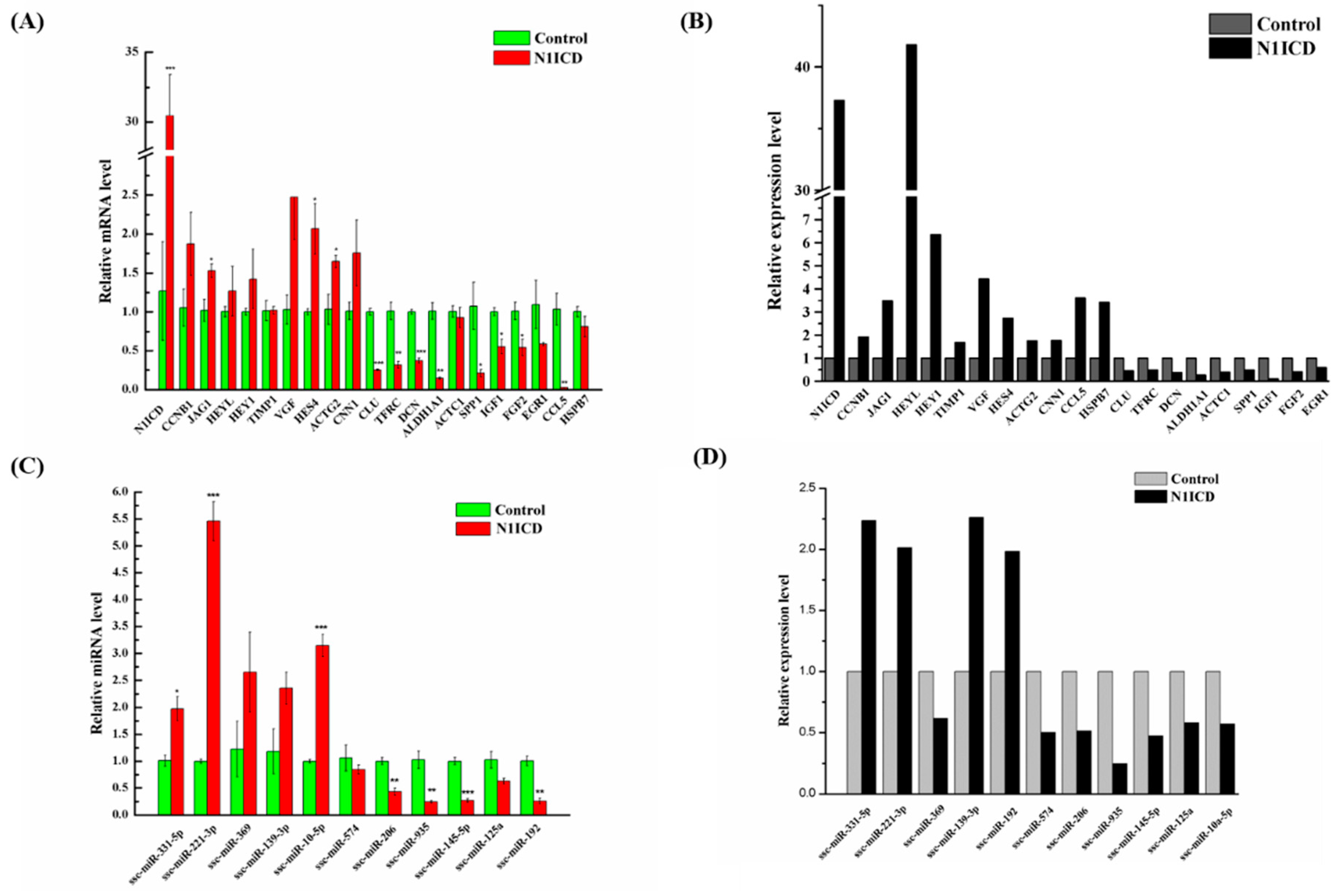
47]. However, this was not so for HES4, which implies that HES4 may be another gene involved in regulating skeletal muscle development. Two myomiRs, miR-133a-3p (about 1.78-fold) and miR-206 (about 1.94-fold), which are known to increase skeletal muscle differentiation, were downregulated [
48,
49,
50,
51]. Indeed, overexpressed N1ICD promotes the Notch signaling pathway and downregulates myomiRs relating to differentiation, which implies promoting PSCs’ proliferation.
To investigate in depth how PSCs proliferation changes in the gene alteration of overexpressed N1ICD, we took advantage of bioinformatics analysis. The GO and KEGG analyses provided substantial evidence that upregulated DEGs are mainly associated with cell cycle regulation, because “cell cycle” was the most enriched in both GO and KEGG analyses. The STRING database network also declares upregulated DEGs mostly related to cell cycle. Numerous genes, including CCNA2, CCNB1, CCNB3, CDC20, CDC25B, E2F transcription factor 1 (E2F1), ESPL1, protein kinase, membrane associated tyrosine/threonine 1 (PKMYT1), PLK1, transforming growth factor beta 1 (TGFB1) and transforming growth factor beta 3 (TGFB3), are enriched in the “cell cycle” pathway. CCNA2, CCNB1, and CCNB3 belong to the cyclin family, whose members regulate cell cycle progression by interacting with CDK kinases [
52]. Notch signaling has been reported to be involved in regulating the cell cycle process. Abnormal high Notch signaling in T-cell acute lymphoblastic leukemia (T-ALL) keeps S-phase kinase associated protein 2 (SKP2) at a high level and lowers p27Kip1, leading to more rapid cell cycle progression [
53]. In adult muscle, Notch inhibition by gamma-secretase inhibitor (GSI) caused prompt upregulation of cyclin-dependent kinase (CDK) inhibitors (p15, p16, p21 and p27), whereas activation of Notch inhibits them [
54]. Our laboratory has demonstrated that knocking down Notch1 reduced the expression of cell cycle-related genes (cyclin B1, D1, D2 and E1), which decreased the proliferation of PSCs [
18]. Similarly, CDC20 and CDC25B as cell-division cycle proteins are also essential to regulate the cell cycle process. The most important function of CDC20 is to activate the anaphase-promoting complex (APC/C). APC/C is a large complex containing 11–13 subunits, which initiates chromatid separation and entrance into anaphase [
55,
56]. CDC25B encodes the enzyme M-phase inducer phosphatase 2 in humans [
57]. All above analyses indicate that the activation of the Notch signaling pathway in PSCs mainly promotes cell proliferation through regulating the cell cycle.
We used the KEGG database to further unveil how the Notch signaling pathway regulates cell cycle. Since the MAPK signaling pathway has been identified to regulate the cell cycle directly in the KEGG database, we suspect that the Notch signaling pathway may regulate the MAPK signaling pathway. Besides, a great many research works demonstrated that there is a close relationship between the Notch signaling pathway and the MAPK signaling pathway based on the result of KEGG analysis. Angiotensin II activates MAPK signaling in ductus arteriosus smooth muscle cells, which was counteracted by gamma-secretase inhibitor DAPT, a Notch signaling inhibitor [
58]. Furthermore, Notch3 protects vascular smooth muscle cell (VSMC) apoptosis by upregulating three pro-survival genes, which is mediated through MAPK signaling [
59]. However, no one has studied if the Notch signaling pathway regulates skeletal satellite cells through mediating the MAPK signaling pathway. Based on the above data, in
Figure 3E, we delineated that the Notch signaling pathway indirectly regulates the cell cycle in PSCs though mediating the MAPK signaling pathway.
To study the regulation of DEGs and DEMs, the TF-mRNA and TF-miRNA networks in P-N1ICD versus P-Control were constructed. Interestingly, SNAI2 and EGR1 as the transcription factors are hub TFs in both the TF-mRNA and TF-miRNA network.
SNAI2 belongs to the Snail gene family, which encodes DNA-binding zinc finger proteins that function as transcription repressors in mammals [
60]. In mice muscle, SNAI1/2 recruit HDAC1/2 (as co-repressor inhibiting CSL in the Notch signaling pathway, which was annotated in the KEGG database) to block myoblast entry into differentiation [
61]. In our data, SNAI2 is also a DEG in P-N1ICD versus P-Control, so the downregulation of SNAI2 may promote PSCs’ proliferation and entry into differentiation. EGR1 is also a zinc-finger transcription factor, which plays a pivotal role in muscle proliferation [
62,
63]. In addition, EGR1 binds to the osteopontin (OPN) promoter and upregulates OPN expression, which promotes vascular smooth muscle cells’ (VSMCs) proliferation [
64]. In our data, the EGR1 upregulation may contribute to PSCs’ proliferation.
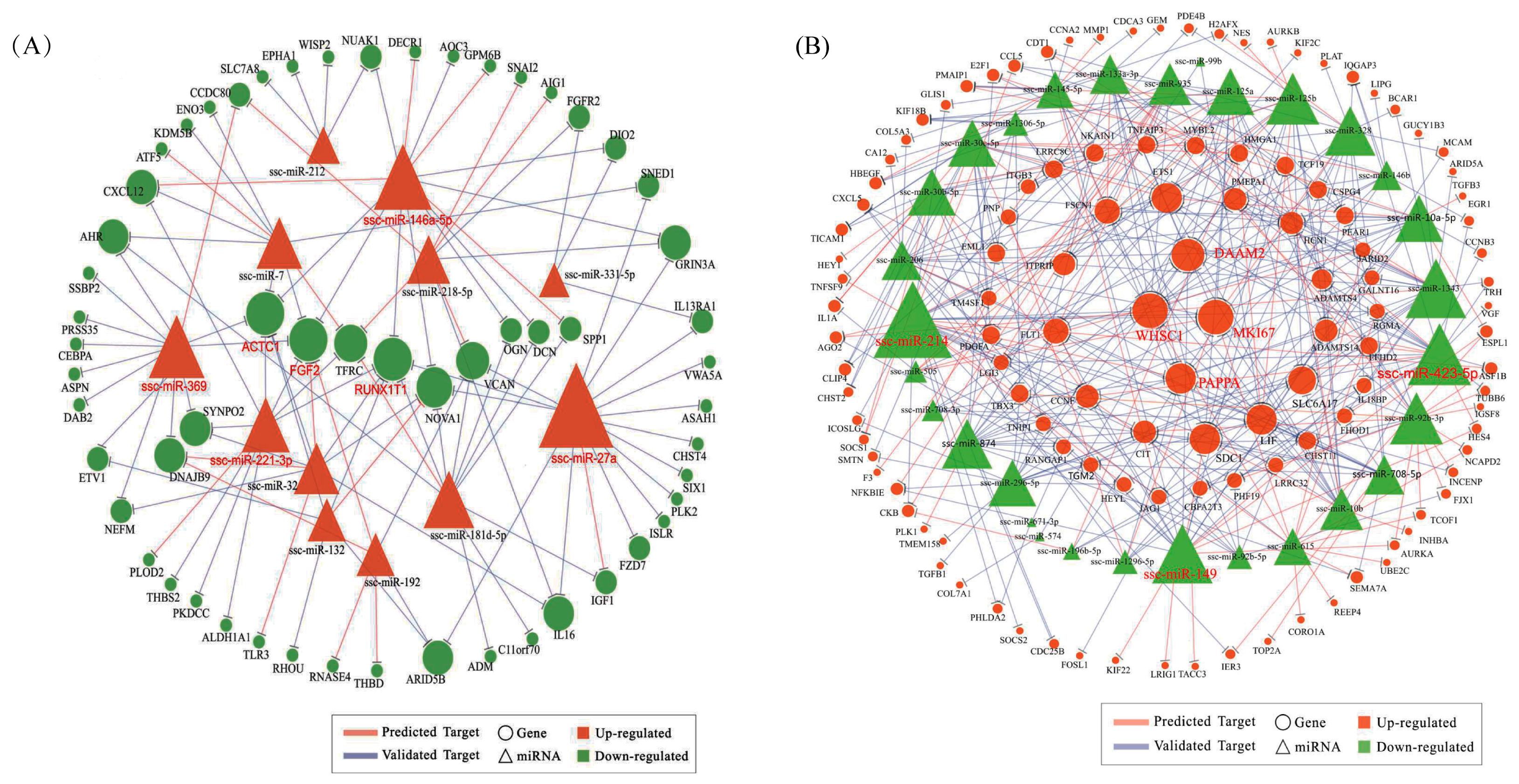
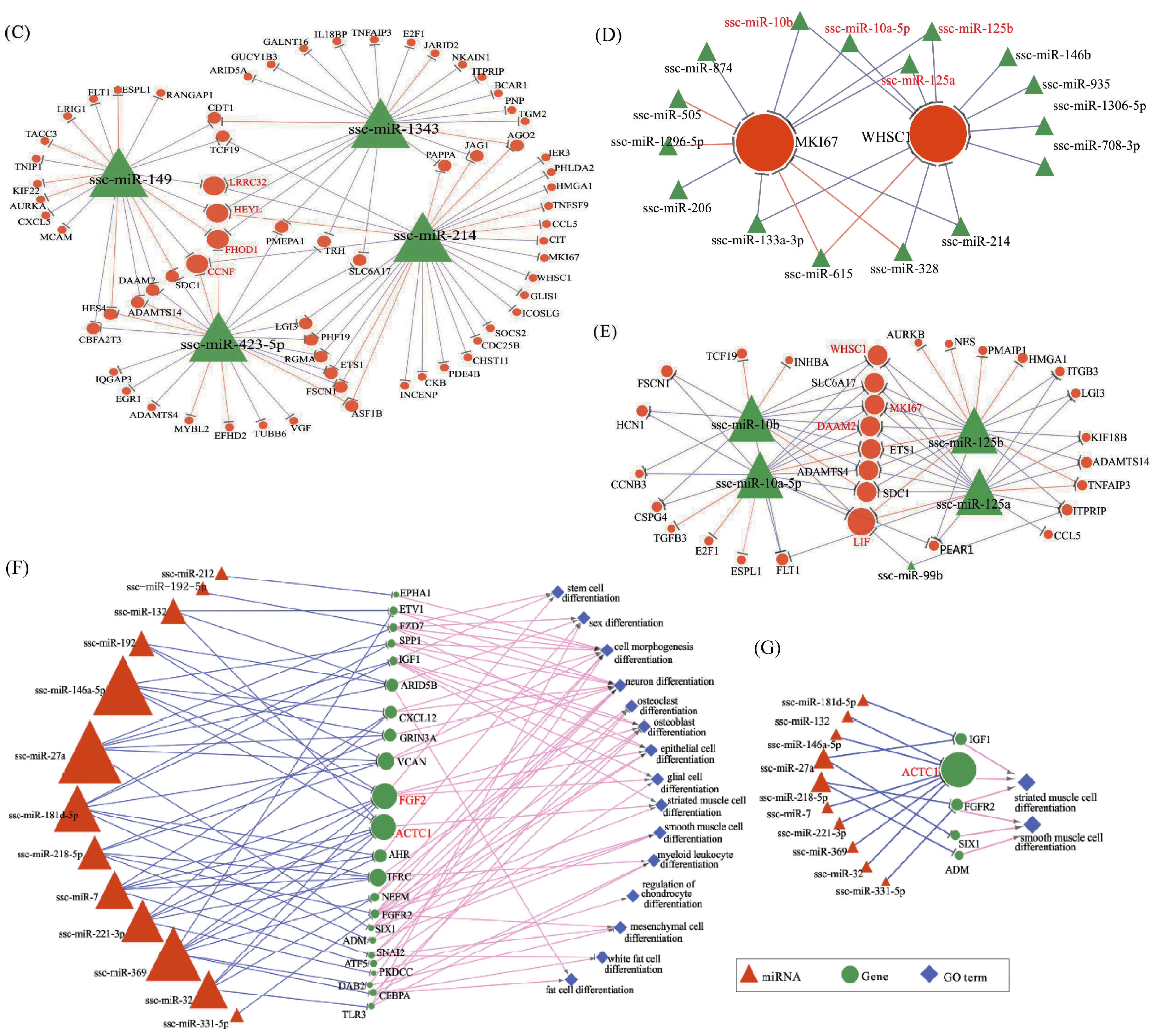
To investigate how miRNAs exactly participate in the gene alteration of overexpressed N1ICD, we constructed miRNA-mRNA networks. Through the integration of miRNA and mRNA expression data and miRNA-mRNA target prediction analysis, many putative miRNA-mRNA interactions were identified. Highly connected nodes in biological networks are known as hubs. Hubs are topologically important to the structure of the network; they are also known to be preferentially associated with various phenotypes of interest [
65]. The relative importance of a hub node, however, can change depending on the biological context. Since hub nodes are known to play important roles in many networks, we also looked for the presence of hub miRNAs. Several downregulated hub miRNAs were identified, including ssc-miR-214, ssc-miR-423-5p, ssc-miR-149 and ssc-miR-1343. Additionally, each of these hub miRNAs targets more than 21 genes. Interestingly, all these hub miRNAs target the HEYL gene. miR-214 mediated Ezh2 protein reduction, which accelerates skeletal muscle cell differentiation [
34]. Ectopic expression of miR-149 in gastric cancer cells inhibits proliferation and cell cycle progression by downregulating zinc finger and BTB domain containing 2 (ZBTB2) [
66]. Though ssc-miR-125a, ssc-miR-125b, ssc-miR-10a-5p, ssc-miR-10b, ssc-miR-935 and ssc-miR-145 are not the hub miRNAs, each of them also targets more than 14 genes and associated with inhibiting proliferation. In our research, miR-125a, miR-125b, miR-10a-5p and miR-10b belong to the miR-10 family, which are most abundant among all significant differential miRNAs and target two hub genes (WHSC1 and MKI67). miR-10 members play key roles in the differentiation process of human mesenchymal stem cells [
67], neuroblastoma [
68] and smooth muscle cells [
69]. Inhibition of miR-10a in smooth muscle cells abrogates their retinoic acid-induced differentiation, likely upregulating HDAC4, an identified target gene of miR-10a [
69]. However, miR-10a alone was unable to drive the differentiation of smooth muscle cells [
69]. miR-125a serves as a tumor suppressor has been shown to inhibit proliferation in different cancers [
70,
71,
72,
73]. Overexpressing miR-125a-5p inhibits C2C12 myoblast proliferation by targeting E2F3 [
74]. miR-125b has also been found to suppress proliferation in different kinds of cancer [
75,
76,
77]. miR-935 suppresses tumorigenesis of gastric signet ring cell carcinoma by targeting Notch1 [
78]. miRNA-145 inhibits VSMC proliferation by targeting CD40 [
79]. VSMC differentiation signals potentially can be fine-tuned or amplified by miR-143/145 through Notch and SRF in parallel pathways [
80]. Similarly, two upregulated hub genes were identified including MKI67 and WHSC1. MKI67 protein (also known as Ki-67) is a cellular marker for proliferation, which appears during the cell cycle (G1, S, G2 and mitosis), but is absent from the resting cells phases (G0) [
81]. In a previous study, WHSC1, a cell-cycle checking point protein, was shown to play important roles in human carcinogenesis [
82]. By contrast, upregulated hub miRNAs include miR-27a, miR-146a-5p, miR-369, miR-181d-5p and miR-221-3p; and each of these hub miRNAs targets more than nine genes. miR-27a depressed the expression of myostatin and promoted porcine myoblast proliferation [
25,
26]. miR-146a promotes C2C12 proliferation by inhibiting the Numb gene, which promotes satellite cell differentiation by inhibiting Notch signaling [
40]. Ectopic expression of miR-221, as well as miR-222 in differentiating myoblasts delayed their withdrawal from the cell cycle and subsequent myogenesis through P27, their target gene [
28]. Meanwhile, two downregulated genes were identified as hub genes, including RUNX1T1 and FGF2. In murine embryonic stem cells, Runx1t1 expression is upregulated during hematopoietic differentiation [
83]. Furthermore, a low level of Runx1t1 expression was detected in undifferentiated human ESCs (embryonic stem cells), and a gradual increase in expression was observed during differentiation [
84]. All these data seem to indicate that RUNX1T1 plays a role during stem cell differentiation, at least in the hematopoietic, neuronal and muscular cells. In 2016, Shelton et al. found that collagenase IV and continued FGF2 supplementation expanded differentiating skeletal muscle progenitors about three-fold every two weeks. All above-mentioned hub genes and miRNAs may regulate the proliferation of N1ICD overexpressed PSCs.
Presently, much research has demonstrated that the Notch signaling pathway also regulates the differentiation of skeletal satellite cells. In C2C12 cells, activation of the Notch signaling pathway significantly inhibits myogenic differentiation and differentiation-associated genes [
85,
86,
87]. Furthermore, in mice primary myoblasts, myogenic cell lineage-specific overexpressed N1ICD decreases the expression of MYOD, and MYOG and inhibits myogenic differentiation [
88]. Besides, inhibition of Notch signaling would be suitable for a rapid temporal switch to activate Wnt signaling, which facilitated satellite cells’ differentiation [
20]. By contrast, overexpressed N1ICD dramatically reduce the expression of MYHC, MYOD and MYOG, which implies the inhibition of PSCs’ differentiation. However, in our sequencing data, only a few genes appeared significantly differential in the differentiation of satellite cells, including EGR1, ACTG2 and CNN1. Knock-down of EGR1 inhibited differentiation of bovine skeletal muscle-derived satellite cells. Therefore, overexpressed N1ICD in PSCs differentiation downregulated the expression of EGR1, which may inhibit PSCs’ differentiation [
89]. However, ACTG2 and CNN1 are not known to be associated with satellite cell differentiation.
Although our study has identified the differentially-expressed genes, miRNAs and the interaction network in NICD1 overexpressed PSCs, the molecular mechanisms of how these genes, miRNAs and related TFs regulate PSCs development need to be further studied.

 ,
, 










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}