Renal Injury during Long-Term Crizotinib Therapy


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
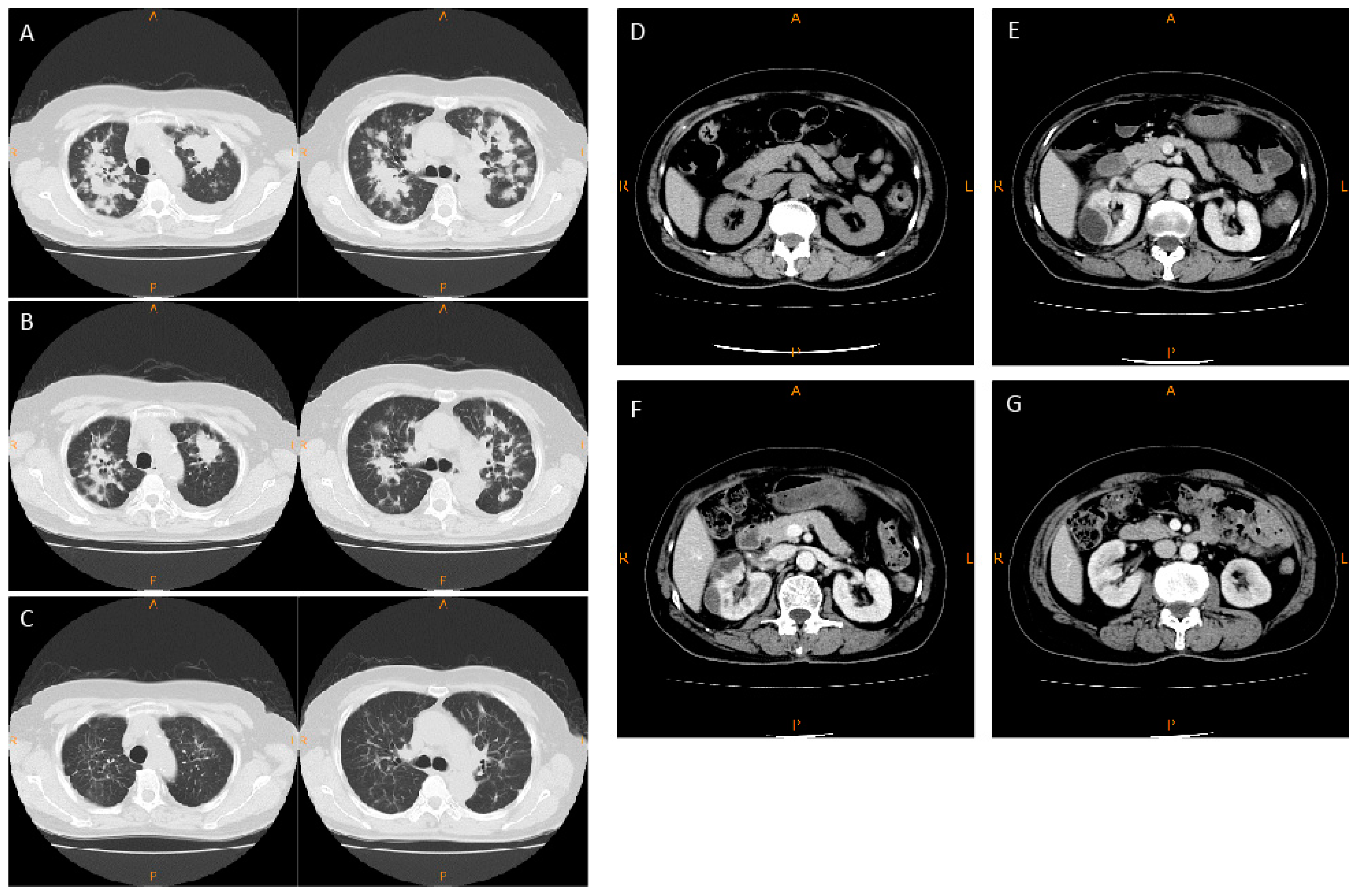
2.1. Case Report
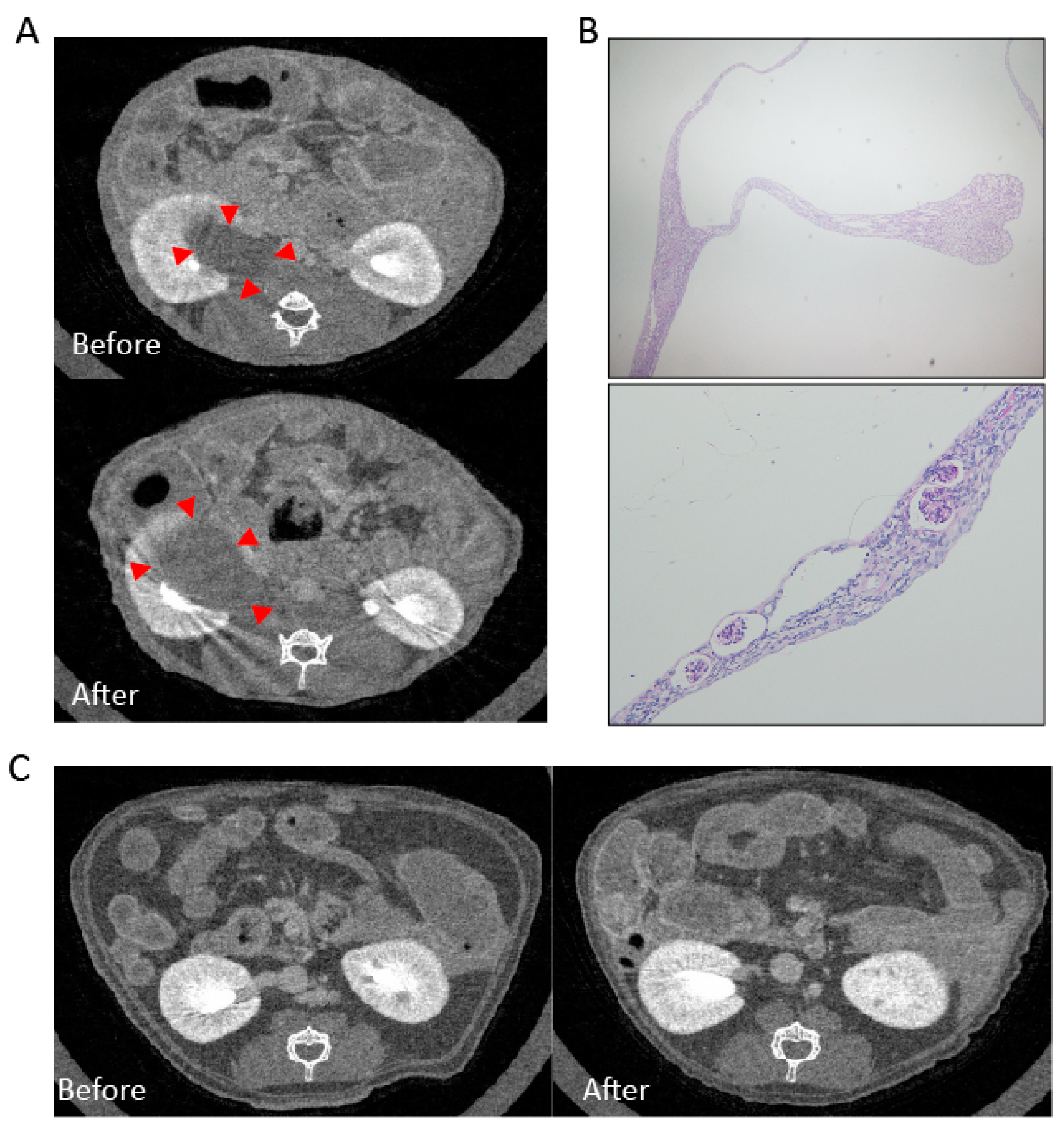
2.2. Experimental Animal Model
2.2.1. Mesangial Expansion in Crizotinib-Treated Mouse
2.2.2. Crizotinib Caused Renal Histopathological Changes
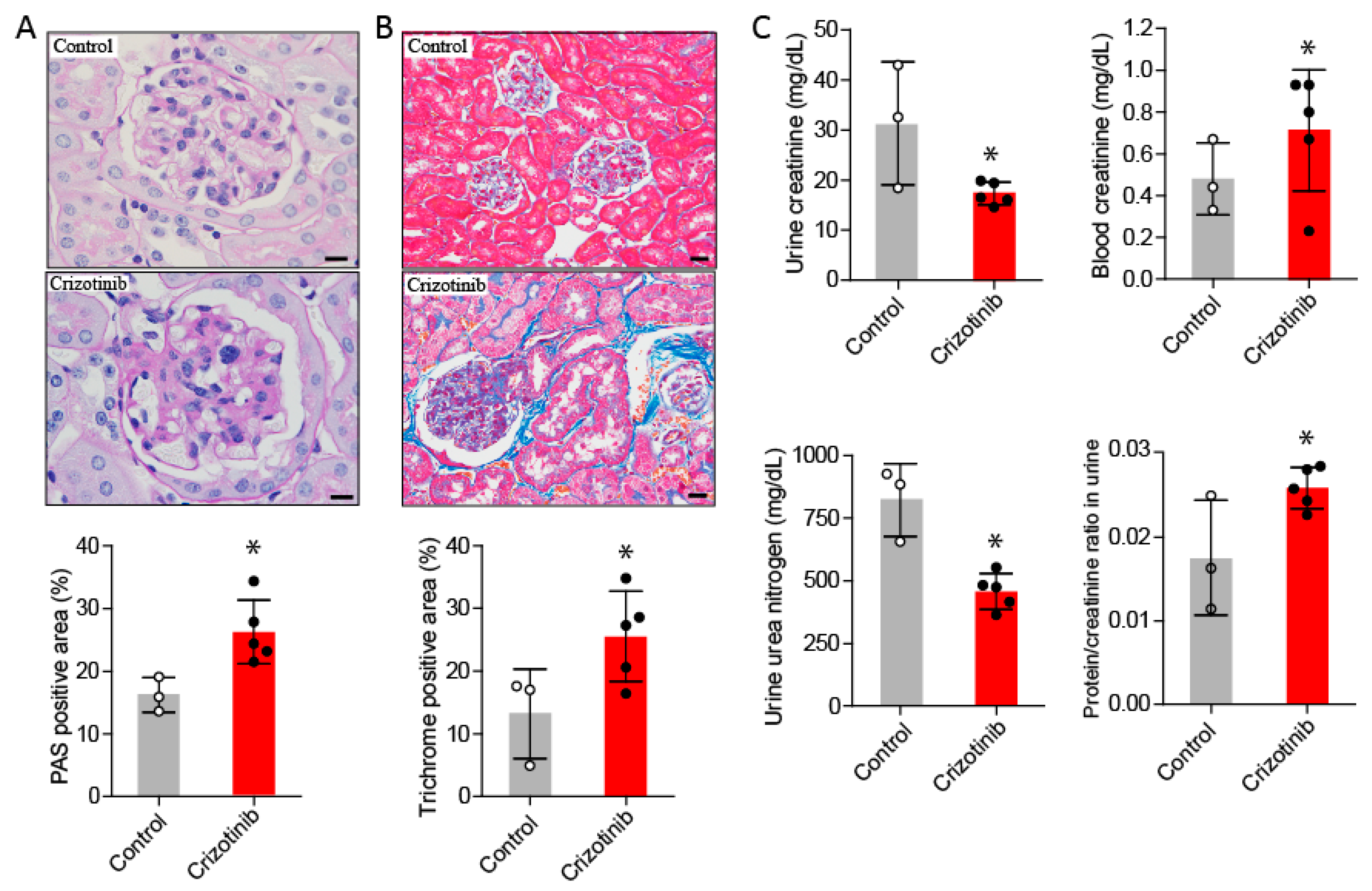
2.2.3. Crizotinib Impaired Renal Function
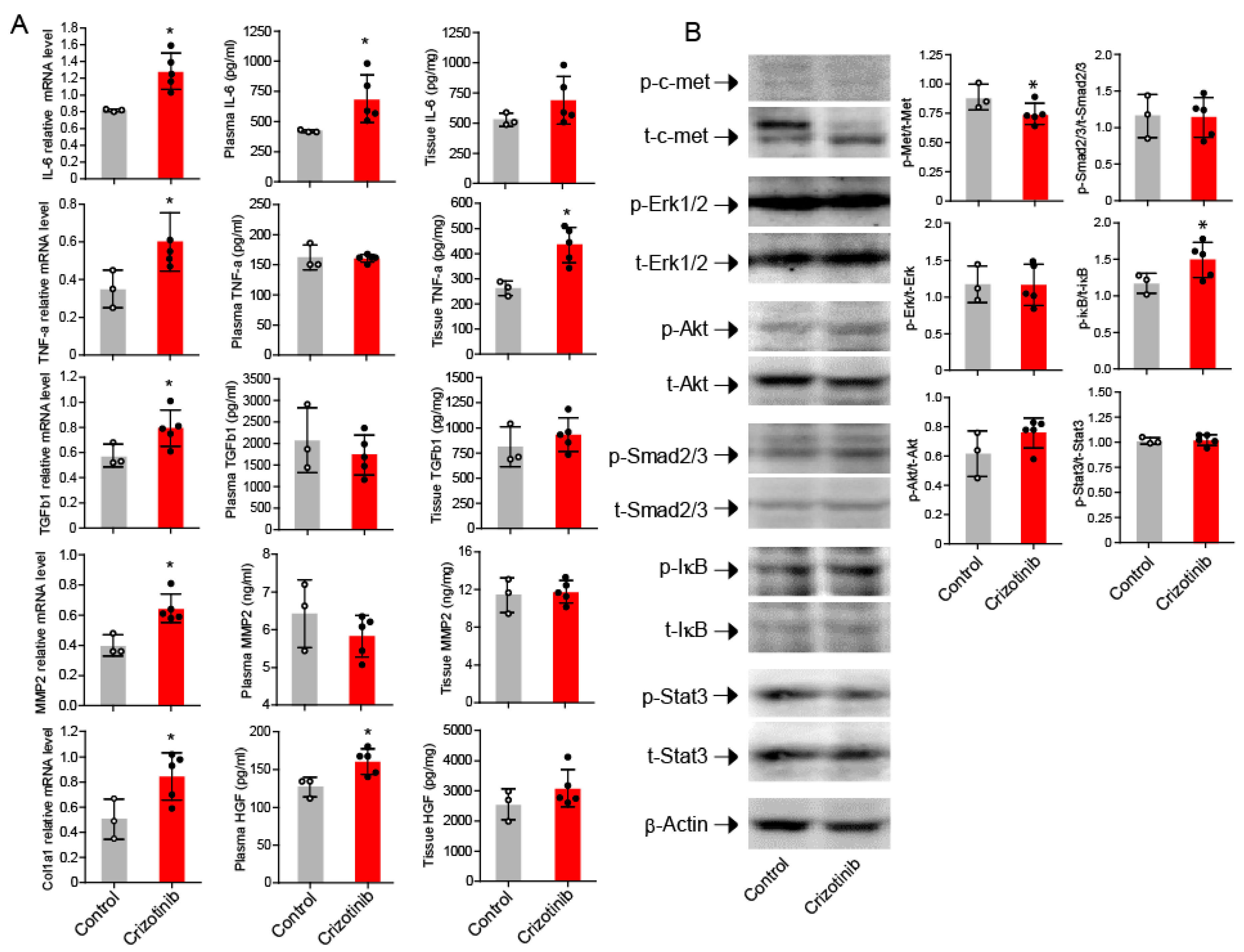
2.3. Crizotinib Associated with Enhanced Inflammatory Markers in the Kidneys
2.4. Activation of NF-κB in the Kidneys after Crizotinib Therapy
3. Discussion
4. Materials and Methods
4.1. Experimental Animal Model
4.2. Micro CT of Kidneys
4.3. Mouse Sacrifice and Sampling
4.4. Biochemical Analysis
4.5. Tissue Preparation and Staining
4.6. Western Blotting
4.7. Reverse Transcription Polymerase Chain Reaction
4.8. Ethical Statement
4.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Christensen, J.G.; Zou, H.Y.; Arango, M.E.; Li, Q.; Lee, J.H.; McDonnell, S.R.; Yamazaki, S.; Alton, G.R.; Mroczkowski, B.; Los, G. Cytoreductive antitumor activity of PF-2341066, a novel inhibitor of anaplastic lymphoma kinase and c-Met, in experimental models of anaplastic large-cell lymphoma. Mol. Cancer Ther. 2007, 6, 3314–3322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zou, H.Y.; Li, Q.; Lee, J.H.; Arango, M.E.; McDonnell, S.R.; Yamazaki, S.; Koudriakova, T.B.; Alton, G.; Cui, J.J.; Kung, P.P.; et al. An orally available small-molecule inhibitor of c-Met, PF-2341066, exhibits cytoreductive antitumor efficacy through antiproliferative and antiangiogenic mechanisms. Cancer Res. 2007, 67, 4408–4417. [Google Scholar] [CrossRef] [PubMed]
- Blackhall, F.H.; Peters, S.; Bubendorf, L.; Dafni, U.; Kerr, K.M.; Hager, H.; Soltermann, A.; O'Byrne, K.J.; Dooms, C.; Sejda, A.; et al. Prevalence and clinical outcomes for patients with ALK-positive resected stage I to III adenocarcinoma: Results from the European Thoracic Oncology Platform Lungscape Project. J. Clin. Oncol. 2014, 32, 2780–2787. [Google Scholar] [CrossRef] [PubMed]
- Rikova, K.; Guo, A.; Zeng, Q.; Possemato, A.; Yu, J.; Haack, H.; Nardone, J.; Lee, K.; Reeves, C.; Li, Y.; et al. Global survey of phosphotyrosine signaling identifies oncogenic kinases in lung cancer. Cell 2007, 131, 1190–1203. [Google Scholar] [CrossRef] [PubMed]
- Soda, M.; Choi, Y.L.; Enomoto, M.; Takada, S.; Yamashita, Y.; Ishikawa, S.; Fujiwara, S.; Watanabe, H.; Kurashina, K.; Hatanaka, H.; et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature 2007, 448, 561–566. [Google Scholar] [CrossRef] [PubMed]
- Facchinetti, F.; Rossi, G.; Bria, E.; Soria, J.C.; Besse, B.; Minari, R.; Friboulet, L.; Tiseo, M. Oncogene addiction in non-small cell lung cancer: Focus on ROS1 inhibition. Cancer Treat. Rev. 2017, 55, 83–95. [Google Scholar] [CrossRef] [PubMed]
- Hanna, N.; Johnson, D.; Temin, S.; Baker, S., Jr.; Brahmer, J.; Ellis, P.M.; Giaccone, G.; Hesketh, P.J.; Jaiyesimi, I.; Leighl, N.B.; et al. Systemic Therapy for Stage IV Non-Small-Cell Lung Cancer: American Society of Clinical Oncology Clinical Practice Guideline Update. J. Clin. Oncol. 2017, 35, 3484–3515. [Google Scholar] [CrossRef] [PubMed]
- Shaw, A.T.; Kim, D.W.; Nakagawa, K.; Seto, T.; Crino, L.; Ahn, M.J.; de Pas, T.; Besse, B.; Solomon, B.J.; Blackhall, F.; et al. Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. N. Engl. J. Med. 2013, 368, 2385–2394. [Google Scholar] [CrossRef] [PubMed]
- Camidge, D.R.; Bang, Y.J.; Kwak, E.L.; Iafrate, A.J.; Varella-Garcia, M.; Fox, S.B.; Riely, G.J.; Solomon, B.; Ou, S.H.; Kim, D.W.; et al. Activity and safety of crizotinib in patients with ALK-positive non-small-cell lung cancer: Updated results from a phase 1 study. Lancet Oncol. 2012, 13, 1011–1019. [Google Scholar] [CrossRef]
- Di Girolamo, M.; Paris, I.; Carbonetti, F.; Onesti, E.C.; Socciarelli, F.; Marchetti, P. Widespread renal polycystosis induced by crizotinib. Tumori 2015, 101, e128–e131. [Google Scholar] [CrossRef] [PubMed]
- Heigener, D.F.; Reck, M. Crizotinib. Recent Results Cancer Res. 2014, 201, 197–205. [Google Scholar] [PubMed]
- Lin, Y.T.; Wang, Y.F.; Yang, J.C.; Yu, C.J.; Wu, S.G.; Shih, J.Y.; Yang, P.C. Development of renal cysts after crizotinib treatment in advanced ALK-positive non-small-cell lung cancer. J. Thorac. Oncol. 2014, 9, 1720–1725. [Google Scholar] [CrossRef] [PubMed]
- Schnell, P.; Bartlett, C.H.; Solomon, B.J.; Tassell, V.; Shaw, A.T.; de Pas, T.; Lee, S.H.; Lee, G.K.; Tanaka, K.; Tan, W.; et al. Complex renal cysts associated with crizotinib treatment. Cancer Med. 2015, 4, 887–896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Souteyrand, P.; Burtey, S.; Barlesi, F. Multicystic kidney disease: A complication of crizotinib. Diagn. Interv. Imaging 2015, 96, 393–395. [Google Scholar] [CrossRef] [PubMed]
- Cameron, L.B.; Jiang, D.H.; Moodie, K.; Mitchell, C.; Solomon, B.; Parameswaran, B.K. Crizotinib Associated Renal Cysts [CARCs]: Incidence and patterns of evolution. Cancer Imaging 2017, 17, 7. [Google Scholar] [CrossRef] [PubMed]
- Halpenny, D.F.; McEvoy, S.; Li, A.; Hayan, S.; Capanu, M.; Zheng, J.; Riely, G.; Ginsberg, M.S. Renal cyst formation in patients treated with crizotinib for non-small cell lung cancer-Incidence, radiological features and clinical characteristics. Lung Cancer 2017, 106, 33–36. [Google Scholar] [CrossRef] [PubMed]
- Klempner, S.J.; Aubin, G.; Dash, A.; Ou, S.H. Spontaneous regression of crizotinib-associated complex renal cysts during continuous crizotinib treatment. Oncologist 2014, 19, 1008–1010. [Google Scholar] [CrossRef] [PubMed]
- Taima, K.; Tanaka, H.; Tanaka, Y.; Itoga, M.; Takanashi, S.; Tasaka, S. Regression of Crizotinib-Associated Complex Cystic Lesions after Switching to Alectinib. Intern. Med. 2017, 56, 2321–2324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horie, S.; Higashihara, E.; Nutahara, K.; Mikami, Y.; Okubo, A.; Kano, M.; Kawabe, K. Mediation of renal cyst formation by hepatocyte growth factor. Lancet 1994, 344, 789–791. [Google Scholar] [CrossRef]
- Maeshima, A.; Zhang, Y.Q.; Furukawa, M.; Naruse, T.; Kojima, I. Hepatocyte growth factor induces branching tubulogenesis in MDCK cells by modulating the activin-follistatin system. Kidney Int. 2000, 58, 1511–1522. [Google Scholar] [CrossRef] [PubMed]
- Weimbs, T.; Olsan, E.E.; Talbot, J.J. Regulation of STATs by polycystin-1 and their role in polycystic kidney disease. JAKSTAT 2013, 2, e23650. [Google Scholar] [CrossRef] [PubMed]
- Weimbs, T.; Talbot, J.J. STAT3 Signaling in Polycystic Kidney Disease. Drug Discov. Today Dis. Mech. 2013, 10, e113–e118. [Google Scholar] [CrossRef] [PubMed]
- Brosnan, E.M.; Weickhardt, A.J.; Lu, X.; Maxon, D.A.; Baron, A.E.; Chonchol, M.; Camidge, D.R. Drug-induced reduction in estimated glomerular filtration rate in patients with ALK-positive non-small cell lung cancer treated with the ALK inhibitor crizotinib. Cancer 2014, 120, 664–674. [Google Scholar] [CrossRef] [PubMed]
- Camidge, D.R.; Brosnan, E.M.; DeSilva, C.; Koo, P.J.; Chonchol, M. Crizotinib effects on creatinine and non-creatinine-based measures of glomerular filtration rate. J. Thorac. Oncol. 2014, 9, 1634–1637. [Google Scholar] [CrossRef] [PubMed]
- Gastaud, L.; Ambrosetti, D.; Otto, J.; Marquette, C.H.; Coutts, M.; Hofman, P.; Esnault, V.; Favre, G. Acute kidney injury following crizotinib administration for non-small-cell lung carcinoma. Lung Cancer 2013, 82, 362–364. [Google Scholar] [CrossRef] [PubMed]
- Martin Martorell, P.; Huerta Alvaro, M.; Solis Salguero, M.A.; Insa Molla, A. Crizotinib and renal insufficiency: A case report and review of the literature. Lung Cancer 2014, 84, 310–313. [Google Scholar] [CrossRef] [PubMed]
- Dai, C.; Saleem, M.A.; Holzman, L.B.; Mathieson, P.; Liu, Y. Hepatocyte growth factor signaling ameliorates podocyte injury and proteinuria. Kidney Int. 2010, 77, 962–973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iekushi, K.; Taniyama, Y.; Azuma, J.; Sanada, F.; Kusunoki, H.; Yokoi, T.; Koibuchi, N.; Okayama, K.; Rakugi, H.; Morishita, R. Hepatocyte growth factor attenuates renal fibrosis through TGF-β1 suppression by apoptosis of myofibroblasts. J. Hypertens. 2010, 28, 2454–2461. [Google Scholar] [CrossRef] [PubMed]
- Kwiecinski, M.; Noetel, A.; Elfimova, N.; Trebicka, J.; Schievenbusch, S.; Strack, I.; Molnar, L.; von Brandenstein, M.; Tox, U.; Nischt, R.; et al. Hepatocyte growth factor (HGF) inhibits collagen I and IV synthesis in hepatic stellate cells by miRNA-29 induction. PLoS ONE 2011, 6, e24568. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; He, D.; Yang, J.; Wang, X. Cordycepin inhibits renal interstitial myofibroblast activation probably by inducing hepatocyte growth factor expression. J. Pharmacol. Sci. 2011, 117, 286–294. [Google Scholar] [CrossRef] [PubMed]
- Shukla, M.N.; Rose, J.L.; Ray, R.; Lathrop, K.L.; Ray, A.; Ray, P. Hepatocyte growth factor inhibits epithelial to myofibroblast transition in lung cells via Smad7. Am. J. Respir. Cell Mol. Biol. 2009, 40, 643–653. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Dai, C.; Liu, Y. Hepatocyte growth factor suppresses renal interstitial myofibroblast activation and intercepts Smad signal transduction. Am. J. Pathol. 2003, 163, 621–632. [Google Scholar] [CrossRef]
- Yi, X.; Li, X.; Zhou, Y.; Ren, S.; Wan, W.; Feng, G.; Jiang, X. Hepatocyte growth factor regulates the TGF-β1-induced proliferation, differentiation and secretory function of cardiac fibroblasts. Int. J. Mol. Med. 2014, 34, 381–390. [Google Scholar] [CrossRef] [PubMed]
- Kulasekaran, P.; Scavone, C.A.; Rogers, D.S.; Arenberg, D.A.; Thannickal, V.J.; Horowitz, J.C. Endothelin-1 and transforming growth factor-β1 independently induce fibroblast resistance to apoptosis via AKT activation. Am. J. Respir. Cell Mol. Biol. 2009, 41, 484–493. [Google Scholar] [CrossRef] [PubMed]
- Hayden, M.S.; Ghosh, S. Regulation of NF-κB by TNF family cytokines. Semin. Immunol. 2014, 26, 253–266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, Y.; Kim, J.M.; Park, H.S.; Yang, A.; Islam, C.; Lakatta, E.G.; Lin, L. AGE-RAGE signal generates a specific NF-κB RelA “barcode” that directs collagen I expression. Sci. Rep. 2016, 6, 18822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urtasun, R.; Lopategi, A.; George, J.; Leung, T.M.; Lu, Y.; Wang, X.; Ge, X.; Fiel, M.I.; Nieto, N. Osteopontin, an oxidant stress sensitive cytokine, up-regulates collagen-I via integrin αVβ3 engagement and PI3K/pAkt/NFκB signaling. Hepatology 2012, 55, 594–608. [Google Scholar] [CrossRef] [PubMed]
- Julien, S.; Puig, I.; Caretti, E.; Bonaventure, J.; Nelles, L.; van Roy, F.; Dargemont, C.; de Herreros, A.G.; Bellacosa, A.; Larue, L. Activation of NF-κB by Akt upregulates Snail expression and induces epithelium mesenchyme transition. Oncogene 2007, 26, 7445–7456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, M.; Ning, X.; Li, R.; Yang, Z.; Yang, X.; Sun, S.; Qian, Q. Signalling pathways involved in hypoxia-induced renal fibrosis. J. Cell. Mol. Med. 2017, 21, 1248–1259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bendinelli, P.; Matteucci, E.; Dogliotti, G.; Corsi, M.M.; Banfi, G.; Maroni, P.; Desiderio, M.A. Molecular basis of anti-inflammatory action of platelet-rich plasma on human chondrocytes: Mechanisms of NF-κB inhibition via HGF. J. Cell. Physiol. 2010, 225, 757–766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romero-Vasquez, F.; Chavez, M.; Perez, M.; Arcaya, J.L.; Garcia, A.J.; Rincon, J.; Rodriguez-Iturbe, B. Overexpression of HGF transgene attenuates renal inflammatory mediators, Na+-ATPase activity and hypertension in spontaneously hypertensive rats. Biochim. Biophys. Acta 2012, 1822, 1590–1599. [Google Scholar] [CrossRef] [PubMed]
- Tamada, S.; Asai, T.; Kuwabara, N.; Iwai, T.; Uchida, J.; Teramoto, K.; Kaneda, N.; Yukimura, T.; Komiya, T.; Nakatani, T.; et al. Molecular mechanisms and therapeutic strategies of chronic renal injury: The role of nuclear factor κB activation in the development of renal fibrosis. J. Pharmacol. Sci. 2006, 100, 17–21. [Google Scholar] [CrossRef] [PubMed]
- Sattler, M.; Salgia, R. c-Met and hepatocyte growth factor: Potential as novel targets in cancer therapy. Curr. Oncol. Rep. 2007, 9, 102–108. [Google Scholar] [CrossRef] [PubMed]
- Oga, T.; Matsuoka, T.; Yao, C.; Nonomura, K.; Kitaoka, S.; Sakata, D.; Kita, Y.; Tanizawa, K.; Taguchi, Y.; Chin, K.; et al. Prostaglandin F2α receptor signaling facilitates bleomycin-induced pulmonary fibrosis independently of transforming growth factor-β. Nat. Med. 2009, 15, 1426–1430. [Google Scholar] [CrossRef] [PubMed]
- Gumusay, O.; Esendagli-Yilmaz, G.; Uner, A.; Cetin, B.; Buyukberber, S.; Benekli, M.; Ilhan, M.N.; Coskun, U.; Gulbahar, O.; Ozet, A. Crizotinib-induced toxicity in an experimental rat model. Wien. Klin. Wochenschr. 2016, 128, 435–441. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.A.; Licata, T.; Lakhani, A.; Garcia, M.V.; Schildhaus, H.U.; Vuaroqueaux, V.; Halmos, B.; Borczuk, A.C.; Chen, Y.A.; Creelan, B.C.; et al. MET-GRB2 Signaling-Associated Complexes Correlate with Oncogenic MET Signaling and Sensitivity to MET Kinase Inhibitors. Clin. Cancer Res. 2017, 23, 7084–7096. [Google Scholar] [CrossRef] [PubMed]
- Urawa, M.; Kobayashi, T.; D’Alessandro-Gabazza, C.N.; Fujimoto, H.; Toda, M.; Roeen, Z.; Hinneh, J.A.; Yasuma, T.; Takei, Y.; Taguchi, O.; et al. Protein S is protective in pulmonary fibrosis. J. Thromb. Haemost. 2016, 14, 1588–1599. [Google Scholar] [CrossRef] [PubMed]




© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yasuma, T.; Kobayashi, T.; D’Alessandro-Gabazza, C.N.; Fujimoto, H.; Ito, K.; Nishii, Y.; Nishihama, K.; Baffour Tonto, P.; Takeshita, A.; Toda, M.; et al. Renal Injury during Long-Term Crizotinib Therapy. Int. J. Mol. Sci. 2018, 19, 2902. https://doi.org/10.3390/ijms19102902
Yasuma T, Kobayashi T, D’Alessandro-Gabazza CN, Fujimoto H, Ito K, Nishii Y, Nishihama K, Baffour Tonto P, Takeshita A, Toda M, et al. Renal Injury during Long-Term Crizotinib Therapy. International Journal of Molecular Sciences. 2018; 19(10):2902. https://doi.org/10.3390/ijms19102902
Chicago/Turabian StyleYasuma, Taro, Tetsu Kobayashi, Corina N. D’Alessandro-Gabazza, Hajime Fujimoto, Kentaro Ito, Yoichi Nishii, Kota Nishihama, Prince Baffour Tonto, Atsuro Takeshita, Masaaki Toda, and et al. 2018. "Renal Injury during Long-Term Crizotinib Therapy" International Journal of Molecular Sciences 19, no. 10: 2902. https://doi.org/10.3390/ijms19102902