Role of KCa3.1 Channels in Modulating Ca2+ Oscillations during Glioblastoma Cell Migration and Invasion

Abstract
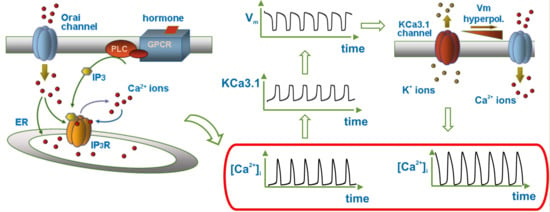
:
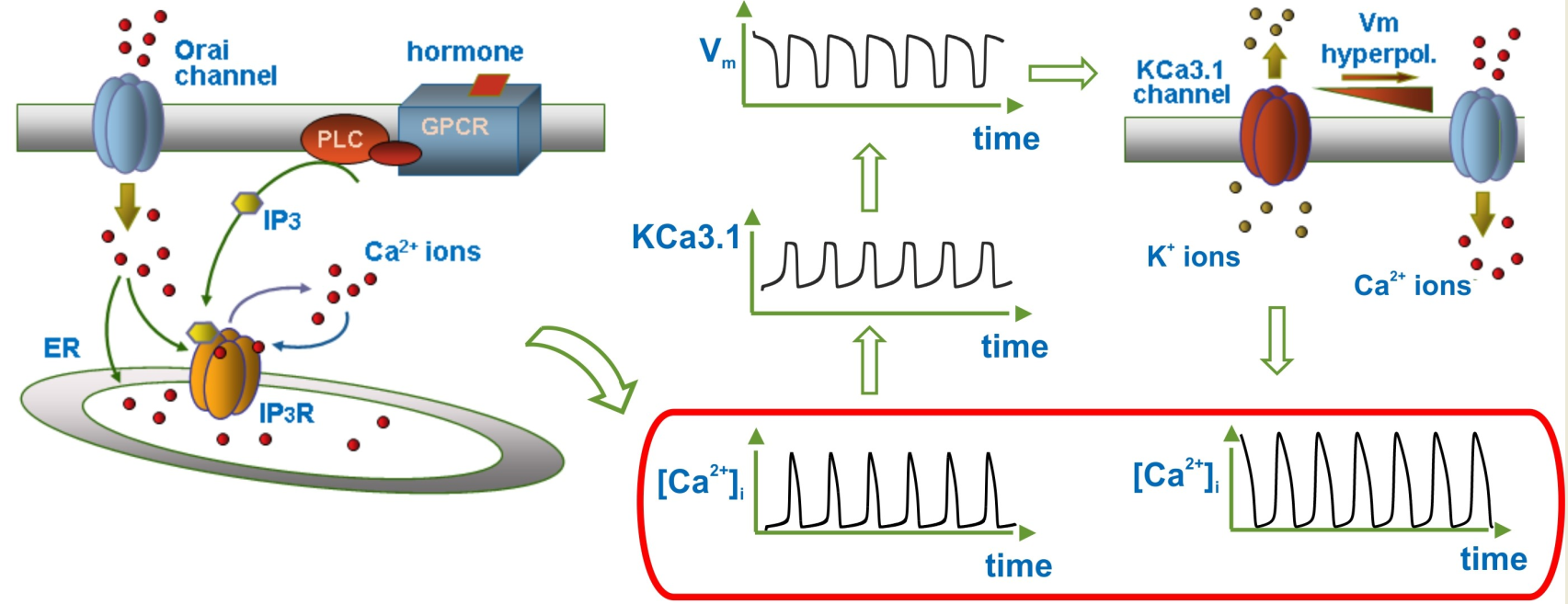{kind=link}
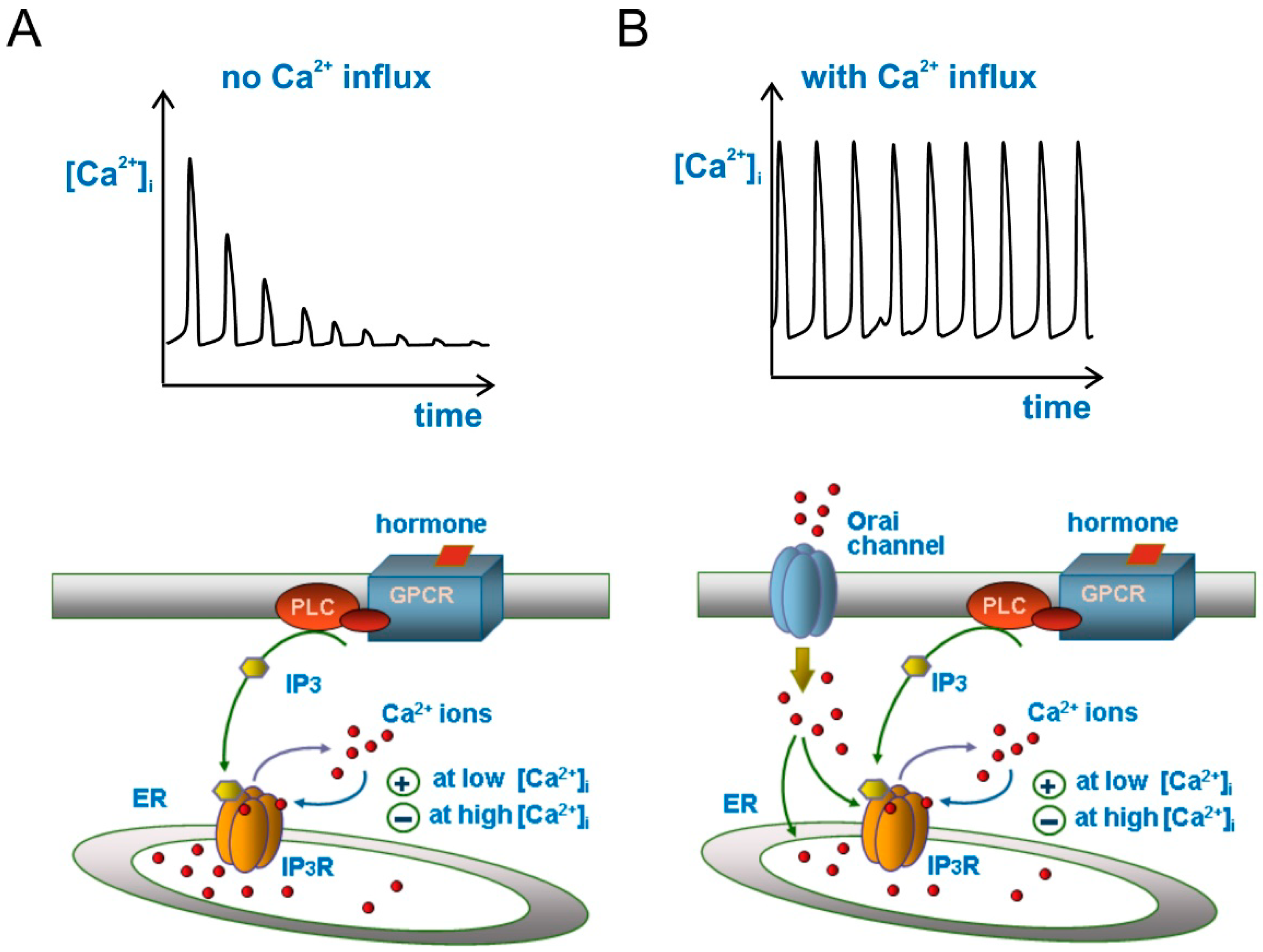{kind=link}
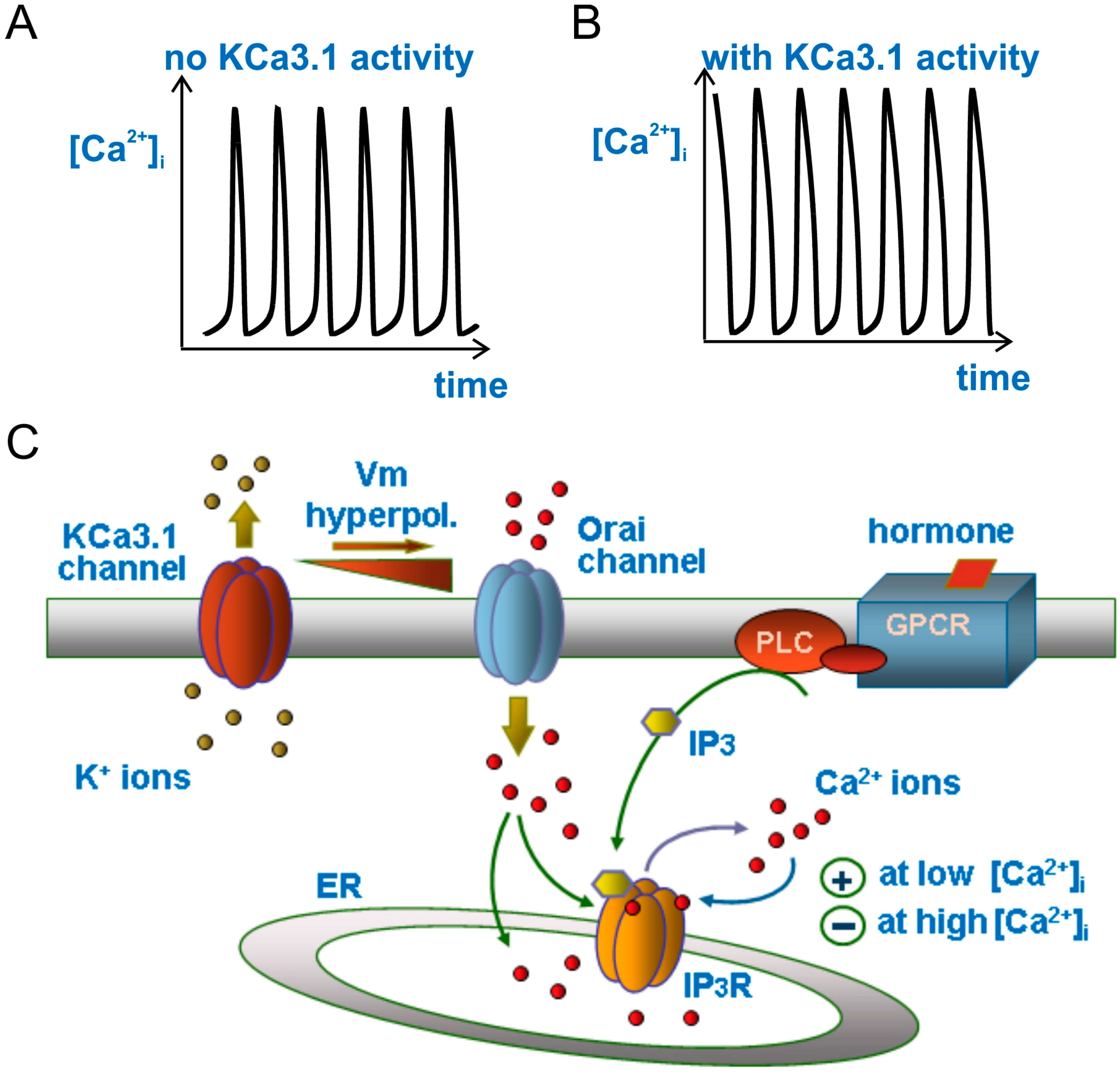{kind=link}
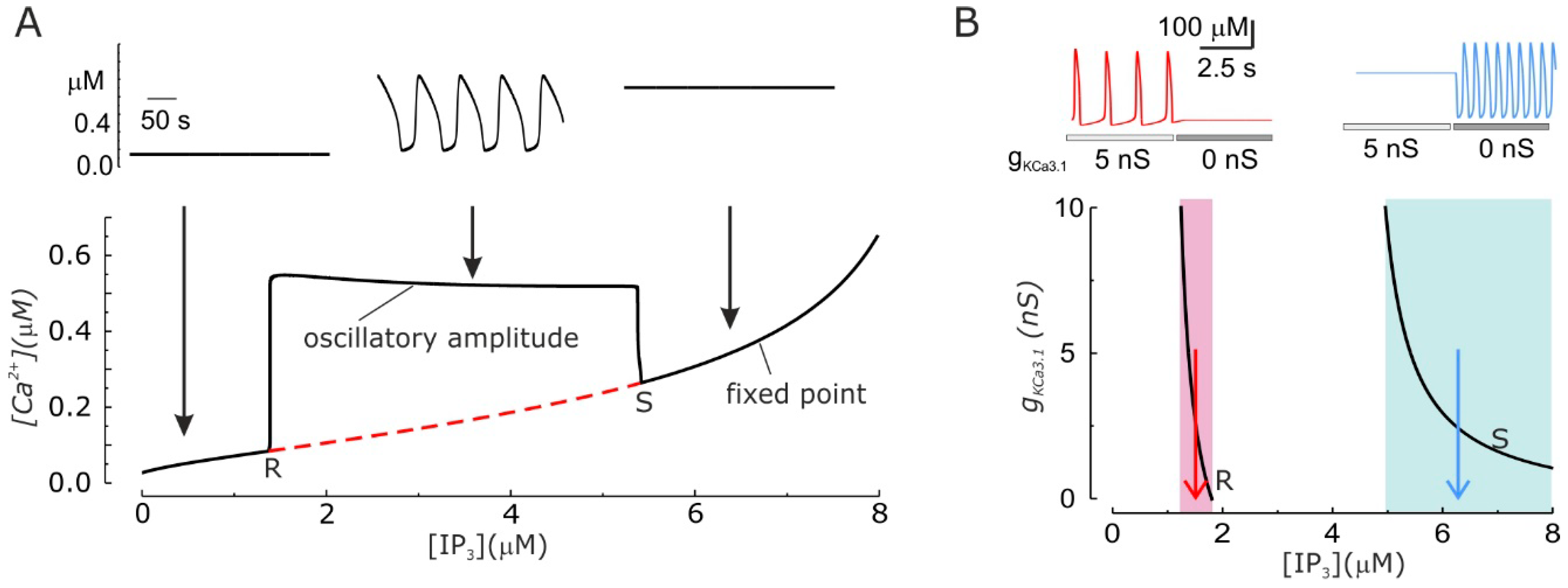{kind=link}
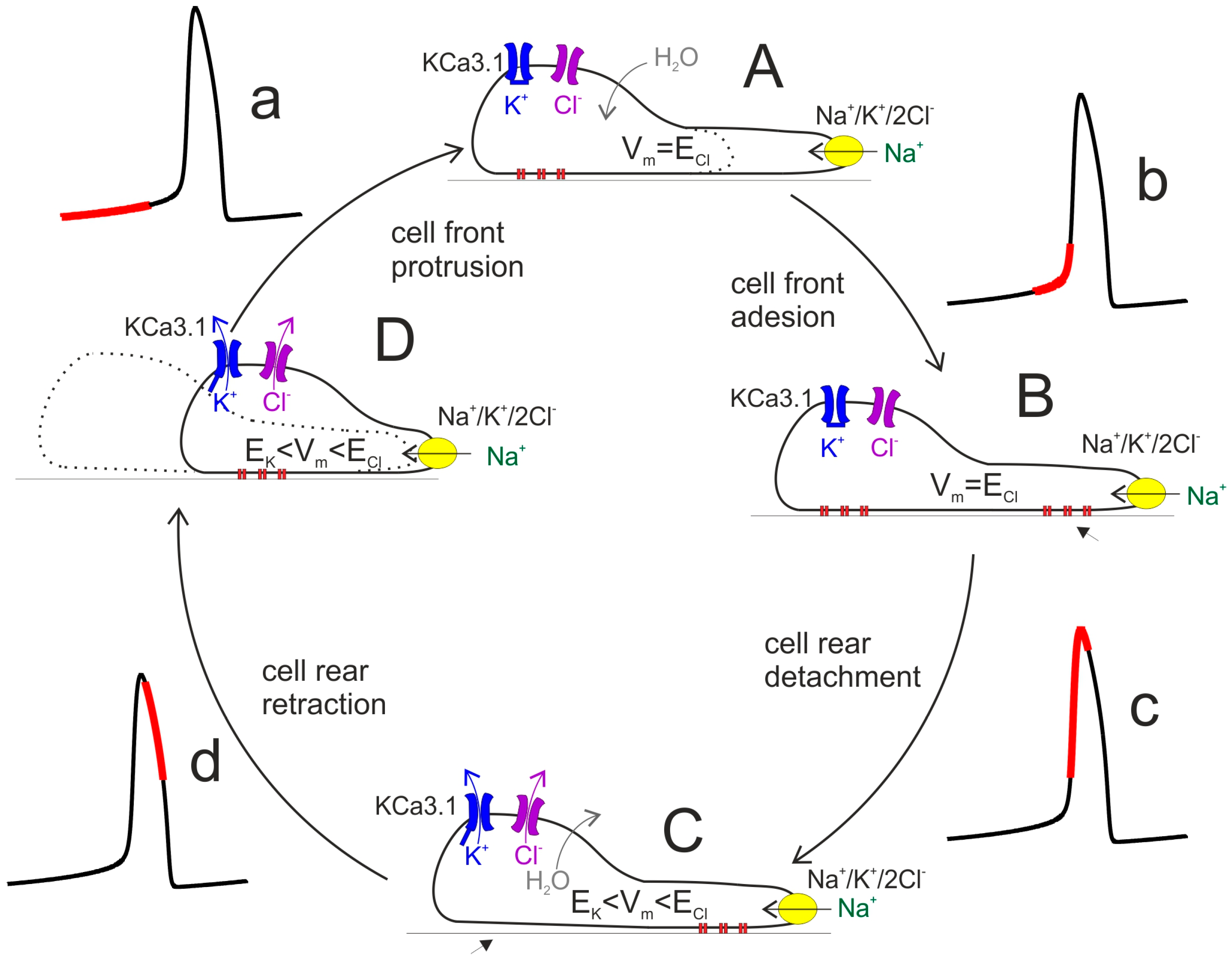{kind=link}
1. The Glioblastoma
2. Glioblastoma Cell Migration and Ca2+ Signaling
2.1. Cell Migration
2.2. Cell Volume Changes Associated with Migration
2.3. Ca2+ Oscillations
2.4. Store-Operated Ca2+ Entry (SOCE)
2.5. Ca2+ Oscillations and Cell Migration
3. The Intermediate Conductance Ca2+-Activated K Channel
3.1. Biophysics, Pharmacology, and Gating of the KCa3.1 Channel
3.2. KCa3.1 Channel Expression and Impact on GBM Migration and Invasion
3.3. Basic Functions of KCa3.1 Channel: Regulation of Cell Ca2+ Signaling
3.4. Modulation of Ca2+ Oscillations by KCa3.1 Channel Activity
3.5. KCa3.1 Channels Switch Ca2+ Oscillations On and Off
3.6. Glioblastoma KCa3.1 Channels Are Activated by Serum-Induced Ca2+ Oscillations and Participate to Cell Migration
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Ridley, A.J.; Schwartz, M.A.; Burridge, K.; Firtel, R.A.; Ginsberg, M.H.; Borisy, G.; Parsons, J.T.; Horwitz, A.R. Cell migration: Integrating signals from front to back. Science 2003, 302, 1704–1709. [Google Scholar] [CrossRef] [PubMed]
- Ridley, A.J. Life at the leading edge. Cell 2011, 145, 1012–1022. [Google Scholar] [CrossRef] [PubMed]
- Carragher, N.O.; Frame, M.C. Focal adhesion and actin dynamics: A place where kinases and proteases meet to promote invasion. Trends Cell Biol. 2004, 14, 241–249. [Google Scholar] [CrossRef] [PubMed]
- Parsons, J.T.; Horwitz, A.R.; Schwartz, M.A. Cell adhesion: Integrating cytoskeletal dynamics and cellular tension. Nat. Rev. Mol. Cell Biol. 2010, 11, 633–643. [Google Scholar] [CrossRef] [PubMed]
- Lund, C.V.; Nguyen, M.T.; Owens, G.C.; Pakchoian, A.J.; Shaterian, A.; Kruse, C.A.; Eliceiri, B.P. Reduced glioma infiltration in Src-deficient mice. J. Neurooncol. 2006, 78, 19–29. [Google Scholar] [CrossRef] [PubMed]
- Wessels, D.; Soll, D.R.; Knecht, D.; Loomis, W.F.; De Lozanne, A.; Spudich, J. Cell motility and chemotaxis in Dictyostelium amebae lacking myosin heavy chain. Dev. Biol. 1988, 128, 164–177. [Google Scholar] [CrossRef]
- Lombardi, M.L.; Knecht, D.A.; Dembo, M.; Lee, J. Traction force microscopy in Dictyostelium reveals distinct roles for myosin II motor and actin-crosslinking activity in polarized cell movement. J. Cell Sci. 2007, 120 Pt 9, 1624–1634. [Google Scholar] [CrossRef]
- Roca-Cusachs, P.; Sunyer, R.; Trepat, X. Mechanical guidance of cell migration: Lessons from chemotaxis. Curr. Opin. Cell Biol. 2013, 25, 543–549. [Google Scholar] [CrossRef] [PubMed]
- Ostman, A.; Heldin, C.H. PDGF receptors as targets in tumor treatment. Adv. Cancer Res. 2007, 97, 247–274. [Google Scholar] [PubMed]
- Lo, C.M.; Wang, H.B.; Dembo, M.; Wang, Y.L. Cell movement is guided by the rigidity of the substrate. Biophys. J. 2000, 79, 144–152. [Google Scholar] [CrossRef]
- Schwab, A.; Fabian, A.; Hanley, P.J.; Stock, C. Role of ion channels and transporters in cell migration. Physiol. Rev. 2012, 92, 1865–1913. [Google Scholar] [CrossRef] [PubMed]
- Catacuzzeno, L.; Aiello, F.; Fioretti, B.; Sforna, L.; Castigli, E.; Ruggieri, P.; Tata, A.M.; Calogero, A.; Franciolini, F. Serum-activated K and Cl currents underlay U87-MG glioblastoma cell migration. J. Cell Physiol. 2011, 226, 1926–1933. [Google Scholar] [CrossRef] [PubMed]
- Cuddapah, V.A.; Habela, C.W.; Watkins, S.; Moore, L.S.; Barclay, T.T.; Sontheimer, H. Kinase activation of ClC-3 accelerates cytoplasmic condensation during mitotic cell rounding. Am. J. Physiol. Cell Physiol. 2012, 302, C527–C538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, C.; Wang, X.; Zheng, M.; Cheng, H. Calcium gradients underlying cell migration. Curr. Opin. Cell Biol. 2012, 24, 254–261. [Google Scholar] [CrossRef] [PubMed]
- Cuthbertson, K.S.; Chay, T.R. Modelling receptor-controlled intracellular calcium oscillators. Cell Calcium 1991, 12, 97–109. [Google Scholar] [CrossRef]
- Gaspers, L.D.; Bartlett, P.J.; Politi, A.; Burnett, P.; Metzger, W.; Johnston, J.; Joseph, S.K.; Höfer, T.; Thomas, A.P. Hormone-induced calcium oscillations depend on cross-coupling with inositol 1,4,5-trisphosphate oscillations. Cell Rep. 2014, 9, 1209–1218. [Google Scholar] [CrossRef] [PubMed]
- Dupont, G.; Combettes, L.; Bird, G.S.; Putney, J.W. Calcium oscillations. Cold Spring Harb. Perspect. Biol. 2011, 3, a004226. [Google Scholar] [CrossRef] [PubMed]
- Iino, M. Biphasic Ca2+ dependence of inositol 1,4,5-trisphosphate-induced Ca release in smooth muscle cells of the guinea pig taenia caeci. J. Gen. Physiol. 1990, 95, 1103–1122. [Google Scholar] [CrossRef] [PubMed]
- Bezprozvanny, I.; Watras, J.; Ehrlich, B.E. Bell-shaped calcium-response curves of Ins(1,4,5)P3- and calcium-gated channels from endoplasmic reticulum of cerebellum. Nature 1991, 351, 751–754. [Google Scholar] [CrossRef] [PubMed]
- Finch, E.A.; Turner, T.J.; Goldin, S.M. Calcium as a coagonist of inositol 1,4,5-trisphosphate-induced calcium release. Science 1991, 252, 443–446. [Google Scholar] [CrossRef] [PubMed]
- Kaznacheyeva, E.; Lupu, V.D.; Bezprozvanny, I. Single-channel properties of inositol (1,4,5)-trisphosphate receptor heterologously expressed in HEK-293 cells. J. Gen. Physiol. 1998, 111, 847–856. [Google Scholar] [CrossRef] [PubMed]
- Liou, J.; Kim, M.L.; Heo, W.D.; Jones, J.T.; Myers, J.W.; Ferrell, J.E., Jr.; Meyer, T. STIM is a Ca2+ sensor essential for Ca2+-store-depletion-triggered Ca2+ influx. Curr. Biol. 2005, 15, 1235–1241. [Google Scholar] [CrossRef] [PubMed]
- Roos, J.; DiGregorio, P.J.; Yeromin, A.V.; Ohlsen, K.; Lioudyno, M.; Zhang, S.; Safrina, O.; Kozak, J.A.; Wagner, S.L.; Cahalan, M.D.; et al. STIM1, an essential and conserved component of store-operated Ca2+ channel function. J. Cell Biol. 2005, 169, 435–445. [Google Scholar] [CrossRef] [PubMed]
- Peinelt, C.; Vig, M.; Koomoa, D.L.; Beck, A.; Nadler, M.J.; Koblan-Huberson, M.; Lis, A.; Fleig, A.; Penner, R.; Kinet, J.P. Amplification of CRAC current by STIM1 and CRACM1 (Orai1). Nat. Cell Biol. 2006, 8, 771–773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moccia, F.; Ruffinatti, F.A.; Zuccolo, E. Intracellular Ca2+ Signals to Reconstruct a Broken Heart: Still a Theoretical Approach? Curr. Drug Targets 2015, 6, 793–815. [Google Scholar] [CrossRef]
- Dubois, C.; Vanden Abeele, F.; Lehen’kyi, V.; Gkika, D.; Guarmit, B.; Lepage, G.; Slomianny, C.; Borowiec, A.S.; Bidaux, G.; Benahmed, M.; et al. Remodeling of channel-forming ORAI proteins determines an oncogenic switch in prostate cancer. Cancer Cell 2014, 26, 19–32. [Google Scholar] [CrossRef] [PubMed]
- Duffy, S.M.; Ashmole, I.; Smallwood, D.T.; Leyland, M.L.; Bradding, P. Orai/CRACM1 and KCa3.1 ion channels interact in the human lung mast cell plasma membrane. Cell Commun. Signal. 2015, 13, 32. [Google Scholar] [CrossRef] [PubMed]
- Rondé, P.; Giannone, G.; Gerasymova, I.; Stoeckel, H.; Takeda, K.; Haiech, J. Mechanism of calcium oscillations in migrating human astrocytoma cells. Biochim. Biophys. Acta 2000, 1498, 273–280. [Google Scholar] [CrossRef]
- Giannone, G.; Rondé, P.; Gaire, M.; Haiech, J.; Takeda, K. Calcium oscillations trigger focal adhesion disassembly in human U87 astrocytoma cells. J. Biol. Chem. 2002, 277, 26364–26371. [Google Scholar] [CrossRef] [PubMed]
- Montana, V.; Sontheimer, H. Bradykinin promotes the chemotactic invasion of primary brain tumors. J. Neurosci. 2011, 31, 4858–4867. [Google Scholar] [CrossRef] [PubMed]
- Totsukawa, G.; Wu, Y.; Sasaki, Y.; Hartshorne, D.J.; Yamakita, Y.; Yamashiro, S.; Matsumura, F. Distinct roles of MLCK and ROCK in the regulation of membrane protrusions and focal adhesion dynamics during cell migration of fibroblasts. J. Cell Biol. 2004, 164, 427–439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.; Tao, T.; Wen, C.; He, W.Q.; Qiao, Y.N.; Gao, Y.Q.; Chen, X.; Wang, P.; Chen, C.P.; Zhao, W.; et al. Myosin light chain kinase (MLCK) regulates cell migration in a myosin regulatory light chain phosphorylation-independent mechanism. J. Biol. Chem. 2014, 289, 28478–28488. [Google Scholar] [CrossRef] [PubMed]
- Sforna, L.; Megaro, A.; Pessia, M.; Franciolini, F.; Catacuzzeno, L. Structure, Gating and Basic Functions of the Ca2+-activated K Channel of Intermediate Conductance. Curr. Neuropharmacol. 2018, 16, 608–617. [Google Scholar] [CrossRef] [PubMed]
- Catacuzzeno, L.; Franciolini, F. Editorial: The Role of Ca2+-activated K+ Channels of Intermediate Conductance in Glioblastoma Malignancy. Curr. Neuropharmacol. 2018, 16, 607. [Google Scholar] [CrossRef] [PubMed]
- Vicente-Manzanares, M.; Koach, M.A.; Whitmore, L.; Lamers, M.L.; Horwitz, A.F. Segregation and activation of myosin IIB creates a rear in migrating cells. J. Cell Biol. 2008, 183, 543–554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaushal, V.; Koeberle, P.D.; Wang, Y.; Schlichter, L.C. The Ca2+-activated K+ channel KCNN4/KCa3.1 contributes to microglia activation and nitric oxide-dependent neurodegeneration. J. Neurosci. 2007, 27, 234–244. [Google Scholar] [CrossRef] [PubMed]
- Ruggieri, P.; Mangino, G.; Fioretti, B.; Catacuzzeno, L.; Puca, R.; Ponti, D.; Miscusi, M.; Franciolini, F.; Ragona, G.; Calogero, A. The inhibition of KCa3.1 channels activity reduces cell motility in glioblastoma derived cancer stem cells. PLoS ONE 2012, 7, e47825. [Google Scholar] [CrossRef] [PubMed]
- D’Alessandro, G.; Catalano, M.; Sciaccaluga, M.; Chece, G.; Cipriani, R.; Rosito, M.; Grimaldi, A.; Lauro, C.; Cantore, G.; Santoro, A.; et al. KCa3.1 channels are involved in the infiltrative behavior of glioblastoma in vivo. Cell Death Dis. 2013, 4, e773. [Google Scholar] [CrossRef] [PubMed]
- Sciaccaluga, M.; Fioretti, B.; Catacuzzeno, L.; Pagani, F.; Bertollini, C.; Rosito, M.; Catalano, M.; D’Alessandro, G.; Santoro, A.; Cantore, G.; et al. CXCL12-induced glioblastoma cell migration requires intermediate conductance Ca2+-activated K+ channel activity. Am. J. Physiol. Cell Physiol. 2010, 299, C175–C184. [Google Scholar] [CrossRef] [PubMed]
- Klumpp, L.; Sezgin, E.C.; Skardelly, M.; Eckert, F.; Huber, S.M. KCa3.1 Channels and Glioblastoma: In Vitro Studies. Curr. Neuropharmacol. 2018, 16, 627–635. [Google Scholar] [CrossRef] [PubMed]
- Turner, K.L.; Honasoge, A.; Robert, S.M.; McFerrin, M.M.; Sontheimer, H. A proinvasive role for the Ca(2+)-activated K(+) channel KCa3.1 in malignant glioma. Glia 2014, 62, 971–981. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.D.; Hanley, P.J.; Rinné, S.; Zuzarte, M.; Daut, J. Calcium-activated K+ channel (KCa3.1) activity during Ca2+ store depletion and store-operated Ca2+ entry in human macrophages. Cell Calcium 2010, 48, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Fioretti, B.; Catacuzzeno, L.; Sforna, L.; Aiello, F.; Pagani, F.; Ragozzino, D.; Castigli, E.; Franciolini, F. Histamine hyperpolarizes human glioblastoma cells by activating the intermediate-conductance Ca2+-activated K+ channel. Am. J. Physiol. Cell Physiol. 2009, 297, C102–C110. [Google Scholar] [CrossRef] [PubMed]
- Ghanshani, S.; Wulff, H.; Miller, M.J.; Rohm, H.; Neben, A.; Gutman, G.A.; Cahalan, M.D.; Chandy, K.G. Up-regulation of the IKCa1 potassium channel during T-cell activation. Molecular mechanism and functional consequences. J. Biol. Chem. 2000, 275, 37137–37149. [Google Scholar] [CrossRef] [PubMed]
- Wulff, H.; Beeton, C.; Chandy, K.G. Potassium channels as therapeutic targets for autoimmune disorders. Curr. Opin. Drug Discov. Dev. 2003, 6, 640–647. [Google Scholar]
- Di, L.; Srivastava, S.; Zhdanova, O.; Ding, Y.; Li, Z.; Wulff, H.; Lafaille, M.; Skolnik, E.Y. Inhibition of the K+ channel KCa3.1 ameliorates T cell-mediated colitis. Proc. Natl. Acad. Sci. USA 2010, 107, 1541–1546. [Google Scholar] [CrossRef] [PubMed]
- Duffy, M.S.; Berger, P.; Cruse, G.; Yang, W.; Bolton, S.J.; Bradding, P. The K+ channel iKCA1 potentiates Ca2+ influx and degranulation in human lung mast cells. J. Allergy Clin. Immunol. 2004, 114, 66–72. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, R.; Schlichter, L.C. Selective activation of KCa3.1 and CRAC channels by P2Y2 receptors promotes Ca(2+) signaling, store refilling and migration of rat microglial cells. PLoS ONE 2013, 8, e62345. [Google Scholar] [CrossRef] [PubMed]
- Catacuzzeno, L.; Fioretti, B.; Franciolini, F. A theoretical study on the role of Ca2+-activated K+ channels in the regulation of hormone-induced Ca2+ oscillations and their synchronization in adjacent cells. J. Theor. Biol. 2012, 309, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Reetz, G.; Reiser, G. [Ca2+]i oscillations induced by bradykinin in rat glioma cells associated with Ca2+ store-dependent Ca2+ influx are controlled by cell volume and by membrane potential. Cell Calcium 1996, 19, 143–156. [Google Scholar] [CrossRef]
- Stegen, B.; Klumpp, L.; Misovic, M.; Edalat, L.; Eckert, M.; Klumpp, D.; Ruth, P.; Huber, S.M. K+ channel signaling in irradiated tumor cells. Eur. Biophys. J. 2016, 45, 585–598. [Google Scholar] [CrossRef] [PubMed]
- Verheugen, J.A.; Vijverberg, H.P. Intracellular Ca2+ oscillations and membrane potential fluctuations in intact human T lymphocytes: Role of K+ channels in Ca2+ signaling. Cell Calcium 1995, 17, 287–300. [Google Scholar] [CrossRef]
- Hashii, M.; Nozawa, Y.; Higashida, H. Bradykinin-induced cytosolic Ca2+ oscillations and inositol tetrakisphosphate-induced Ca2+ influx in voltage-clamped ras-transformed NIH/3T3 fibroblasts. J. Biol. Chem. 1993, 268, 19403–19410. [Google Scholar] [PubMed]
- Rane, S.G. A Ca2(+)-activated K+ current in ras-transformed fibroblasts is absent from nontransformed cells. Am. J. Physiol. 1991, 260, C104–C112. [Google Scholar] [CrossRef] [PubMed]
- Seitz, R.J.; Wechsler, W. Immunohistochemical demonstration of serum proteins in human cerebral gliomas. Acta Neuropathol. 1987, 73, 145–152. [Google Scholar] [CrossRef] [PubMed]
- Steinle, M.; Palme, D.; Misovic, M.; Rudner, J.; Dittmann, K.; Lukowski, R.; Ruth, P.; Huber, S.M. Ionizing radiation induces migration of glioblastoma cells by activating BK K+ channels. Radiother. Oncol. 2011, 101, 122–126. [Google Scholar] [CrossRef] [PubMed]




© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Catacuzzeno, L.; Franciolini, F. Role of KCa3.1 Channels in Modulating Ca2+ Oscillations during Glioblastoma Cell Migration and Invasion. Int. J. Mol. Sci. 2018, 19, 2970. https://doi.org/10.3390/ijms19102970
Catacuzzeno L, Franciolini F. Role of KCa3.1 Channels in Modulating Ca2+ Oscillations during Glioblastoma Cell Migration and Invasion. International Journal of Molecular Sciences. 2018; 19(10):2970. https://doi.org/10.3390/ijms19102970
Chicago/Turabian StyleCatacuzzeno, Luigi, and Fabio Franciolini. 2018. "Role of KCa3.1 Channels in Modulating Ca2+ Oscillations during Glioblastoma Cell Migration and Invasion" International Journal of Molecular Sciences 19, no. 10: 2970. https://doi.org/10.3390/ijms19102970
APA StyleCatacuzzeno, L., & Franciolini, F. (2018). Role of KCa3.1 Channels in Modulating Ca2+ Oscillations during Glioblastoma Cell Migration and Invasion. International Journal of Molecular Sciences, 19(10), 2970. https://doi.org/10.3390/ijms19102970