Non-Canonical Regulation of Type I Collagen through Promoter Binding of SOX2 and Its Contribution to Ameliorating Pulmonary Fibrosis by Butylidenephthalide

 ,
,
Abstract
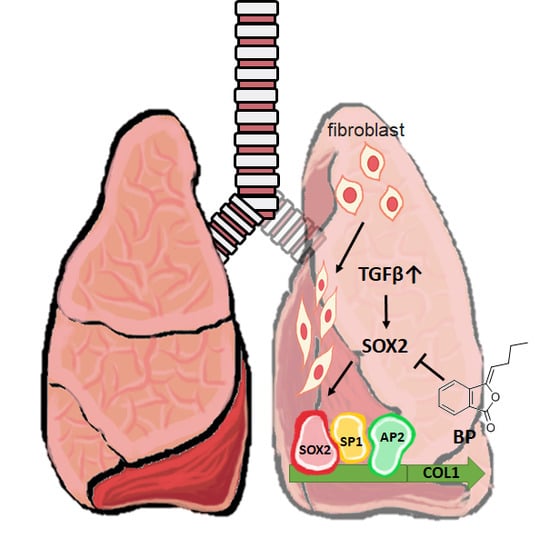
:
1. Introduction
2. Results
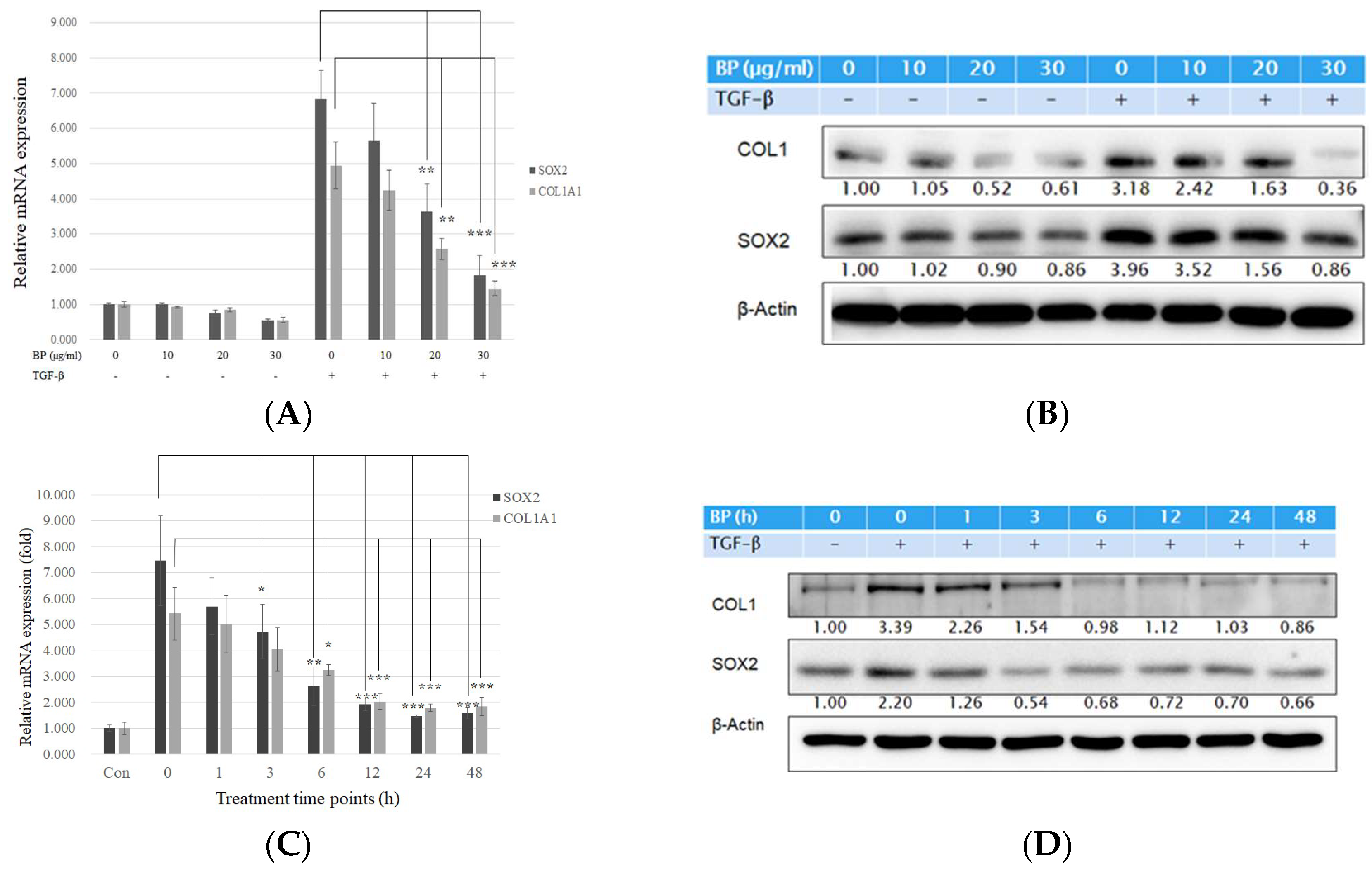
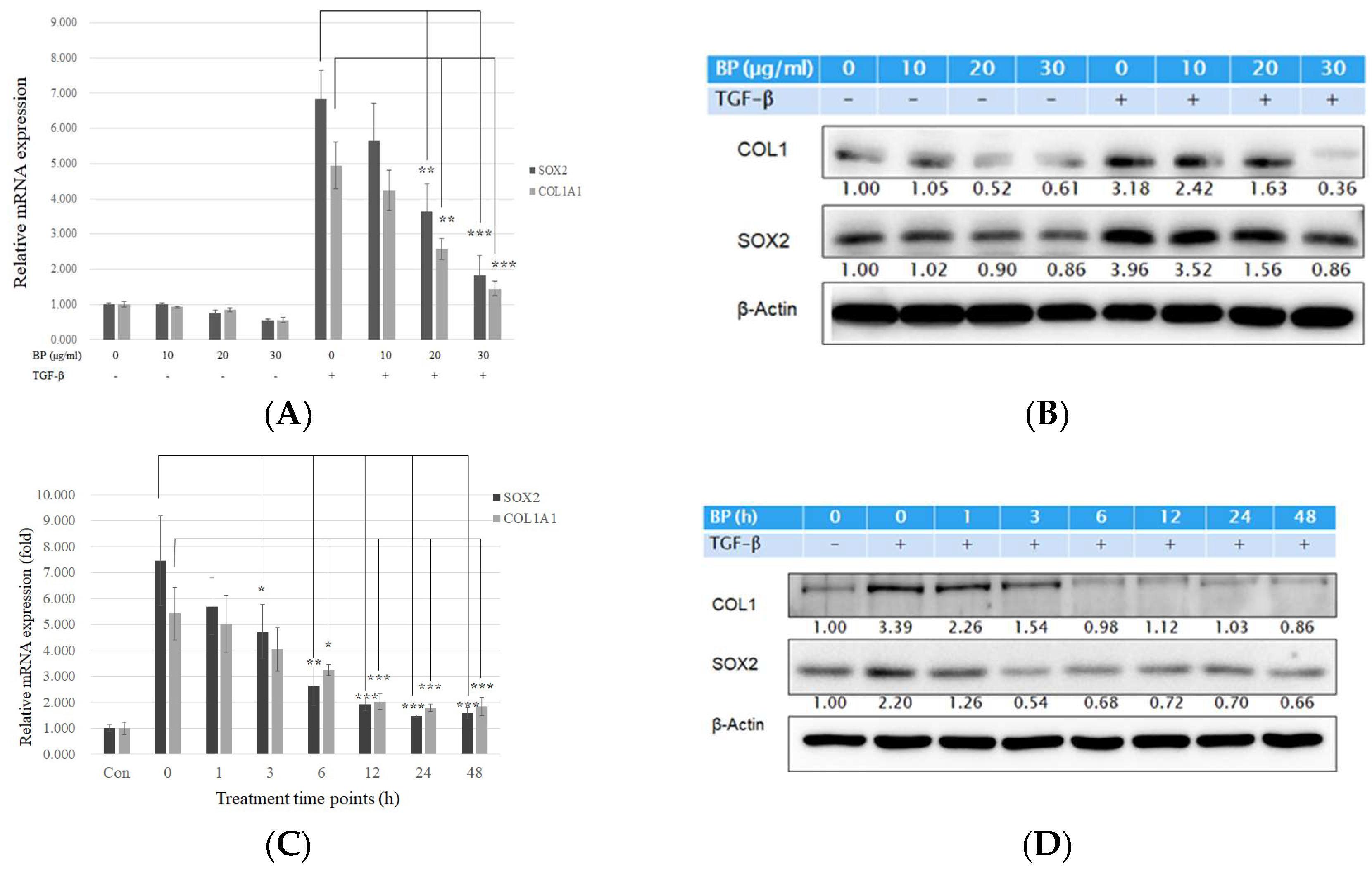
2.1. BP Treatment in Human Lung Fibroblasts Attenuated Collagen Expression Driven by Exogenous TGF-β
2.2. BP Did Not Regulate the Smad and Non-Smad Pathways to Block the Effects of TGF-β
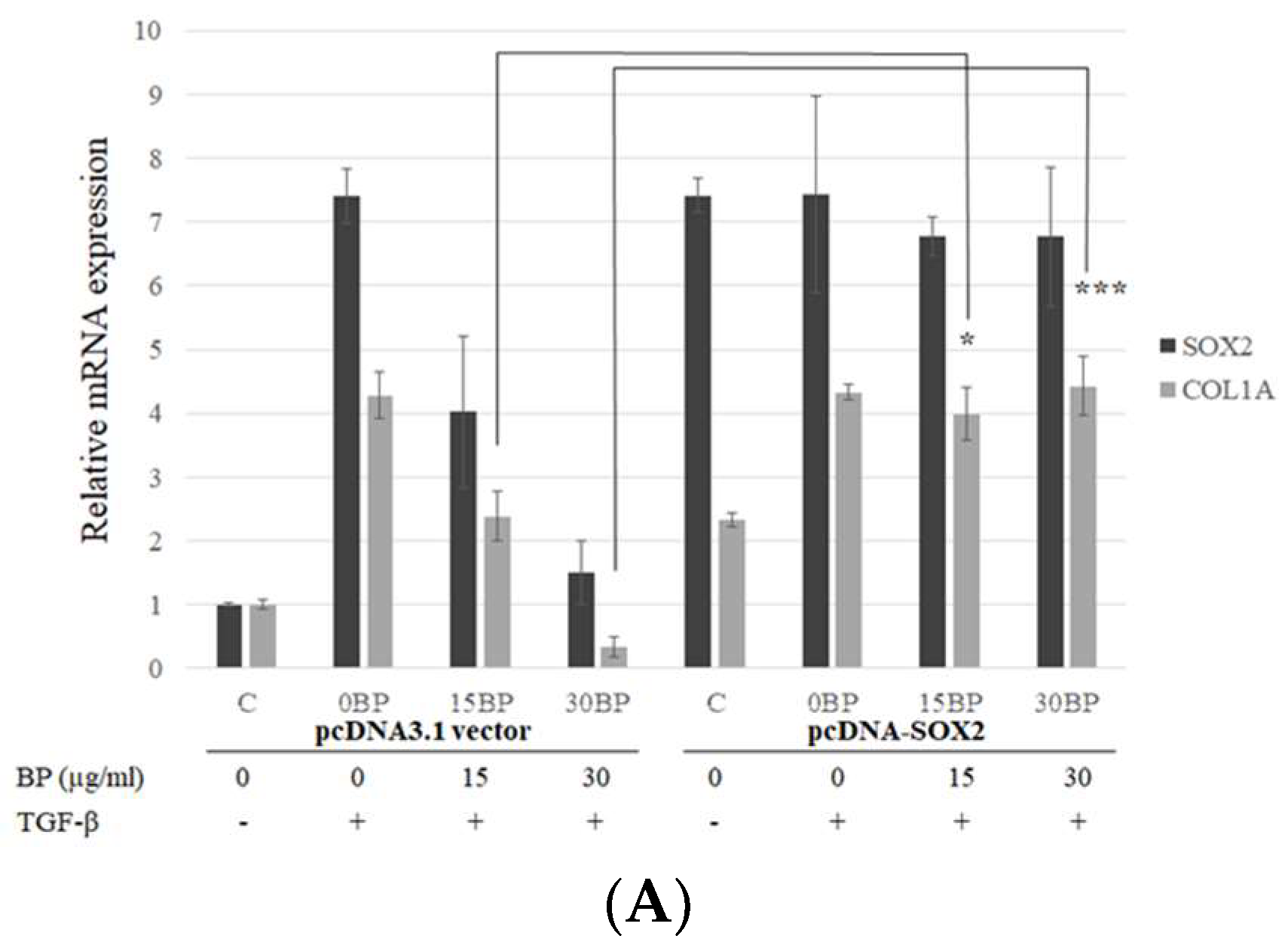
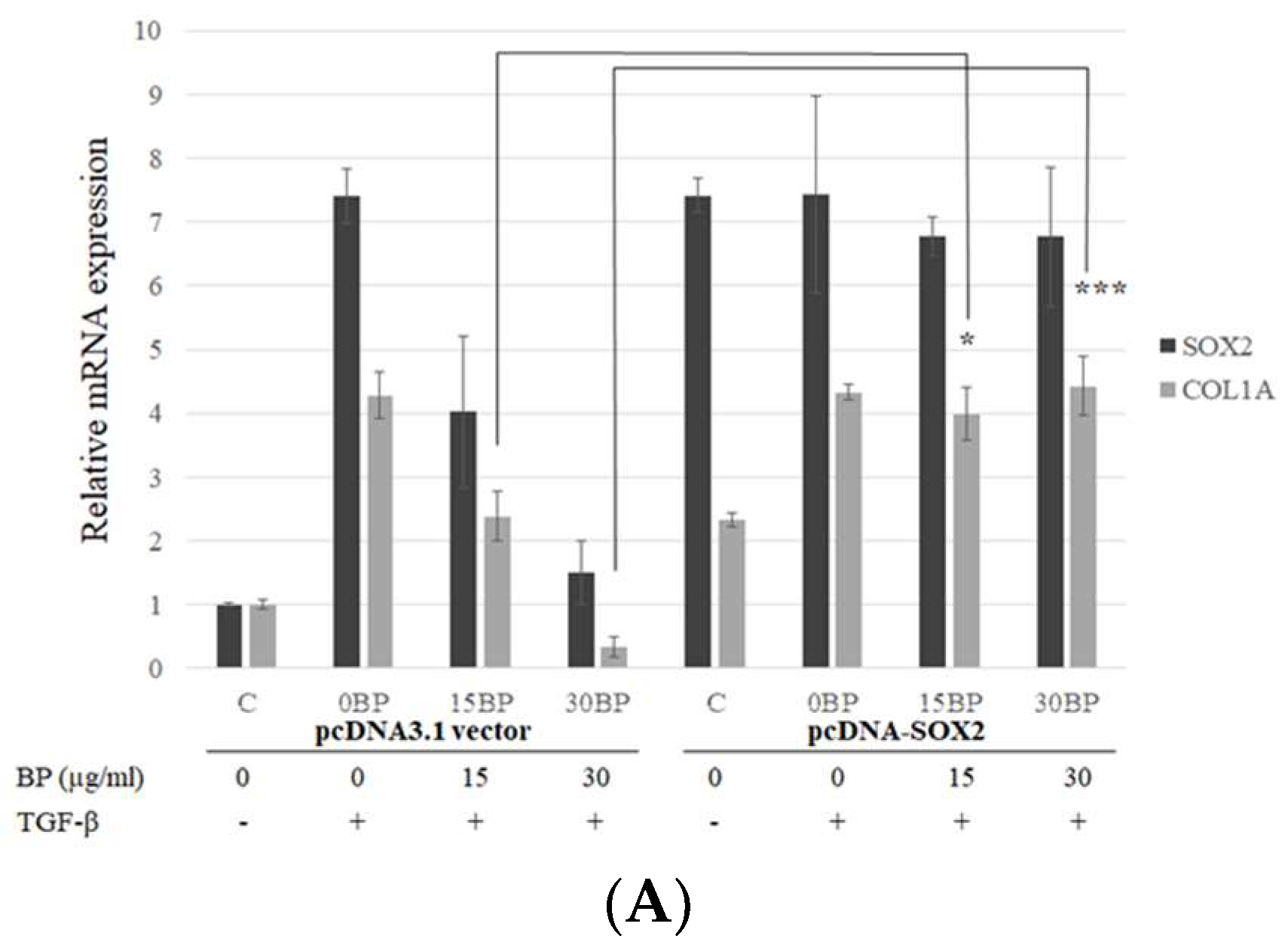
2.3. SOX2-Overexpression Prevented Collagen Reduction in BP Treatment
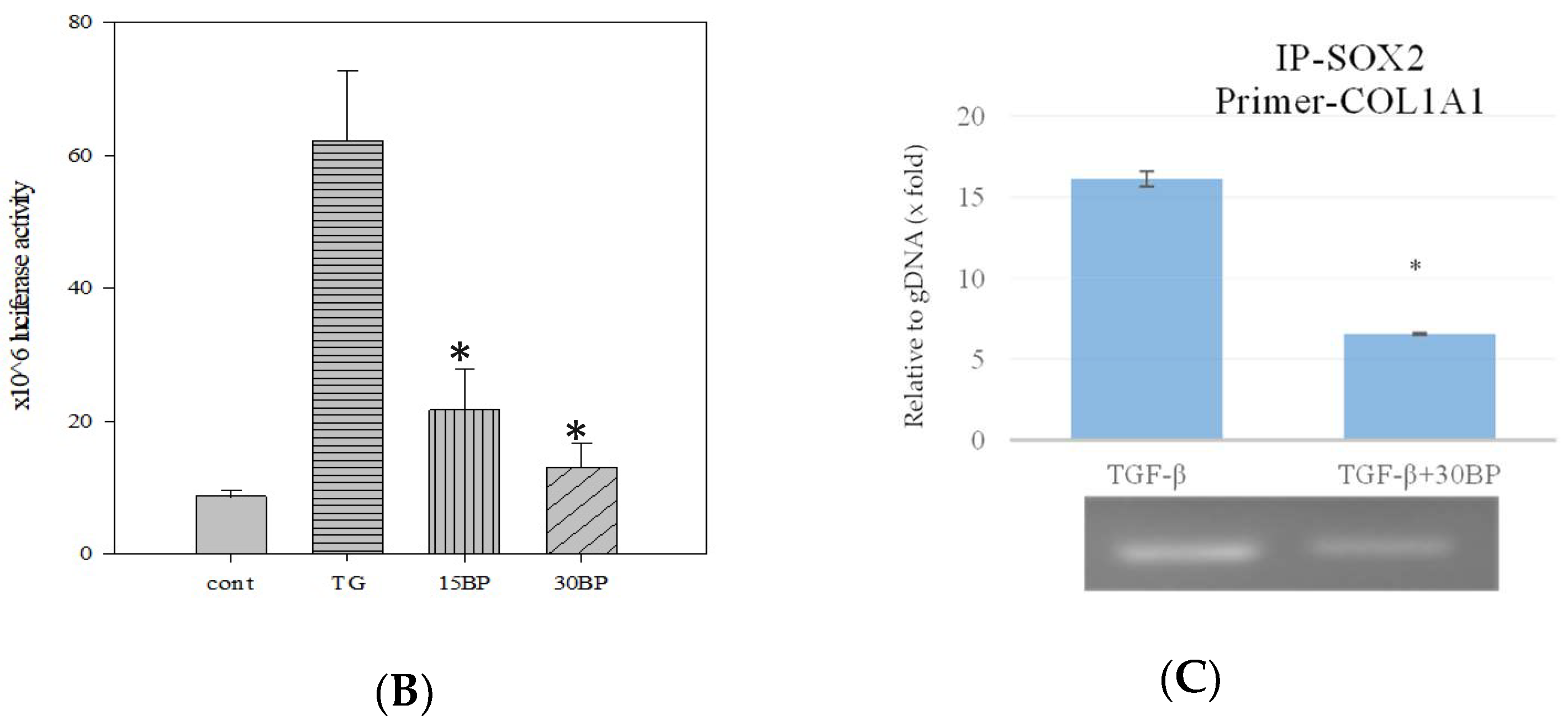
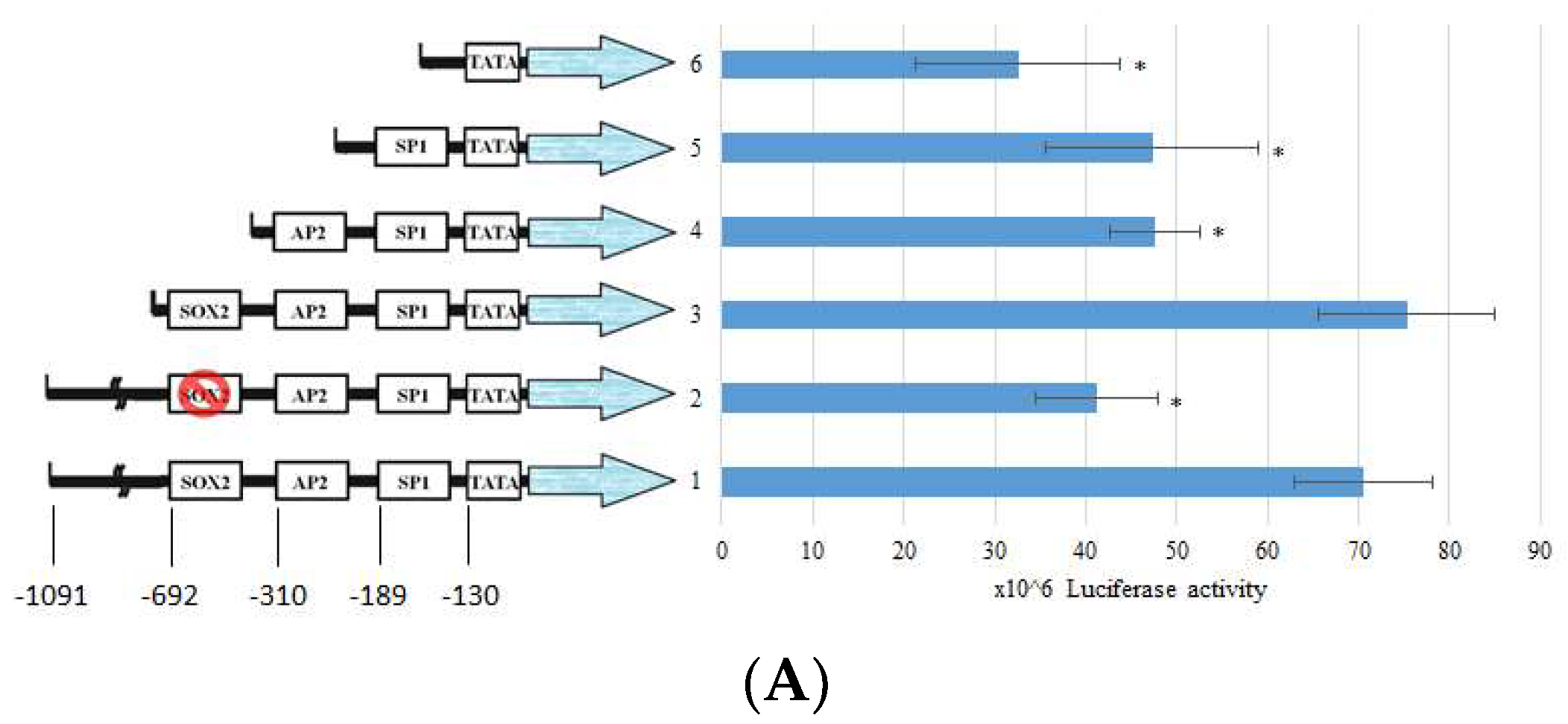
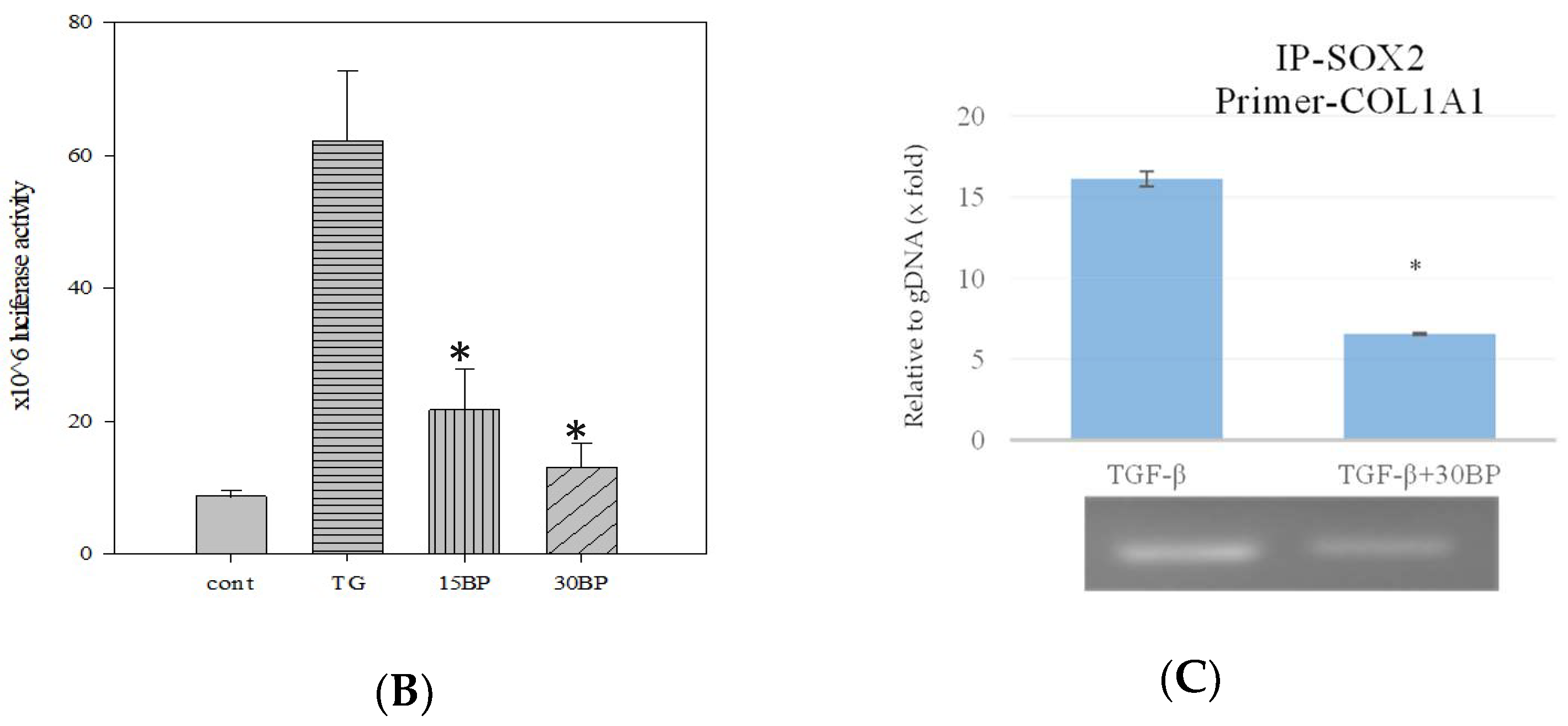
2.4. BP Decreased SOX2 Specific Promoter Binding on Collagen
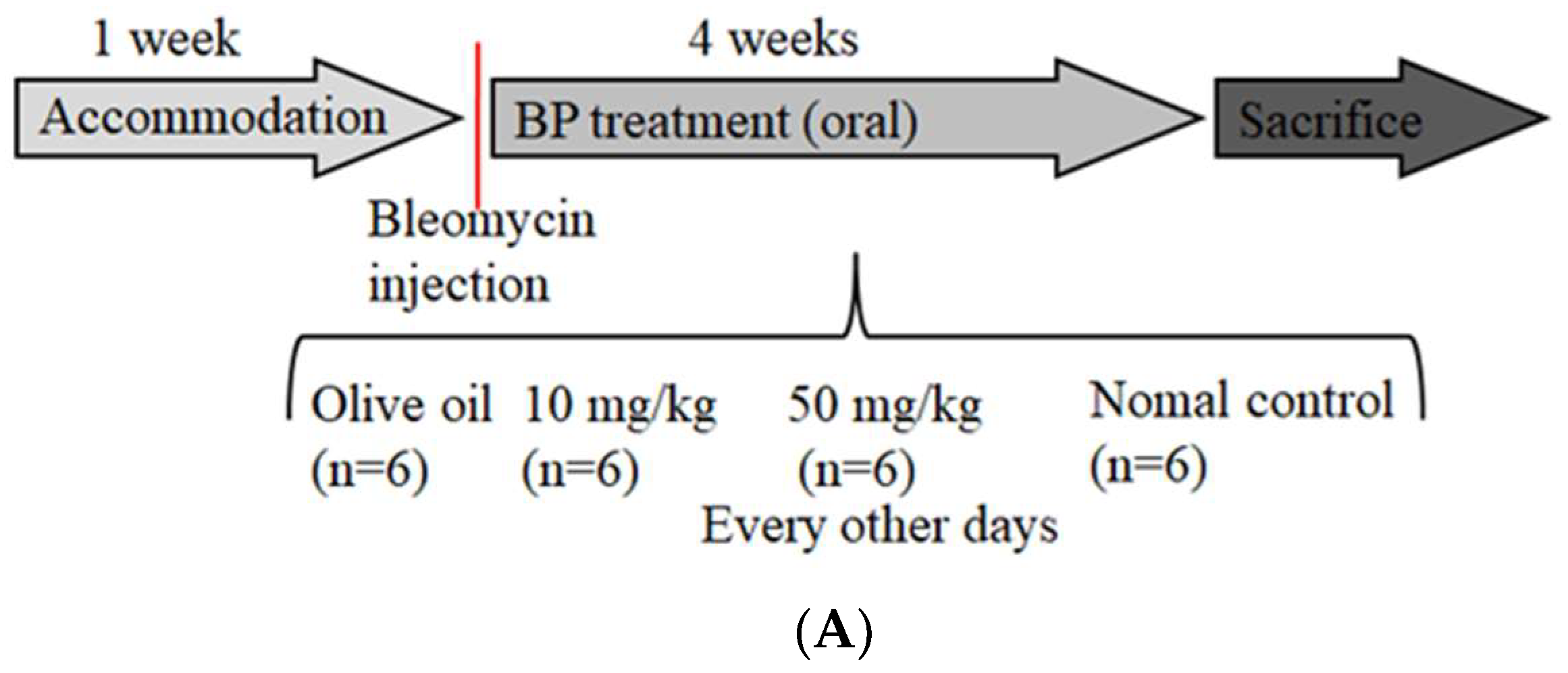
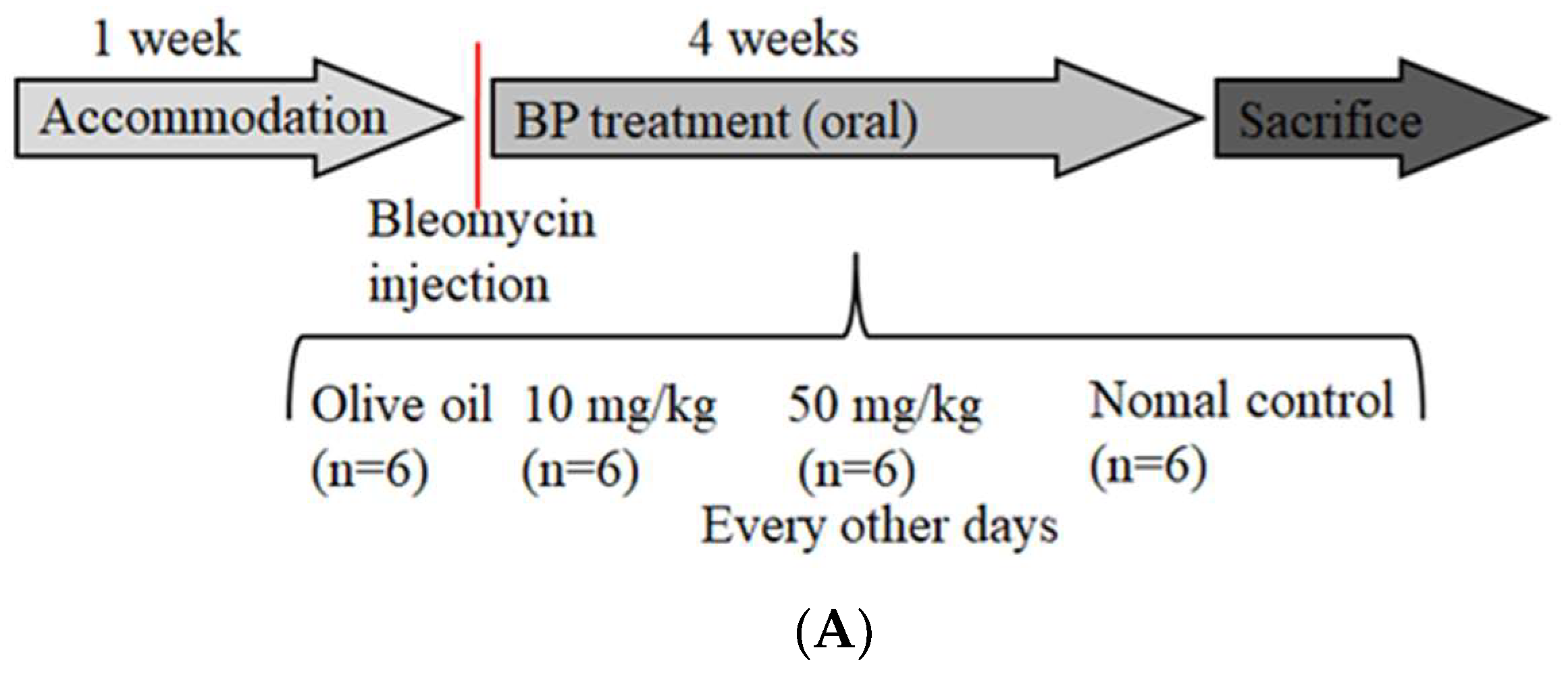
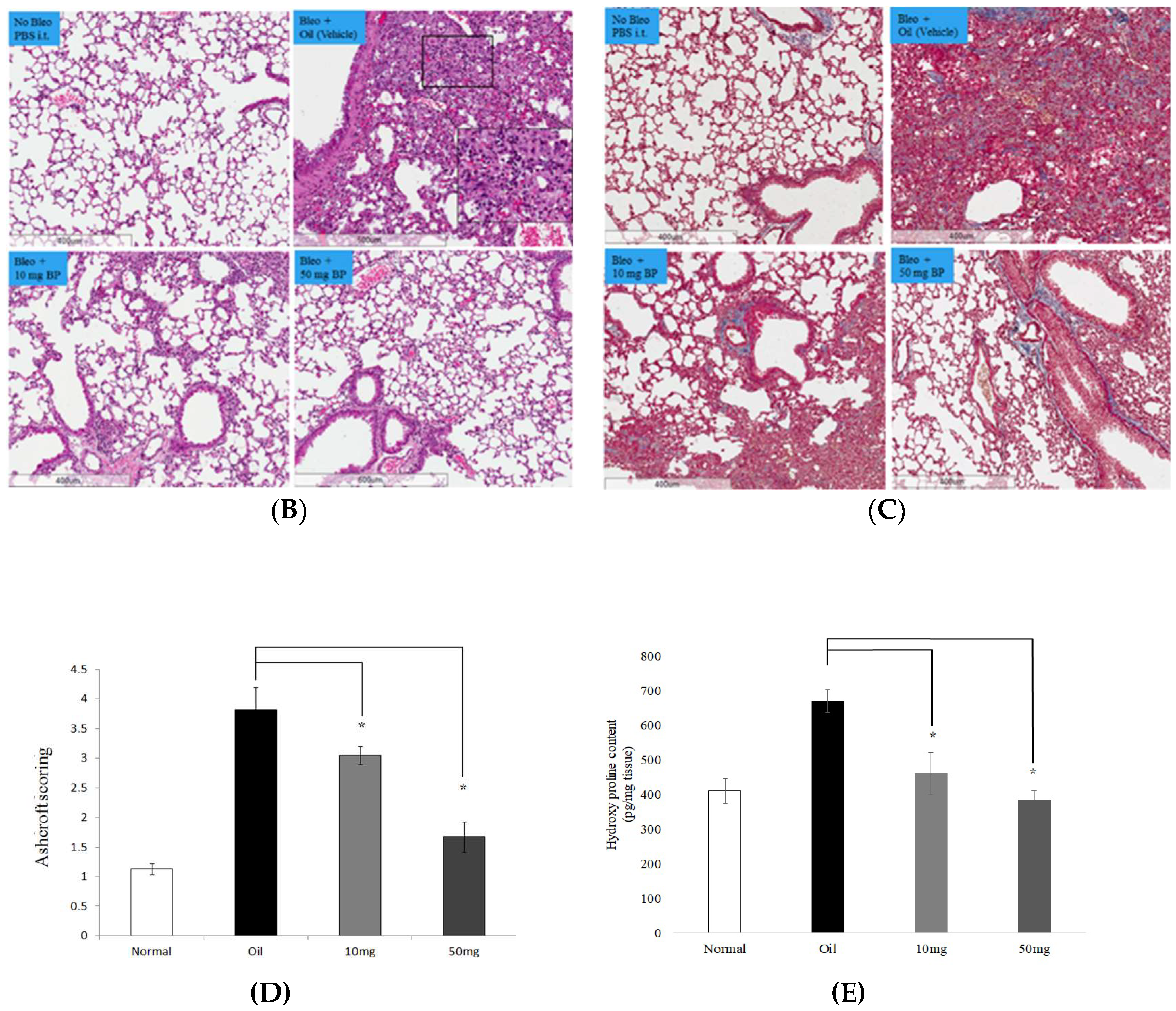
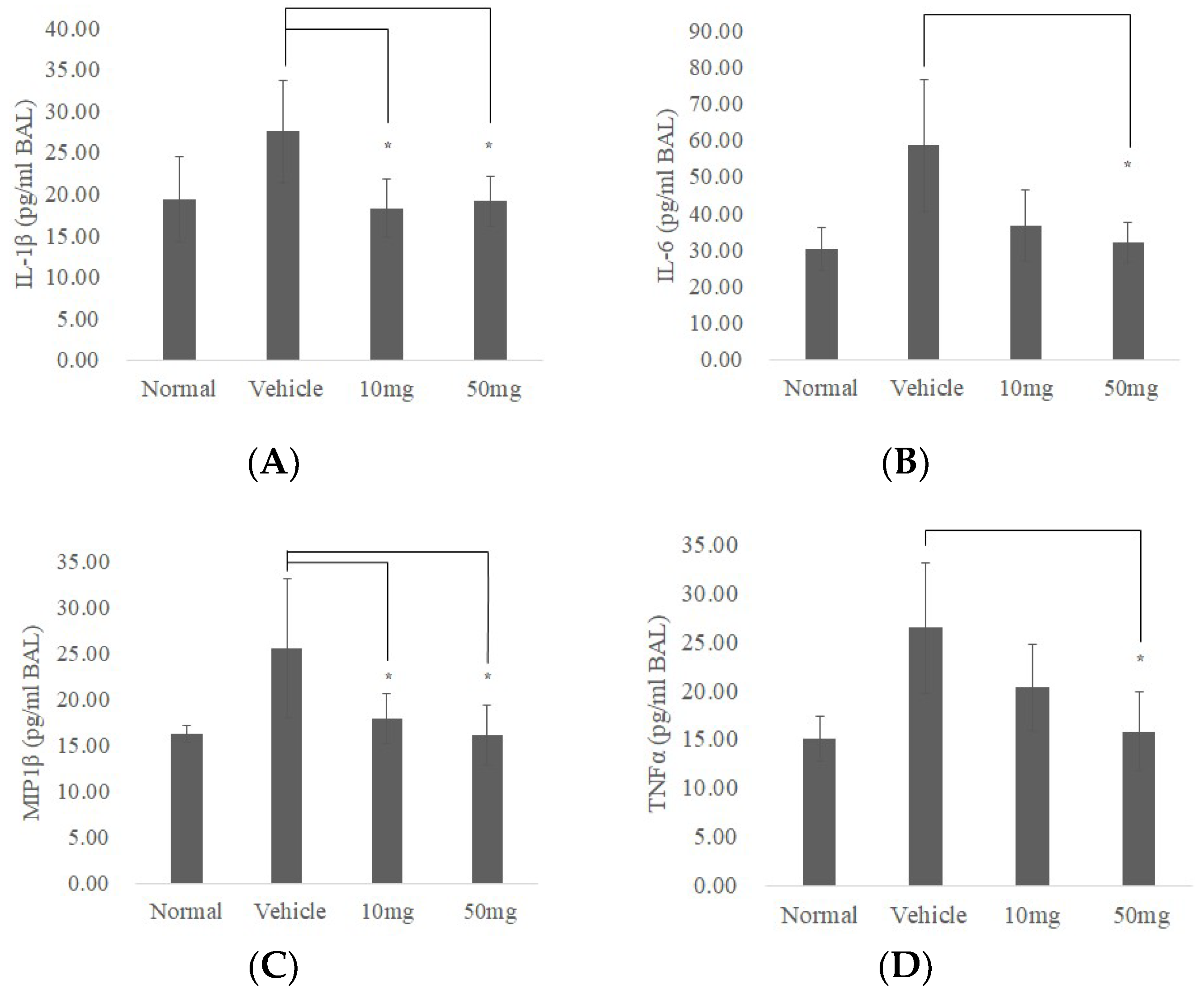
2.5. BP Reduced bleomycin-Induced Pulmonary Fibrosis in Mice
2.6. BP Restored Pulmonary Function in Bleomycin-Treated Mice
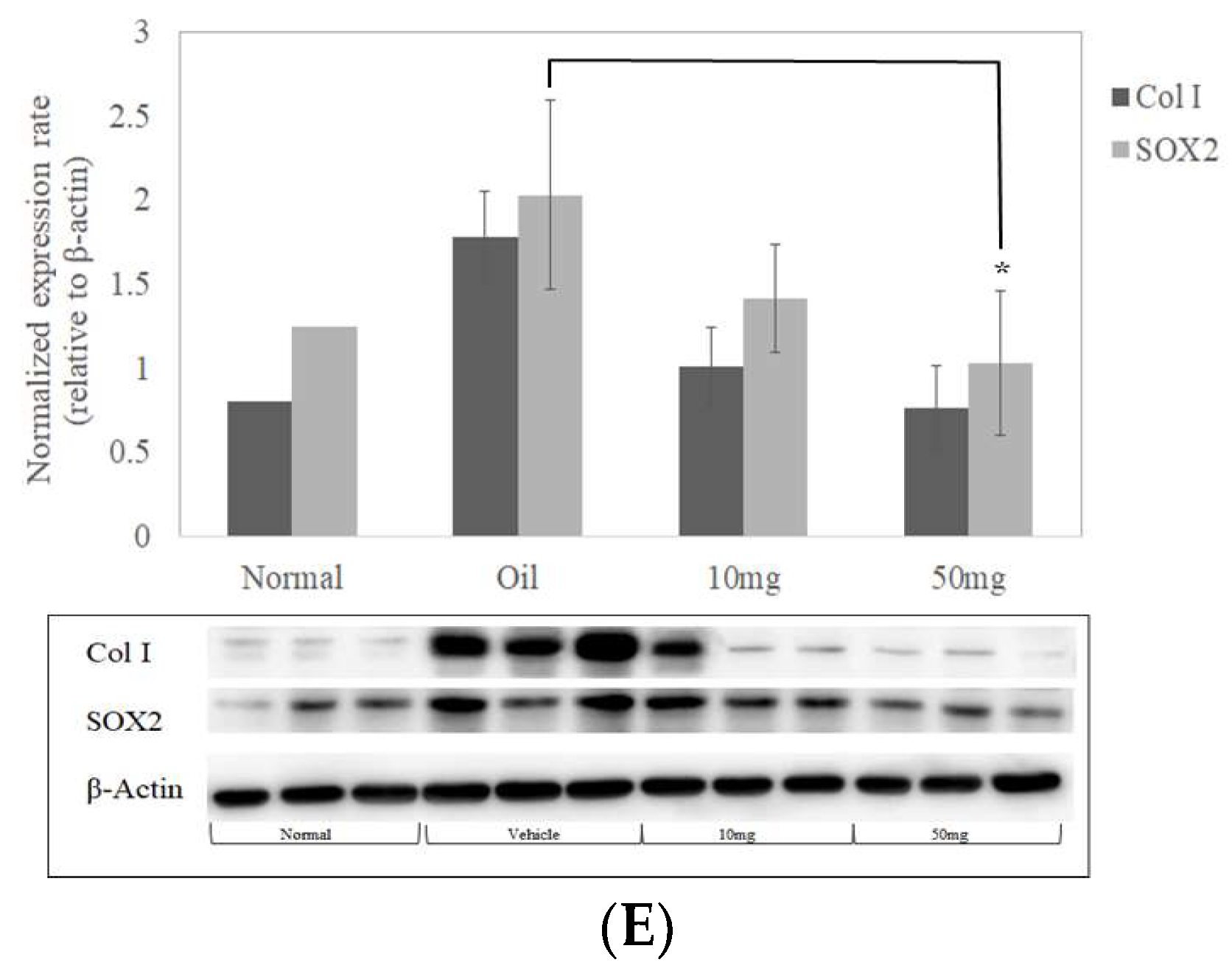
2.7. SOX2 and Collagen Expression Reduced in BP-Treated Lung Tissues
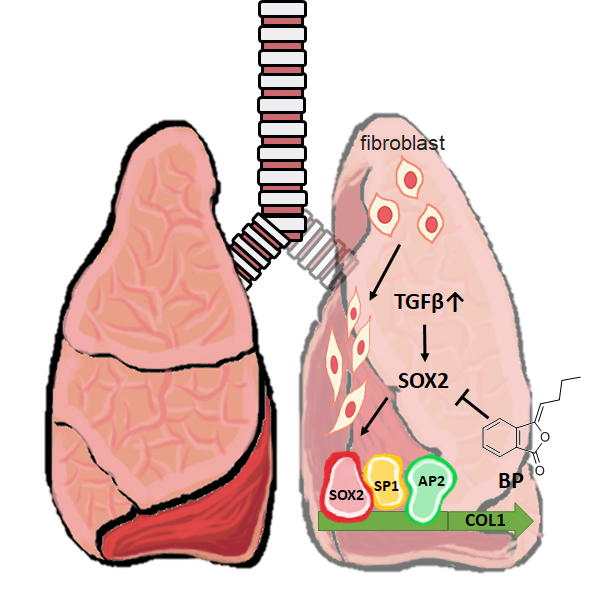
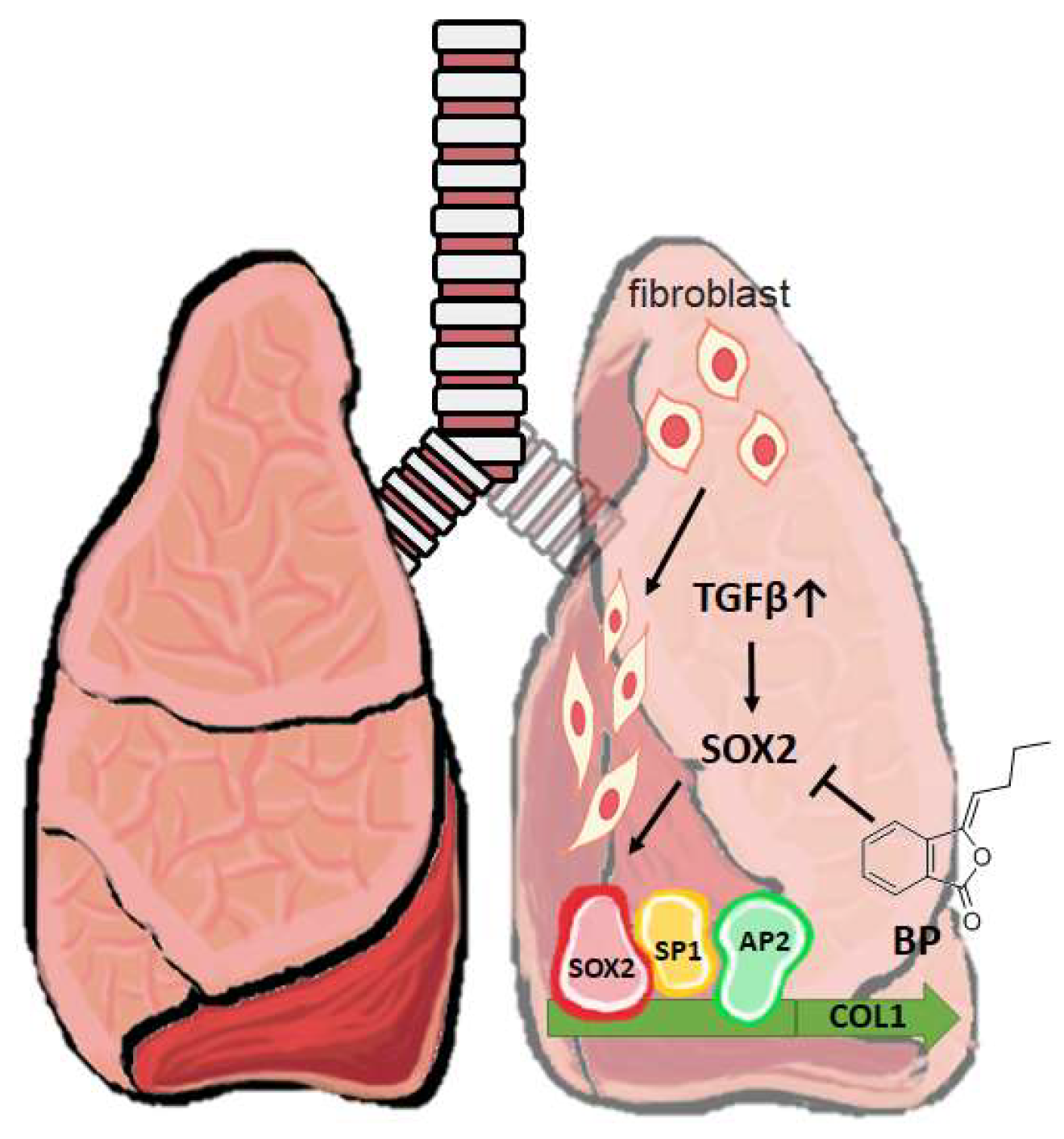
3. Discussion
4. Material and Methods
4.1. Chemical and Treatment
4.2. Cell Culture and Transfection
4.3. RT-PCR and Western Blot Analysis
4.4. Promoter Construct and Assay
4.5. Chromatin Immunoprecipitation Assay
4.6. Bleomycin-Induced Pulmonary Fibrosis and Tissue Collection
4.7. Pulmonary Function Test in Mice
4.8. Pathologic Morphology Staining and Evaluation
4.9. Statistical Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| Term | Definition |
| ECM | extracellular matrix |
| SOX2 | sex determining region Y (SRY)-box 2 |
| COL1 | type I collagen |
| COL1A1 | type I collagen α1 |
| TGF-β | transforming growth factor beta |
| BP | butylidenephthalide |
| NHLF | normal lung fibroblast cell lines |
| E-cad | E-cadherin |
| i.t. | intratracheal |
| WBP | whole body plethysmography |
| BAL | bronchoalveolar lavage |
References
- Raghu, G.; Collard, H.R.; Egan, J.J.; Martinez, F.J.; Behr, J.; Brown, K.K.; Colby, T.V.; Cordier, J.F.; Flaherty, K.R.; Lasky, J.A.; et al. An official ATS/ERS/JRS/ALAT statement: Idiopathic pulmonary fibrosis: Evidence-based guidelines for diagnosis and management. Am. J. Respir. Crit. Care Med. 2011, 183, 788–824. [Google Scholar] [CrossRef] [PubMed]
- Du Bois, R.M. An earlier and more confident diagnosis of idiopathic pulmonary fibrosis. Eur. Respir. Rev. 2012, 21, 141–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Datta, A.; Scotton, C.J.; Chambers, R.C. Novel therapeutic approaches for pulmonary fibrosis. Br. J. Pharmacol 2011, 163, 141–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, X.M.; Nikolic-Paterson, D.J.; Lan, H.Y. TGF-beta: The master regulator of fibrosis. Nat. Rev. Nephrol. 2016, 12, 325–338. [Google Scholar] [CrossRef] [PubMed]
- Cutroneo, K.R.; White, S.L.; Phan, S.H.; Ehrlich, H.P. Therapies for bleomycin induced lung fibrosis through regulation of TGF-beta1 induced collagen gene expression. J. Cell Physiol. 2007, 211, 585–589. [Google Scholar] [CrossRef] [PubMed]
- Willis, B.C.; Liebler, J.M.; Luby-Phelps, K.; Nicholson, A.G.; Crandall, E.D.; du Bois, R.M.; Borok, Z. Induction of epithelial-mesenchymal transition in alveolar epithelial cells by transforming growth factor-beta1: Potential role in idiopathic pulmonary fibrosis. Am. J. Pathol. 2005, 166, 1321–1332. [Google Scholar] [CrossRef]
- Palcy, S.; Bolivar, I.; Goltzman, D. Role of activator protein 1 transcriptional activity in the regulation of gene expression by transforming growth factor beta1 and bone morphogenetic protein 2 in ROS 17/2.8 osteoblast-like cells. J. Bone Miner. Res. 2000, 15, 2352–2361. [Google Scholar] [CrossRef] [PubMed]
- King, T.E., Jr.; Bradford, W.Z.; Castro-Bernardini, S.; Fagan, E.A.; Glaspole, I.; Glassberg, M.K.; Gorina, E.; Hopkins, P.M.; Kardatzke, D.; Lancaster, L.; et al. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N. Engl. J. Med. 2014, 370, 2083–2092. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.E. Non-Smad pathways in TGF-beta signaling. Cell Res. 2009, 19, 128–139. [Google Scholar] [CrossRef] [PubMed]
- Exposito, J.Y.; Valcourt, U.; Cluzel, C.; Lethias, C. The fibrillar collagen family. Int. J. Mol. Sci. 2010, 11, 407–426. [Google Scholar] [CrossRef] [PubMed]
- Kaarteenaho-Wiik, R.; Lammi, L.; Lakari, E.; Kinnula, V.L.; Risteli, J.; Ryhanen, L.; Paakko, P. Localization of precursor proteins and mRNA of type I and III collagens in usual interstitial pneumonia and sarcoidosis. J. Mol. Histol. 2005, 36, 437–446. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Herault, Y.; Pavlovic, G.; Leask, A. Skin progenitor cells contribute to bleomycin-induced skin fibrosis. Arthritis. Rheumatol. 2014, 66, 707–713. [Google Scholar] [CrossRef] [PubMed]
- Plantier, L.; Crestani, B.; Wert, S.E.; Dehoux, M.; Zweytick, B.; Guenther, A.; Whitsett, J.A. Ectopic respiratory epithelial cell differentiation in bronchiolised distal airspaces in idiopathic pulmonary fibrosis. Thorax 2011, 66, 651–657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basu-Roy, U.; Ambrosetti, D.; Favaro, R.; Nicolis, S.K.; Mansukhani, A.; Basilico, C. The transcription factor Sox2 is required for osteoblast self-renewal. Cell Death Differ. 2010, 17, 1345–1353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tompkins, D.H.; Besnard, V.; Lange, A.W.; Wert, S.E.; Keiser, A.R.; Smith, A.N.; Lang, R.; Whitsett, J.A. Sox2 is required for maintenance and differentiation of bronchiolar Clara, ciliated, and goblet cells. PLoS ONE 2009, 4, e8248. [Google Scholar] [CrossRef] [PubMed]
- Chuang, H.M.; Su, H.L.; Li, C.; Lin, S.Z.; Yen, S.Y.; Huang, M.H.; Ho, L.I.; Chiou, T.W.; Harn, H.J. The Role of Butylidenephthalide in Targeting the Microenvironment Which Contributes to Liver Fibrosis Amelioration. Front Pharmacol. 2016, 7, 112–125. [Google Scholar] [CrossRef] [PubMed]
- Yen, S.Y.; Chuang, H.M.; Huang, M.H.; Lin, S.Z.; Chiou, T.W.; Harn, H.J. n-Butylidenephthalide Regulated Tumor Stem Cell Genes EZH2/AXL and Reduced Its Migration and Invasion in Glioblastoma. Int. J. Mol. Sci. 2017, 18, 372. [Google Scholar] [CrossRef] [PubMed]
- Bonniaud, P.; Margetts, P.J.; Kolb, M.; Schroeder, J.A.; Kapoun, A.M.; Damm, D.; Murphy, A.; Chakravarty, S.; Dugar, S.; Higgins, L.; et al. Progressive transforming growth factor beta1-induced lung fibrosis is blocked by an orally active ALK5 kinase inhibitor. Am. J. Respir. Crit. Care Med. 2005, 171, 889–898. [Google Scholar] [CrossRef] [PubMed]
- Raghu, G.; Masta, S.; Meyers, D.; Narayanan, A.S. Collagen synthesis by normal and fibrotic human lung fibroblasts and the effect of transforming growth factor-beta. Am. Rev. Respir. Dis. 1989, 140, 95–100. [Google Scholar] [CrossRef] [PubMed]
- Leask, A.; Abraham, D.J. TGF-beta signaling and the fibrotic response. FASEB J. 2004, 18, 816–827. [Google Scholar] [CrossRef] [PubMed]
- Baarsma, H.A.; Engelbertink, L.H.; van Hees, L.J.; Menzen, M.H.; Meurs, H.; Timens, W.; Postma, D.S.; Kerstjens, H.A.; Gosens, R. Glycogen synthase kinase-3 (GSK-3) regulates TGF-beta(1)-induced differentiation of pulmonary fibroblasts. Br. J. Pharmacol. 2013, 169, 590–603. [Google Scholar] [CrossRef] [PubMed]
- Weina, K.; Wu, H.; Knappe, N.; Orouji, E.; Novak, D.; Bernhardt, M.; Huser, L.; Larribere, L.; Umansky, V.; Gebhardt, C.; et al. TGF-beta induces SOX2 expression in a time-dependent manner in human melanoma cells. Pigment Cell Melanoma Res. 2016, 29, 453–458. [Google Scholar] [CrossRef] [PubMed]
- Buttner, C.; Skupin, A.; Rieber, E.P. Transcriptional activation of the type I collagen genes COL1A1 and COL1A2 in fibroblasts by interleukin-4: Analysis of the functional collagen promoter sequences. J. Cell Physiol. 2004, 198, 248–258. [Google Scholar] [CrossRef] [PubMed]
- Kamachi, Y.; Kondoh, H. Sox proteins: Regulators of cell fate specification and differentiation. Development 2013, 140, 4129–4144. [Google Scholar] [CrossRef] [PubMed]
- Emad, A.; Emad, V. Elevated levels of MCP-1, MIP-alpha and MIP-1 beta in the bronchoalveolar lavage (BAL) fluid of patients with mustard gas-induced pulmonary fibrosis. Toxicology 2007, 240, 60–69. [Google Scholar] [CrossRef] [PubMed]
- Schafer, M.J.; White, T.A.; Iijima, K.; Haak, A.J.; Ligresti, G.; Atkinson, E.J.; Oberg, A.L.; Birch, J.; Salmonowicz, H.; Zhu, Y.; et al. Cellular senescence mediates fibrotic pulmonary disease. Nat. Commun. 2017, 8, 14532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wynn, T.A. Common and unique mechanisms regulate fibrosis in various fibroproliferative diseases. J. Clin. Investig. 2007, 117, 524–529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lodato, M.A.; Ng, C.W.; Wamstad, J.A.; Cheng, A.W.; Thai, K.K.; Fraenkel, E.; Jaenisch, R.; Boyer, L.A. SOX2 co-occupies distal enhancer elements with distinct POU factors in ESCs and NPCs to specify cell state. PLoS Genet. 2013, 9, e1003288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamanaka, R.B.; Nigdelioglu, R.; Meliton, A.Y.; Tian, Y.; Witt, L.J.; O’Leary, E.; Sun, K.A.; Woods, P.S.; Wu, D.; Ansbro, B.; et al. Inhibition of Phosphoglycerate Dehydrogenase Attenuates Bleomycin-induced Pulmonary Fibrosis. Am. J. Respir. Cell Mol. Biol. 2018, 58, 585–593. [Google Scholar] [CrossRef] [PubMed]
- Ganzert, S.; Moller, K.; Steinmann, D.; Schumann, S.; Guttmann, J. Pressure-dependent stress relaxation in acute respiratory distress syndrome and healthy lungs: An investigation based on a viscoelastic model. Crit. Care 2009, 13, R199. [Google Scholar] [CrossRef] [PubMed]
- Hamelmann, E.; Schwarze, J.; Takeda, K.; Oshiba, A.; Larsen, G.L.; Irvin, C.G.; Gelfand, E.W. Noninvasive measurement of airway responsiveness in allergic mice using barometric plethysmography. Am. J. Respir. Crit. Care Med. 1997, 156, 766–775. [Google Scholar] [CrossRef] [PubMed]
- Swaney, J.S.; Chapman, C.; Correa, L.D.; Stebbins, K.J.; Bundey, R.A.; Prodanovich, P.C.; Fagan, P.; Baccei, C.S.; Santini, A.M.; Hutchinson, J.H.; et al. A novel, orally active LPA(1) receptor antagonist inhibits lung fibrosis in the mouse bleomycin model. Br. J. Pharmacol 2010, 160, 1699–1713. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.; Kasam, R.K.; Sontake, V.; Wynn, T.A.; Madala, S.K. Repetitive intradermal bleomycin injections evoke T-helper cell 2 cytokine-driven pulmonary fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2017, 313, L796–L806. [Google Scholar] [CrossRef] [PubMed]
- Raghu, G.; Anstrom, K.J.; King, T.E., Jr.; Lasky, J.A.; Martinez, F.J. Prednisone, azathioprine, and N-acetylcysteine for pulmonary fibrosis. N. Engl. J. Med. 2012, 366, 1968–1977. [Google Scholar] [PubMed]
- Chiou, G.Y.; Chien, C.S.; Wang, M.L.; Chen, M.T.; Yang, Y.P.; Yu, Y.L.; Chien, Y.; Chang, Y.C.; Shen, C.C.; Chio, C.C.; et al. Epigenetic regulation of the miR142-3p/interleukin-6 circuit in glioblastoma. Mol. Cell 2013, 52, 693–706. [Google Scholar] [CrossRef] [PubMed]
- Tsai, N.M.; Chen, Y.L.; Lee, C.C.; Lin, P.C.; Cheng, Y.L.; Chang, W.L.; Lin, S.Z.; Harn, H.J. The natural compound n-butylidenephthalide derived from Angelica sinensis inhibits malignant brain tumor growth in vitro and in vivo. J. Neurochem. 2006, 99, 1251–1262. [Google Scholar] [CrossRef] [PubMed]
- Ashcroft, T.; Simpson, J.M.; Timbrell, V. Simple method of estimating severity of pulmonary fibrosis on a numerical scale. J. Clin. Pathol. 1988, 41, 467–470. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variable | Normal | 10 mg/kg | 50 mg/kg | Vehicle |
|---|---|---|---|---|
| Frequency (breaths/m) | 452.5 ± 2.8 | 447.4 ± 75.4 | 460.1 ± 27.1 | 393.1 ± 41 |
| Tidal volume (mL) | 0.046 ± 0.001 | 0.046 ± 0.002 | 0.046 ± 0.001 | 0.048 ± 0.002 |
| Accumulated volume (mL) | 247.23 ± 201.77 | 141.03 ± 99.02 * | 288.96 ± 7.5 * | 124.42 ± 48.14 |
| Minute volume (mL/m) | 20.51 ± 0.06 | 19.94 ± 2.62 * | 20.11 ± 2.59 * | 18.08 ± 1.36 |
| Inspiratory time (s) | 0.0569 ± 0.0023 | 0.0623 ± 0.0086 | 0.0587 ± 0.008 | 0.0587 ± 0.0037 |
| Expiratory time (s) | 0.092 ± 0.003 | 0.101 ± 0.029 | 0.096 ± 0.027 | 0.078 ± 0.041 |
| Peak inspiratory (mL/s) | 1.313 ± 0.008 | 1.271 ± 0.076 | 1.269 ± 0.139 | 1.361 ± 0.048 |
| Peak expiratory (mL/s) | 1.103 ± 0.008 | 1.046 ± 0.07 | 1.037 ± 0.066 | 0.848 ± 0.411 |
| Relaxation time (s) | 0.0598 ± 0.0043 | 0.0586 ± 0.0168 * | 0.0501 ± 0.0106 * | 0.0815 ± 0.0075 |
| End inspiratory pause (ms) | 5.514 ± 1.089 | 5.296 ± 0.916 | 4.955 ± 0.377 | 6.047 ± 1.293 |
| End expiratory pause (ms) | 7.1129 ± 2.339 | 21.35 ± 24.67 | 14.08 ± 9.7 * | 37.9 ± 16.21 |
| Enhanced pause (Penh) | 0.62 ± 0.002 | 0.608 ± 0.068 * | 0.582 ± 0.105 * | 0.742 ± 0.091 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chuang, H.-M.; Ho, L.-I.; Huang, M.-H.; Huang, K.-L.; Chiou, T.-W.; Lin, S.-Z.; Su, H.-L.; Harn, H.-J. Non-Canonical Regulation of Type I Collagen through Promoter Binding of SOX2 and Its Contribution to Ameliorating Pulmonary Fibrosis by Butylidenephthalide. Int. J. Mol. Sci. 2018, 19, 3024. https://doi.org/10.3390/ijms19103024
Chuang H-M, Ho L-I, Huang M-H, Huang K-L, Chiou T-W, Lin S-Z, Su H-L, Harn H-J. Non-Canonical Regulation of Type I Collagen through Promoter Binding of SOX2 and Its Contribution to Ameliorating Pulmonary Fibrosis by Butylidenephthalide. International Journal of Molecular Sciences. 2018; 19(10):3024. https://doi.org/10.3390/ijms19103024
Chicago/Turabian StyleChuang, Hong-Meng, Li-Ing Ho, Mao-Hsuan Huang, Kun-Lun Huang, Tzyy-Wen Chiou, Shinn-Zong Lin, Hong-Lin Su, and Horng-Jyh Harn. 2018. "Non-Canonical Regulation of Type I Collagen through Promoter Binding of SOX2 and Its Contribution to Ameliorating Pulmonary Fibrosis by Butylidenephthalide" International Journal of Molecular Sciences 19, no. 10: 3024. https://doi.org/10.3390/ijms19103024