Differentially Expressed Genes Associated with the Cabbage Yellow-Green-Leaf Mutant in the ygl-1 Mapping Interval with Recombination Suppression

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials
2.2. Identification of Recombination Suppression in the ygl-1 Gene-Mapping Interval
2.3. BSA, RNA Isolation, and Library Construction
3. Data Analysis
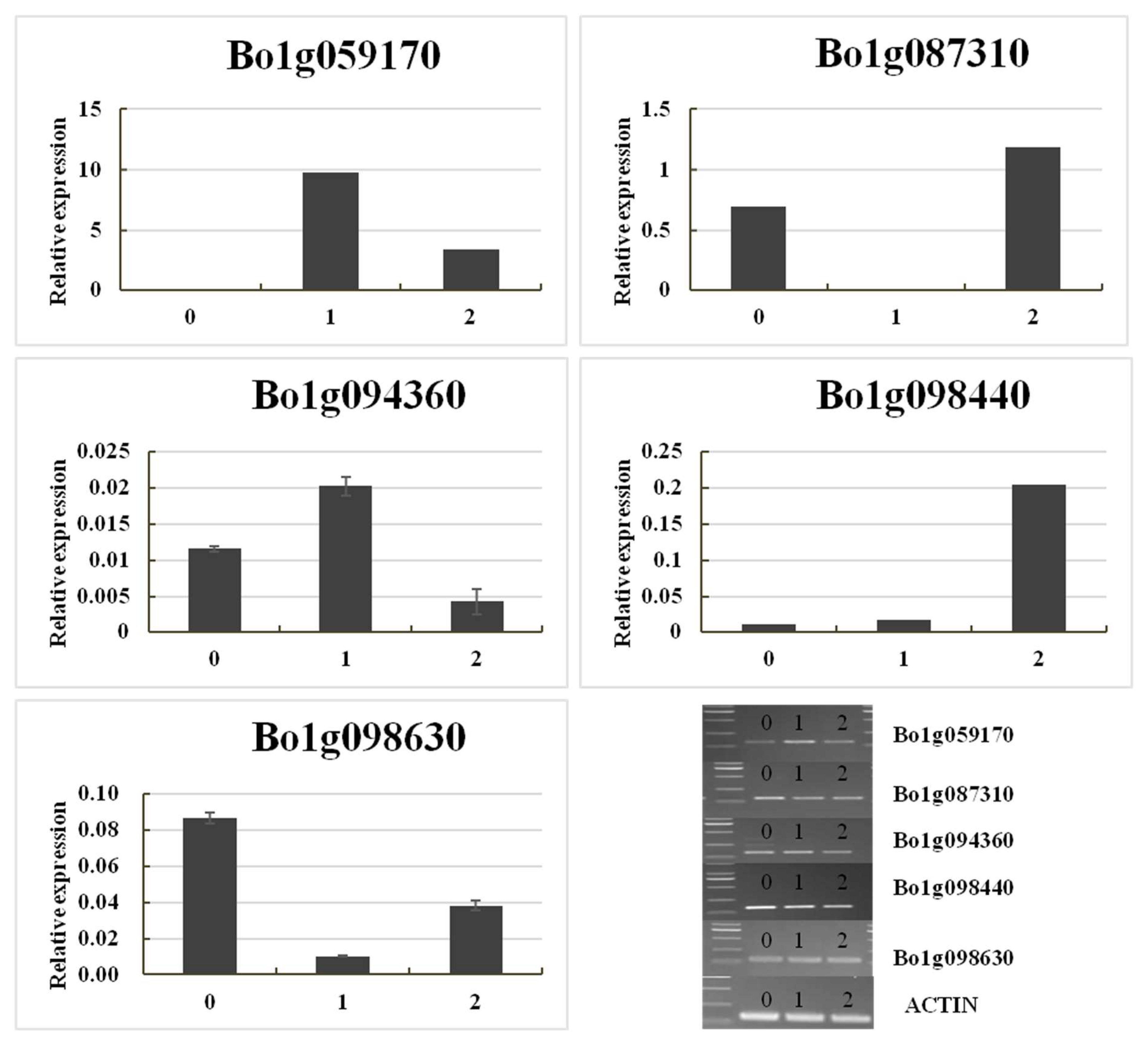
Gene Expression Validation
4. Results
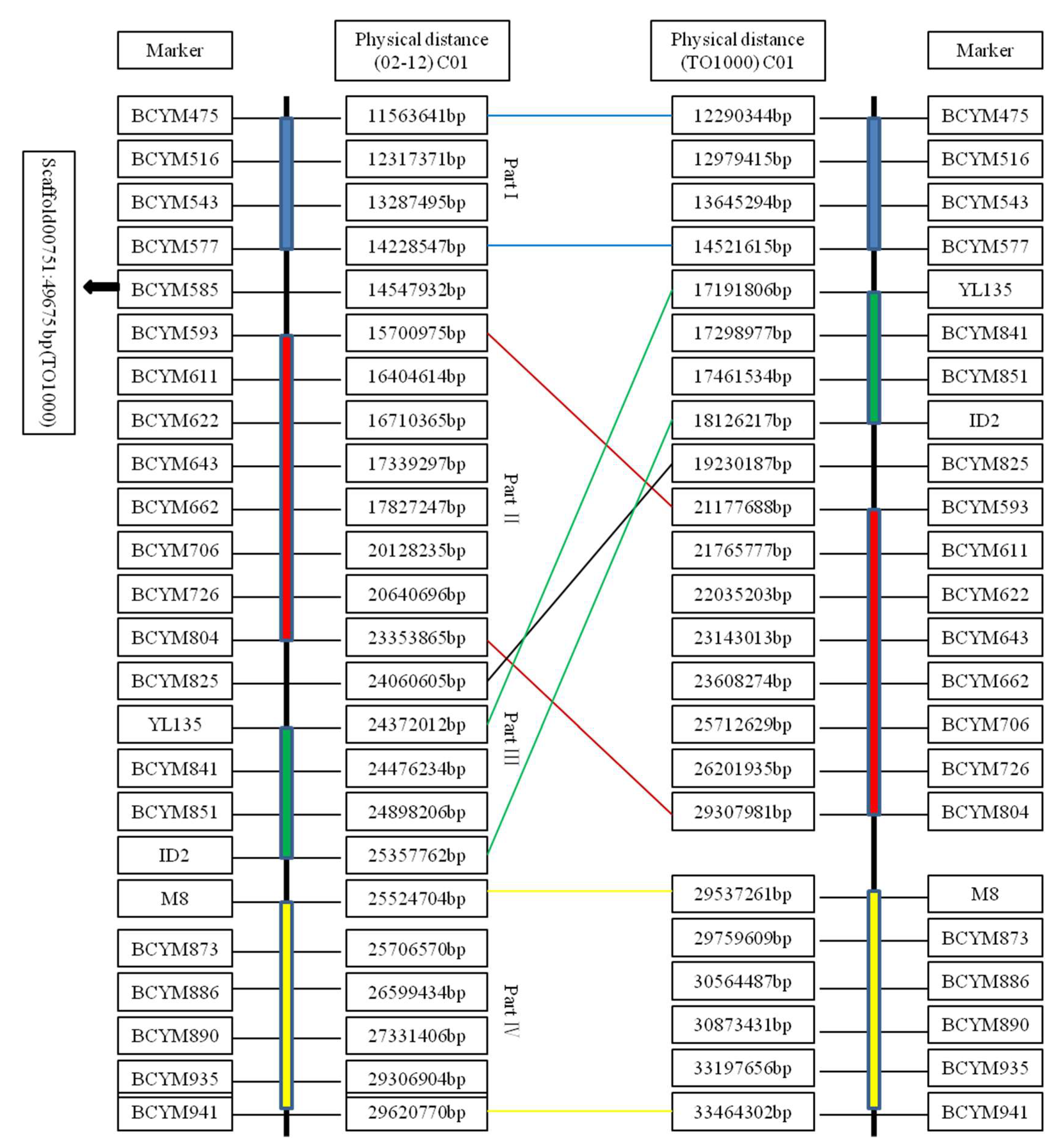
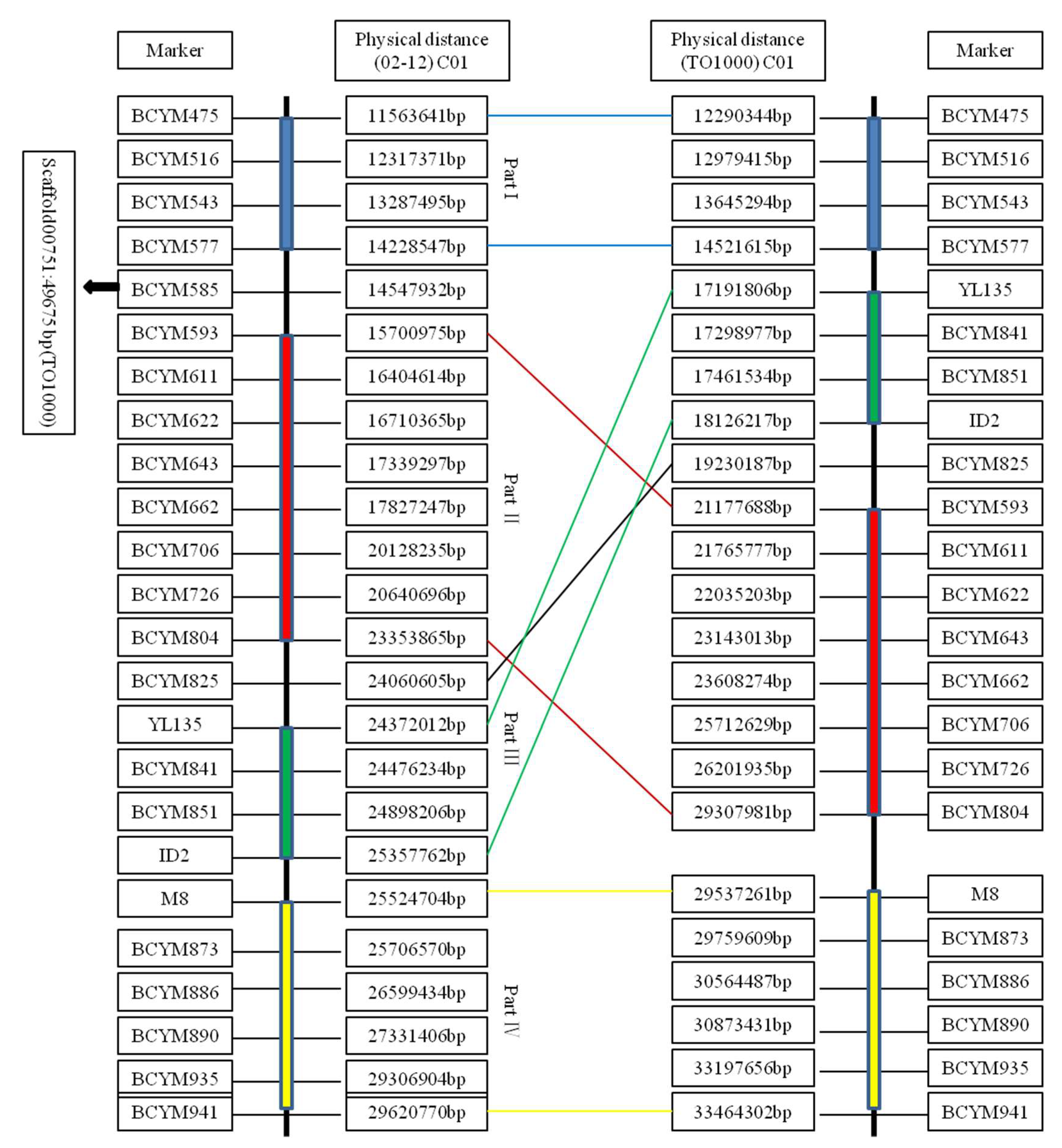
4.1. Identification of the Recombination-Suppressed Region
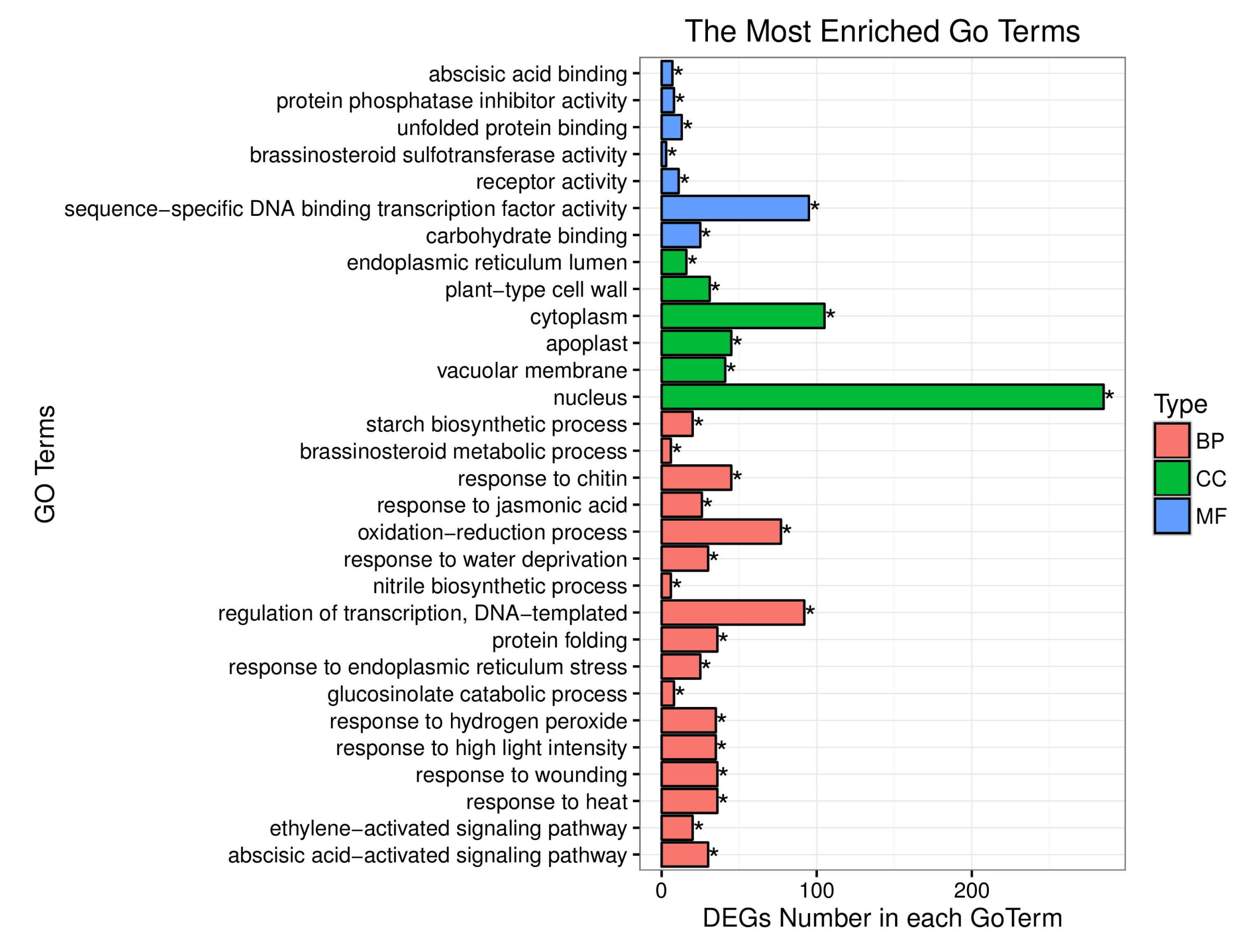
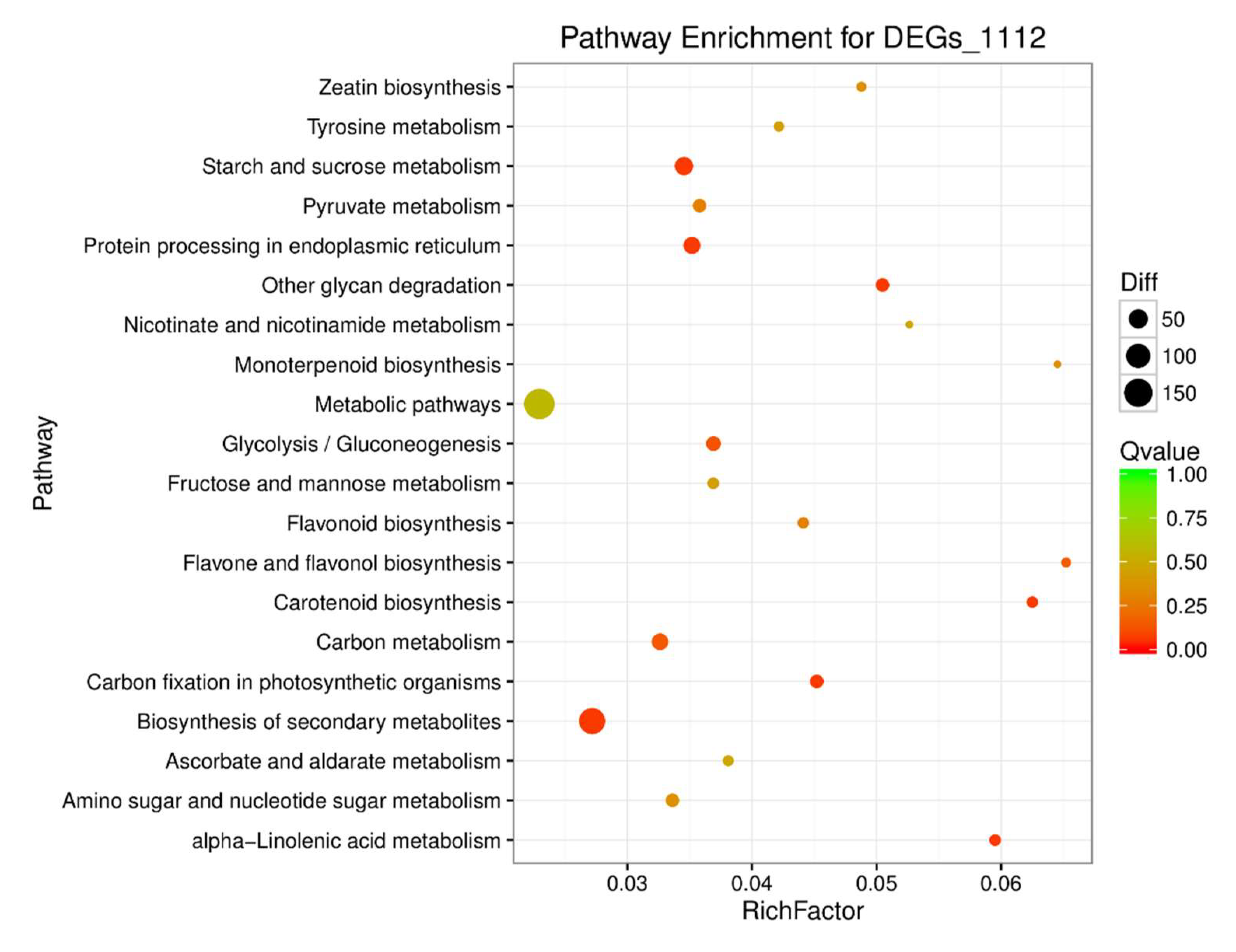
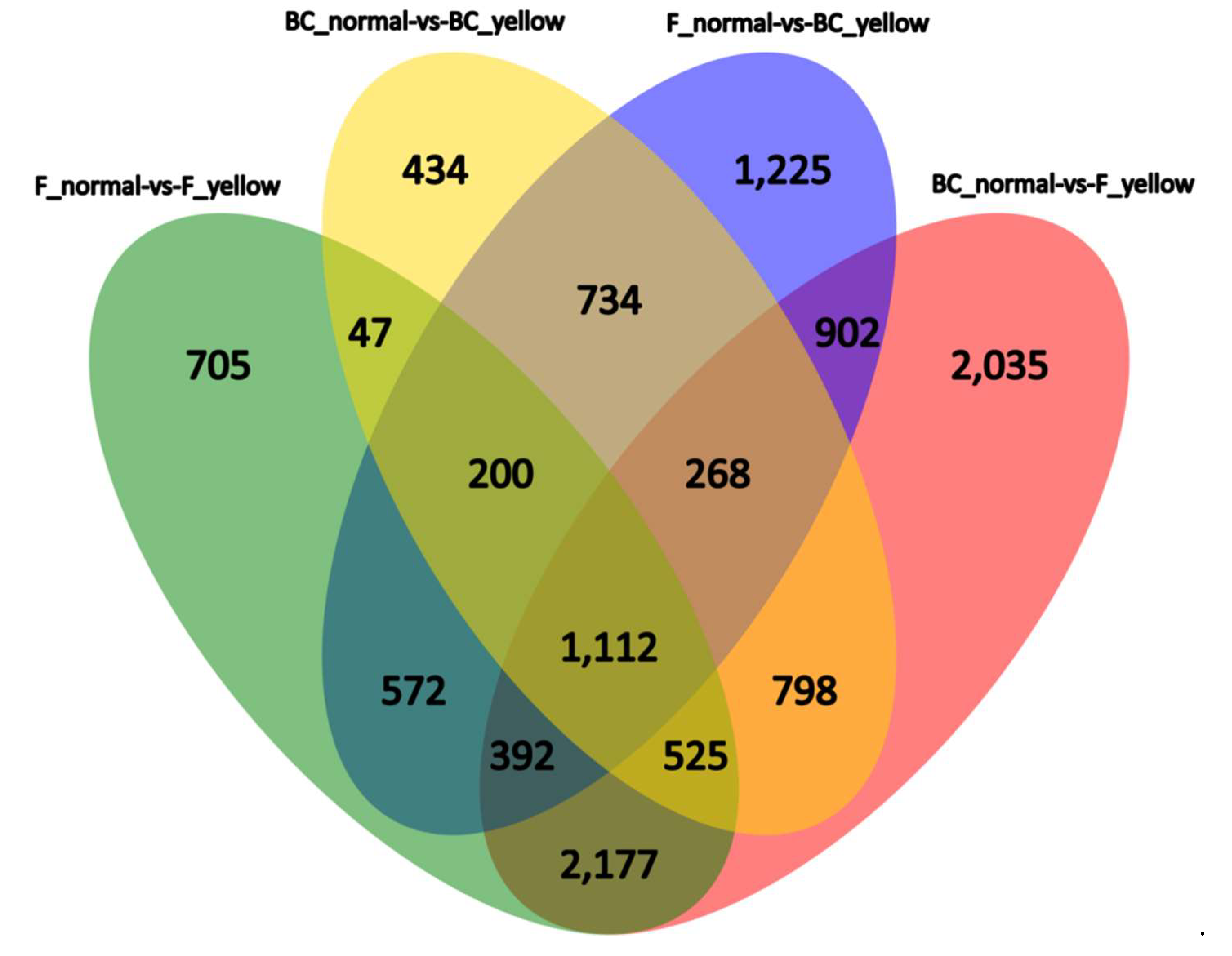
4.2. BSR Analysis, DEGs between the Yellow-Green-Leaf and Normal-Leaf Pools
4.3. DEGs Involved in B. oleracea Chlorophyll Synthesis
5. Discussion
5.1. Efficiency of BSR in DEG Detection
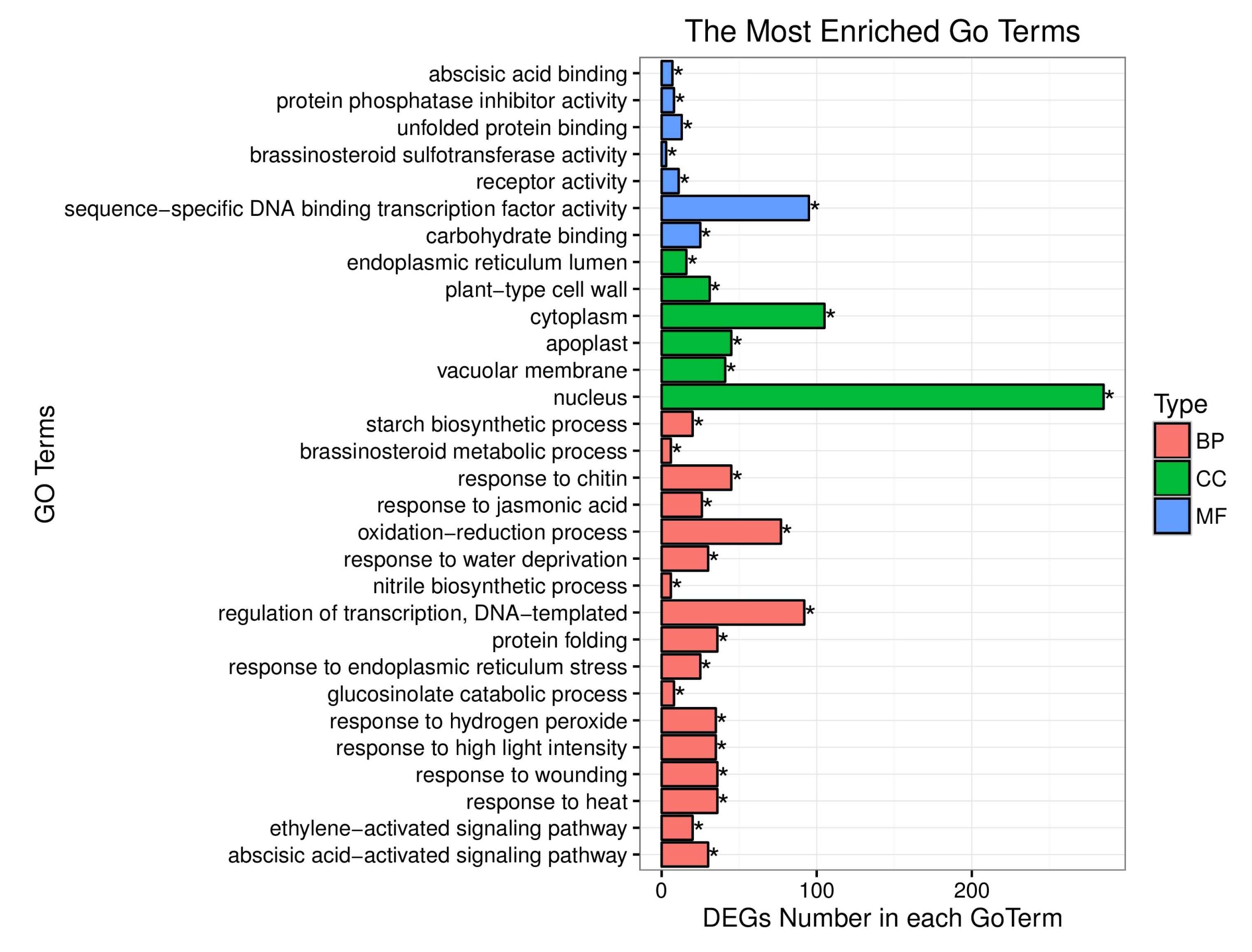
5.2. DEGs Analysis Associated with the Yellow-Green-Leaf in a Recombination-Suppressed Region via RNA-Seq
5.3. Assembly Error in the Reference Genome
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Carol, P.; Stevenson, D. Mutations in the Arabidopsis gene IMMUTANS cause a variegated phenotype by inactivating a chloroplast terminal oxidase assoeiated with phytoene desaturation. Plant Cell. 1999, 11, 57–68. [Google Scholar] [CrossRef] [PubMed]
- Svensson, J.T.; Crosatti, C. Transcriptome analysis of cold acclimation in barley albina and xantha mutants. Plant Physiol. 2006, 141, 257–270. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Zeng, X. Genetic characterisation and fine mapping of a chlorophyll-deficient mutant (BnaC.ygl) in Brassica napus. Mol. Breed. 2014, 34, 603–614. [Google Scholar] [CrossRef]
- Chen, H.; Cheng, Z. A knockdown mutation of YELLOW—GREEN LEAF2, blocks chlorophyll biosynthesis in rice. Plant Cell Rep. 2013, 32, 1855–1867. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Hu, Y. Mutation of FdC2 gene encoding a ferredoxin-like protein with C-terminal extension causes yellow-green-leaf phenotype in rice. Plant Sci. 2015, 238, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Sun, X. Map-based cloning and characterization of the novel yellow-green-leaf gene ys83 in rice (Oryza sativa). Plant Physiol. Biochem. 2016, 111, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Yang, C. Genetics and fine mapping of a yellow-green-leaf gene (ygl-1) in cabbage (Brassica oleracea var. capitata L.). Mol. Breed. 2016, 36, 1–8. [Google Scholar] [CrossRef]
- Whitaker, T.W. Genetic and Chlorophyll Studies of a Yellow-Green Mutant in Muskmelon. Plant Physiol. 1952, 27, 263–268. [Google Scholar] [CrossRef] [PubMed]
- Zhong, X.M.; Sun, S.F. Research on photosynthetic physiology of a yellow-green mutant line in maize. Photosynthetica 2015, 53, 1–8. [Google Scholar] [CrossRef]
- Yang, C.; Zhang, Y.Y. Photosynthetic Physiological Characteristics and Chloroplast Ultrastructure of Yellow Leaf Mutant YL-1 in Cabbage. Acta Hortic. Sin. 2014, 41, 1133–1144. [Google Scholar]
- Ladygin, V.G. Spectral features and structure of chloroplasts under an early block of chlorophyll synthesis. Biophysics 2006, 51, 635–644. [Google Scholar] [CrossRef]
- Runge, S.; Cleve, B.V.; Lebedev, N.; Armstrong, G.; Apel, K. Isolation and classification of chlorophyll-deficient xantha mutants of Arabidopsis thaliana. Planta 1995, 197, 490–500. [Google Scholar] [CrossRef] [PubMed]
- Chantret, N.; Sourdille, P. Location and mapping of the powdery mildew resistance gene MIRE and detection of a resistance QTL by bulked segregant analysis (BSA) with microsatellites in wheat. Theor. Appl. Genet. 2000, 100, 1217–1224. [Google Scholar] [CrossRef]
- Futschik, A.; Schlötterer, C. The next generation of molecular markers from massively parallel sequencing of pooled DNA samples. Genetics 2010, 186, 207–218. [Google Scholar] [CrossRef] [PubMed]
- Zeng, F.; Yi, B.; Tu, J.; Fu, T. Identification of AFLP and SCAR markers linked to the male fertility restorer gene of pol, CMS (Brassica napus L.). Euphytica 2009, 165, 363–369. [Google Scholar] [CrossRef]
- Wang, Y.; Thomas, C.E.; Dean, R.A. Genetic mapping of a Fusarium wilt resistance gene (Fom-2) in melon (Cucumis melo L.). Mol. Breed. 2000, 6, 379–389. [Google Scholar] [CrossRef]
- Subudhi, P.K.; Borkakati, R.P.; Virmani, S.S.; Huang, N. Molecular mapping of a thermosensitive genetic male sterility gene in rice using bulked segregant analysis. Genome 1997, 40, 188–194. [Google Scholar] [CrossRef] [PubMed]
- Cheema, K.K.; Grewal, N.K. A novel bacterial blight resistance gene from Oryza nivara mapped to 38 kb region on chromosome 4L and transferred to Oryza sativa L. Gen. Res. 2008, 90, 397–407. [Google Scholar] [CrossRef] [PubMed]
- Mackay, I.J.; Caligari, P.D. Efficiencies of F2 and backcross generations for bulked segregant analysis using dominant markers. Crop Sci. 2000, 40, 626–630. [Google Scholar] [CrossRef]
- Zhang, G.; Guo, G.W. Deep RNA sequencing at single base-pair resolution reveals high complexity of the rice transcriptome. Genome Res. 2010, 20, 646–654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, H.K.; Hong, S.E. Deep RNA Sequencing Reveals Novel Cardiac Transcriptomic Signatures for Physiological and Pathological Hypertrophy. PLoS ONE 2012, 7, e35552. [Google Scholar] [CrossRef] [PubMed]
- Jarvie, T.; Harkins, T. Transcriptome sequencing with the Genome Sequencer FLX system. Nat. Methods 2008, 5. [Google Scholar] [CrossRef]
- Wang, R.; Sun, L. Bulk segregant RNA-seq reveals expression and positional candidate genes and allele-specific expression for disease resistance against enteric septicemia of catfish. BMC Genomics 2013, 14, 929–939. [Google Scholar] [CrossRef] [PubMed]
- Ramirez-Gonzalez, R.H.; Segovia, V. RNA-Seq bulked segregant analysis enables the identification of high-resolution genetic markers for breeding in hexaploid wheat. Plant Biotech. J. 2014, 13, 613–624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parkin, I.A.; Koh, C. Transcriptome and methylome profiling reveals relics of genome dominance in the mesopolyploid Brassica oleracea. Genome Biol. 2014, 15, R77. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef] [PubMed]
- Krzywinski, M.; Schein, J. Circos: An information aesthetic for comparative genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashburner, M.; Ashburner, M. Gene ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S. The KEGG resource for deciphering the genome. Nucleic Acids Res. 2004, 32, D277–D280. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Zhang, Y. Transcriptome sequencing and de novo analysis of a recessive genic male sterile line in cabbage (Brassica oleracea L. var. capitata). Mol. Breed. 2016, 36, 117–119. [Google Scholar] [CrossRef]
- Piippo, M.; Allahverdiyeva, Y. Chloroplast-mediated regulation of nuclear genes in Arabidopsis thaliana in the absence of light stress. Physiol. Gen. 2006, 25, 142–152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sohani, M.M.; Schenk, P.M.; Schultz, C.J.; Schmidt, O. Phylogenetic and transcriptional analysis of a strictosidine synthase-like gene family in Arabidopsis thaliana, reveals involvement in plant defence responses. Plant Biol. 2009, 11, 105–117. [Google Scholar] [CrossRef] [PubMed]
- Jyothi, T.; Duan, H.; Liu, L.; Schuler, M.A. Bicistronic and fused monocistronic transcripts are derived from adjacent loci in the Arabidopsis genome. RNA 2005, 11, 128–138. [Google Scholar] [Green Version]
- Kim, S.; Kim, C.W.; Park, M.; Choi, D. Identification of candidate genes associated with fertility restoration of cytoplasmic male-sterility in onion (Allium cepa L.) using a combination of bulked segregant analysis and RNA-seq. Theor. Appl. Genet. 2015, 128, 2289–2299. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Zhou, Q.; Dong, L.; Wang, H.; Liu, F.; Weng, J.; Li, X.; Xie, C. Genetic architecture of the maize kernel row number revealed by combining QTL mapping using a high-density genetic map and bulked segregant RNA sequencing. BMC Genomics 2016, 17, 915. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Peng, G.; Liu, X.; Deora, A.; Falk, K.C.; Gossen, B.D.; McDonald, M.R.; Yu, F. Fine Mapping of a Clubroot Resistance Gene in Chinese Cabbage Using SNP Markers Identified from Bulked Segregant RNA Sequencing. Front. Plant Sci. 2017, 8, 1448–1459. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xie, J.; Zhang, H. Mapping stripe rust resistance gene YrZH22, in Chinese wheat cultivar Zhoumai 22 by bulked segregant RNA-Seq (BSR-Seq) and comparative genomics analyses. Theor. Appl. Genet. 2017, 130, 2191–2201. [Google Scholar] [CrossRef] [PubMed]
- Trick, M.; Adamski, N.M.; Mugford, S.G.; Jiang, C.C.; Febrer, M.; Uauy, C. Combining SNP discovery from next-generation sequencing data with bulked segregant analysis (BSA) to fine-map genes in polyploid wheat. BMC Plant Biol. 2012, 12, 14–19. [Google Scholar] [CrossRef] [PubMed]
- Lei, S.; Yao, X. Towards map-based cloning: Fine mapping of a recessive genic male-sterile gene (BnMs2) in Brassica napus L. and syntenic region identification based on the Arabidopsis thaliana genome sequences. Theor. Appl. Genet. 2007, 115, 643–651. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, M.; Pu, Z.J. Map-based cloning of a candidate gene conferring Fusarium yellows resistance in Brassica oleracea. Theor. Appl. Genet. 2015, 128, 119–130. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Ma, Y. Map-based cloning of the dominant genic male sterile Ms-cd1 gene in cabbage (Brassica oleracea). Theor. Appl. Genet. 2016, 5, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Huang, Q. Inheritance of chlorophyll-deficient mutant L638-y in Brassica juncea L. and molecular markers for chlorophyll deficient gene gr1. J. Northwest A F Univ. 2012, 12, 17–19. [Google Scholar]
- Wang, Y.; He, Y. Fine mapping of a dominant gene conferring chlorophyll-deficiency in Brassica napus. Sci Rep. 2016, 6, 314–319. [Google Scholar] [CrossRef] [PubMed]
- Sherman, J.D.; Stack, S.M. Two-dimensional spreads of synaptonemal complexes from Solanaceous plants. VI. Highresolution recombination nodule map for tomato (Lycopersicon esculentum). Genetics 1995, 141, 683–708. [Google Scholar] [PubMed]
- Wei, F.; Gobelman-Werner, K. The Mla (powdery mildew) resistance cluster is associated with three NBS-LRR gene families and suppressed recombination within a 240-kb DNA interval on chromosome 5S (1HS) of barley. Genetics 1999, 153, 1929–1948. [Google Scholar] [PubMed]
- Ten, H.R.; Robbins, T.P. Localization of T-DNA Insertions in Petunia by Fluorescence in Situ Hybridization: Physical Evidence for Suppression of Recombination. Plant Cell 1996, 8, 823–830. [Google Scholar] [Green Version]
- Stirling, B.; Newcombe, G. Suppressed recombination around the MXC3 locus, a major gene for resistance to poplar leaf rust. Theor. Appl. Genet. 2001, 103, 1129–1137. [Google Scholar] [CrossRef]
- Neu, C.; Stein, N.; Keller, B. Genetic mapping of the Lr20-Pm1 resistance locus reveals suppressed recombination on chromosome arm 7AL in hexaploid wheat. Genome 2002, 45, 737–744. [Google Scholar] [CrossRef] [PubMed]
- Jessup, R.W.; Burson, B.L. Disomic Inheritance, Suppressed Recombination, and Allelic Interactions Govern Apospory in Buffelgrass as Revealed by Genome Mapping. Crop Sci. 2002, 42, 1688–1694. [Google Scholar] [CrossRef]
- Lee, J.; Izzah, N.K. Genotyping-by-sequencing map permits identification of clubroot resistance QTLs and revision of the reference genome assembly in cabbage (Brassica oleracea L.). DNA Res. 2015, 14, S113. [Google Scholar] [CrossRef] [PubMed]
- Lv, H.; Wang, Q. Whole-Genome Mapping Reveals Novel QTL Clusters Associated with Main Agronomic Traits of Cabbage (Brassica oleracea var. capitata L.). Front. Plant Sci. 2016, 7, 989–999. [Google Scholar] [CrossRef] [PubMed]
- Schweiger, W.; Schweiger, W.; Steiner, B.; Vautrin, S.; Nussbaumer, T.; Siegwart, G.; Zamini, M.; Jungreithmeier, F.; Gratl, V.; Lemmens, M.; et al. Suppressed recombination and unique candidate genes in the divergent haplotype encoding Fhb1, a major Fusarium head blight resistance locus in wheat. Theor. Appl. Genet. 2016, 129, 1607–1623. [Google Scholar] [CrossRef] [PubMed]
- Lv, W.; Du, B. BAC and RNA sequencing reveal the brown planthopper resistance gene BPH15, in a recombination cold spot that mediates a unique defense mechanism. BMC Genomics 2014, 15, 674–679. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Liu, Y.; Yang, X.; Tong, C.; Edwards, D.; Parkin, I.A. The Brassica oleracea genome reveals the asymmetrical evolution of polyploid genomes. Nat. Commun. 2014. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.P.; Gao, B.Z.; Han, F.Q.; Fang, Z.Y.; Yang, L.M.; Zhuang, M.; Lv, H.H.; Liu, Y.M.; Li, Z.S.; Cai, C.C.; et al. Genetics and fine mapping of a purple leaf gene, BoPr, in ornamental kale (Brassica oleracea L. var. acephala). BMC Genomics 2017, 18, 230–239. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| YL-1 × 01-20 | YL-1 × 11-192 | ||
|---|---|---|---|
| Primers | Genetic Distance (cM) | Primers | Genetic Distance (cM) |
| T2-3 | 9.21 | T2-3 | 13.3 |
| T2-5 | 6.90 | - | - |
| T1-1 | 6.28 | - | - |
| T1-14 | 4.39 | T1-14 | 6.5 |
| T1-18 | 3.97 | T1-18 | 4.4 |
| T1-26 | 2.51 | T1-26 | 2.3 |
| T1-28 | 1.46 | T1-28 | 1.5 |
| T1-30 | 1.05 | T1-30 | 1.3 |
| T1-34 | 0.63 | T1-34 | 0.3 |
| T1-36 | 0.42 | T1-36 | 0.00 |
| T1-58 | 0.42 | T1-58 | 0.7 |
| T2-6 | 0.42 | T2-6 | 1.04 |
| T2-10 | 0.63 | T2-10 | 1.04 |
| T2-14 | 0.63 | T2-14 | 1.04 |
| T2-16 | 3.14 | T2-16 | 2.61 |
| T2-18 | 5.02 | T2-18 | 6.02 |
| No. of DEGs | No. of Up-Regulated DEGs | Percentage (%) | No. of Down-Regulated DEGs | Percentage (%) | |
|---|---|---|---|---|---|
| BC_normal vs. BC_yellow | 4118 | 2384 | 58 | 1734 | 42 |
| BC_normal vs. F_yellow | 8009 | 4894 | 60 | 3315 | 40 |
| F_normal vs. F_yellow | 5730 | 3226 | 56 | 2504 | 44 |
| F_normal vs. BC_yellow | 5405 | 2844 | 53 | 2561 | 47 |
| Gene ID a | Physical Distance (TO1000) | F Normal b | F_Yellow b | BC_Normal b | BC_Yellow b | Diff c | A.T. Annotation d |
|---|---|---|---|---|---|---|---|
| Bo1g087310 | C1:25381300–25383803 | 1837.98 | 156.85 | 1920.64 | 287.42 | Down | Calreticulins-1, response to oxidative stress, response to cadmiumion, response to salt stress, calciumion homeostasis; |
| Bo1g094360 | C1:27829353-27834745 | 48.65 | 10.53 | 29.70 | 2.04 | Down | d-alanine-d-alanine ligase activity |
| Bo1g098630 | C1:29261755-29263303 | 4002.89 | 475.81 | 1119.81 | 125.36 | Down | GPT2: glucose-6-phosphate/phosphate translocator 2 |
| Bo1g059170 | C1:18110687-18112080 | 167.35 | 828.45 | 277.80 | 858.19 | Up | SSL4: strictosidine synthase-like 4 |
| Bo1g098440 | C1:29037892-29038492 | 129.41 | 285.27 | 120.70 | 427.05 | Up | Protein of unknown function, DUF538 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, X.; Yu, H.; Han, F.; Li, Z.; Fang, Z.; Yang, L.; Zhuang, M.; Lv, H.; Liu, Y.; Li, Z.; et al. Differentially Expressed Genes Associated with the Cabbage Yellow-Green-Leaf Mutant in the ygl-1 Mapping Interval with Recombination Suppression. Int. J. Mol. Sci. 2018, 19, 2936. https://doi.org/10.3390/ijms19102936
Liu X, Yu H, Han F, Li Z, Fang Z, Yang L, Zhuang M, Lv H, Liu Y, Li Z, et al. Differentially Expressed Genes Associated with the Cabbage Yellow-Green-Leaf Mutant in the ygl-1 Mapping Interval with Recombination Suppression. International Journal of Molecular Sciences. 2018; 19(10):2936. https://doi.org/10.3390/ijms19102936
Chicago/Turabian StyleLiu, Xiaoping, Hailong Yu, Fengqing Han, Zhiyuan Li, Zhiyuan Fang, Limei Yang, Mu Zhuang, Honghao Lv, Yumei Liu, Zhansheng Li, and et al. 2018. "Differentially Expressed Genes Associated with the Cabbage Yellow-Green-Leaf Mutant in the ygl-1 Mapping Interval with Recombination Suppression" International Journal of Molecular Sciences 19, no. 10: 2936. https://doi.org/10.3390/ijms19102936