Deletion of RasGRF1 Attenuated Interstitial Fibrosis in Streptozotocin-Induced Diabetic Cardiomyopathy in Mice through Affecting Inflammation and Oxidative Stress

 ,
,  and
and
Abstract
:1. Introduction
2. Results
2.1. Establishment of Diabetic Mouse Model
2.2. The Express of RasGRF1 Is Increased in Cardiac Fibroblasts of Diabetic Mice
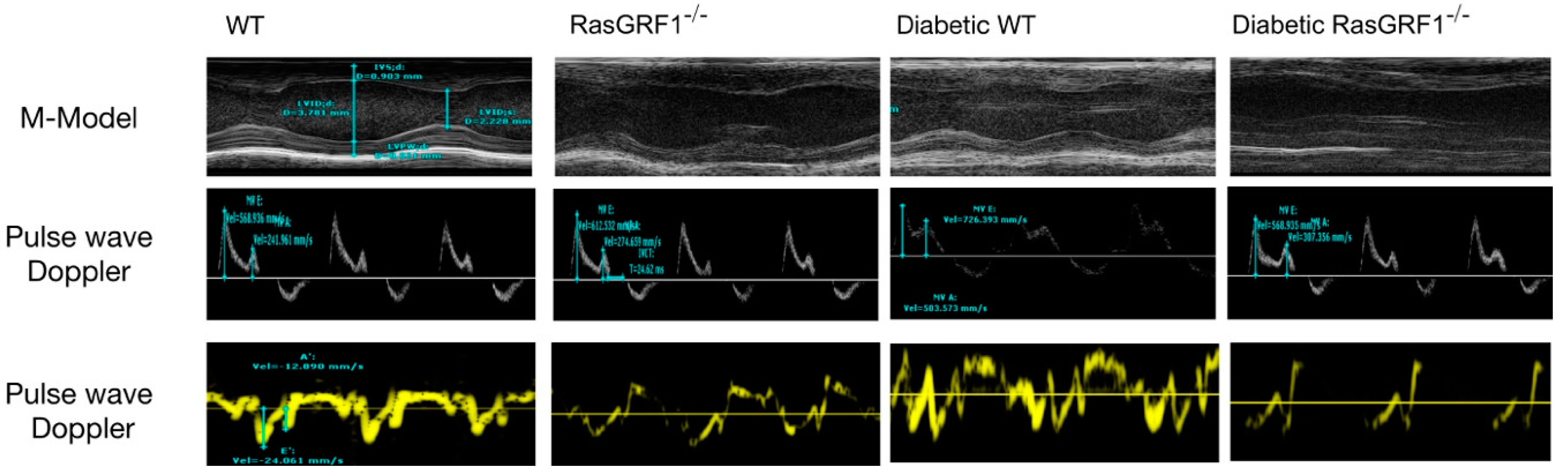
2.3. RasGRF1 Deficiency Mice Lowered the Circulation IL-6 Level and Improved Diastolic Function in Diabetic Mice
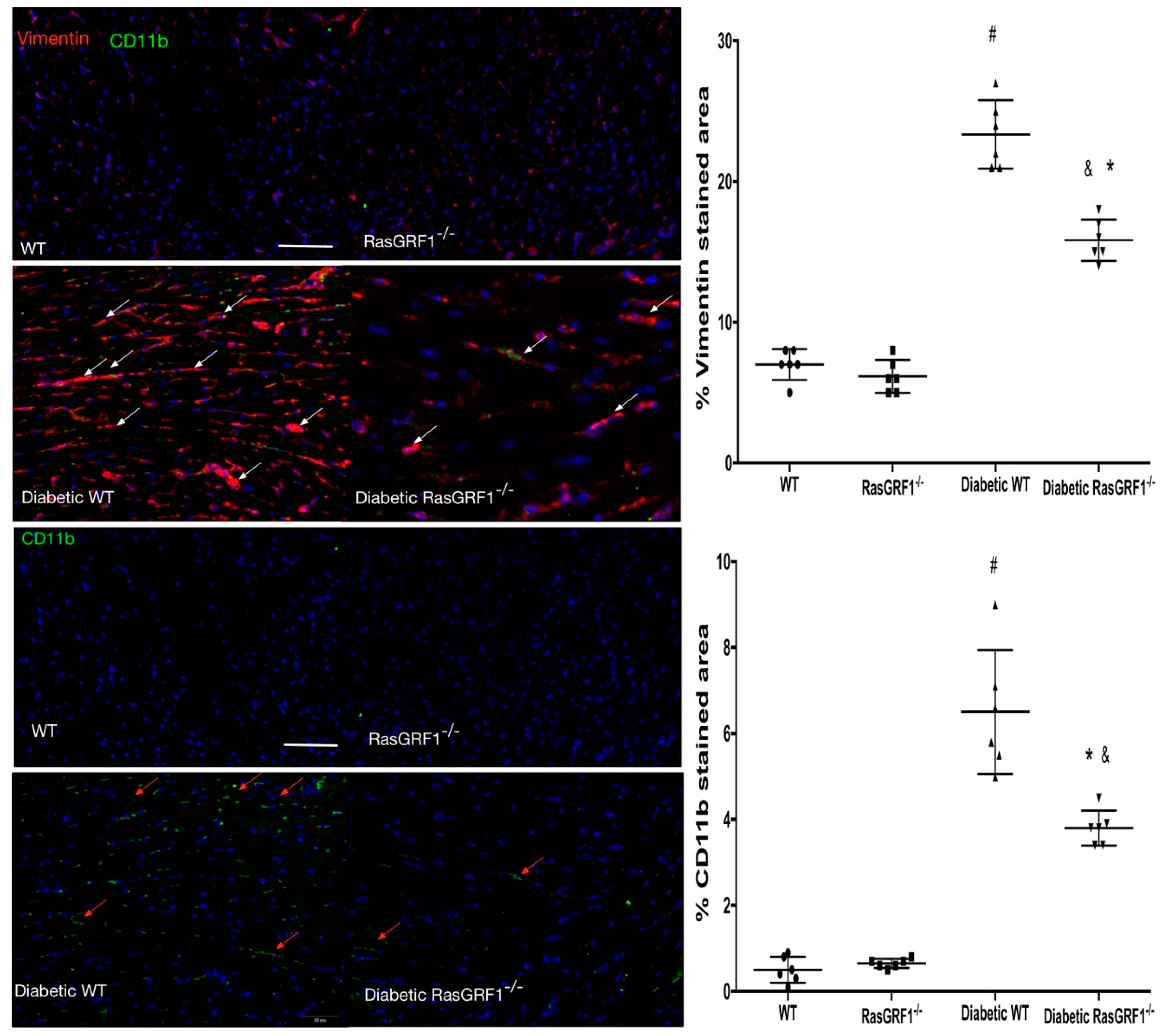
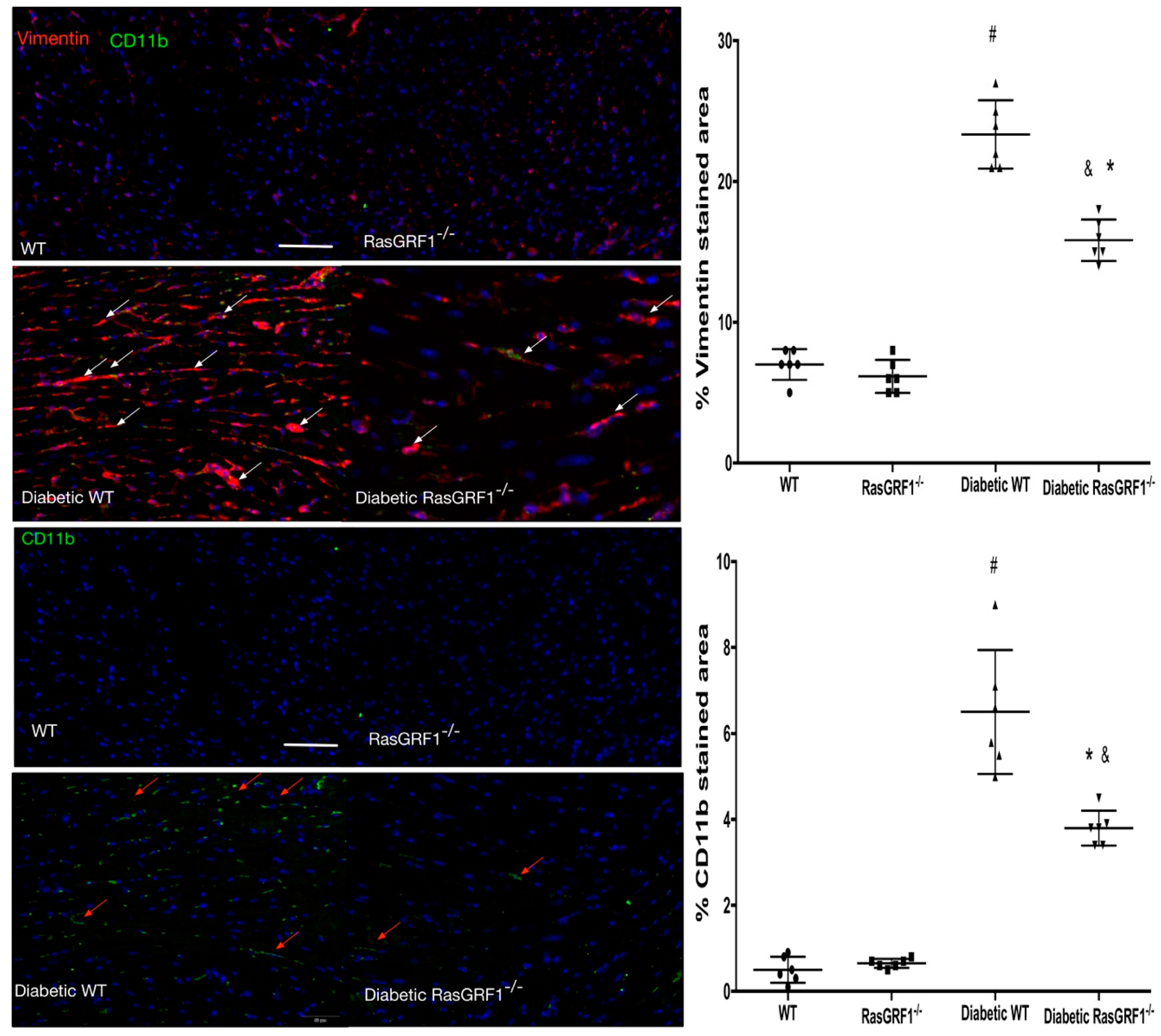
2.4. Deficiency of RasGRF1 Attenuated Cardiac Fibrosis in Diabetic Mice
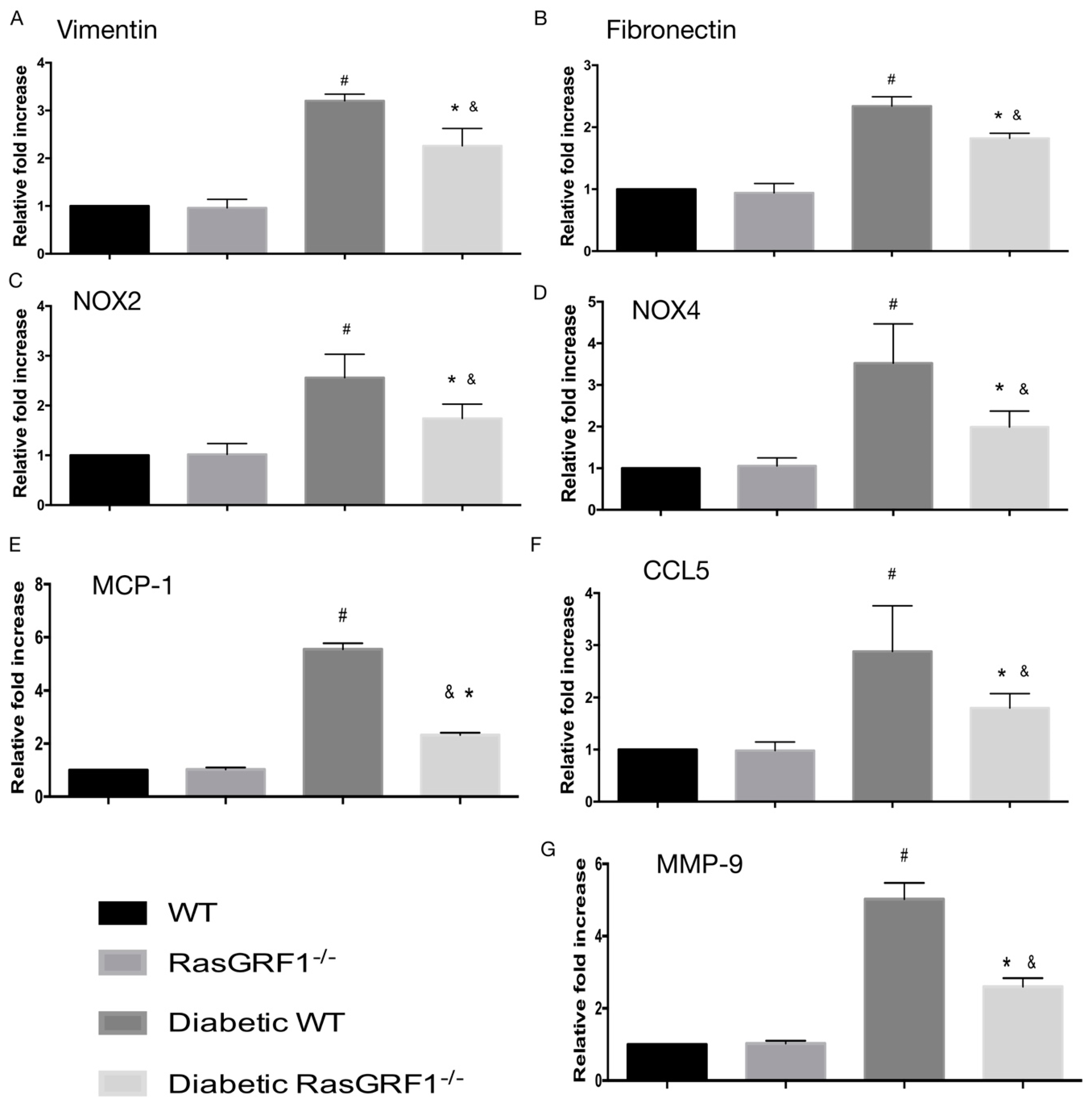
2.5. Deficiency of RasGRF1 Prevented Inflammatory Cytokine and Extra-Matrix Deposition Production in the Hearts of Diabetic Mice
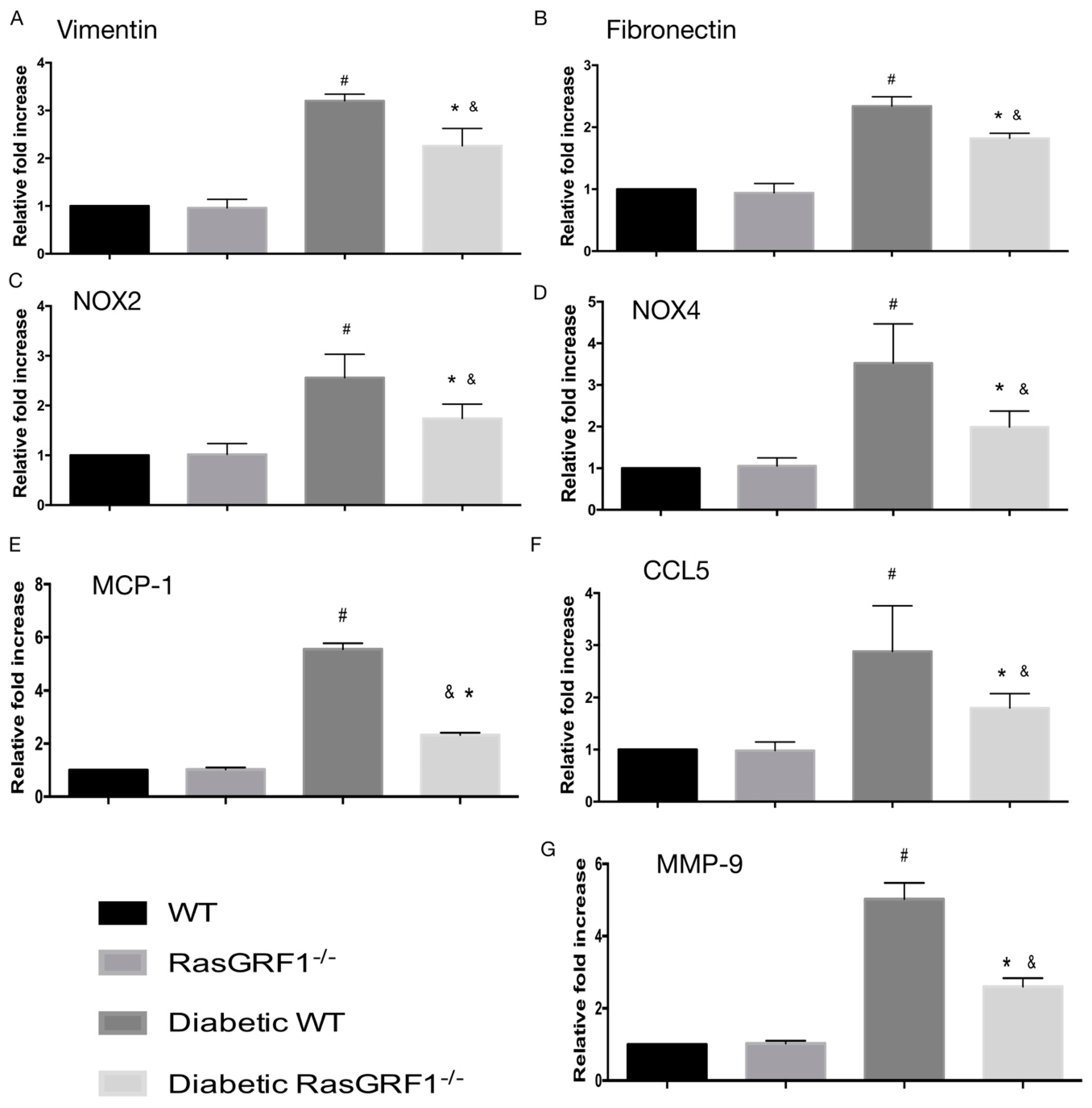
2.6. Effects of Deficiency of RasGRF1 on the mRNA Expression of Inflammatory Cytokines, Oxidative Stress, and Extracellular Matrix in Diabetic Mice
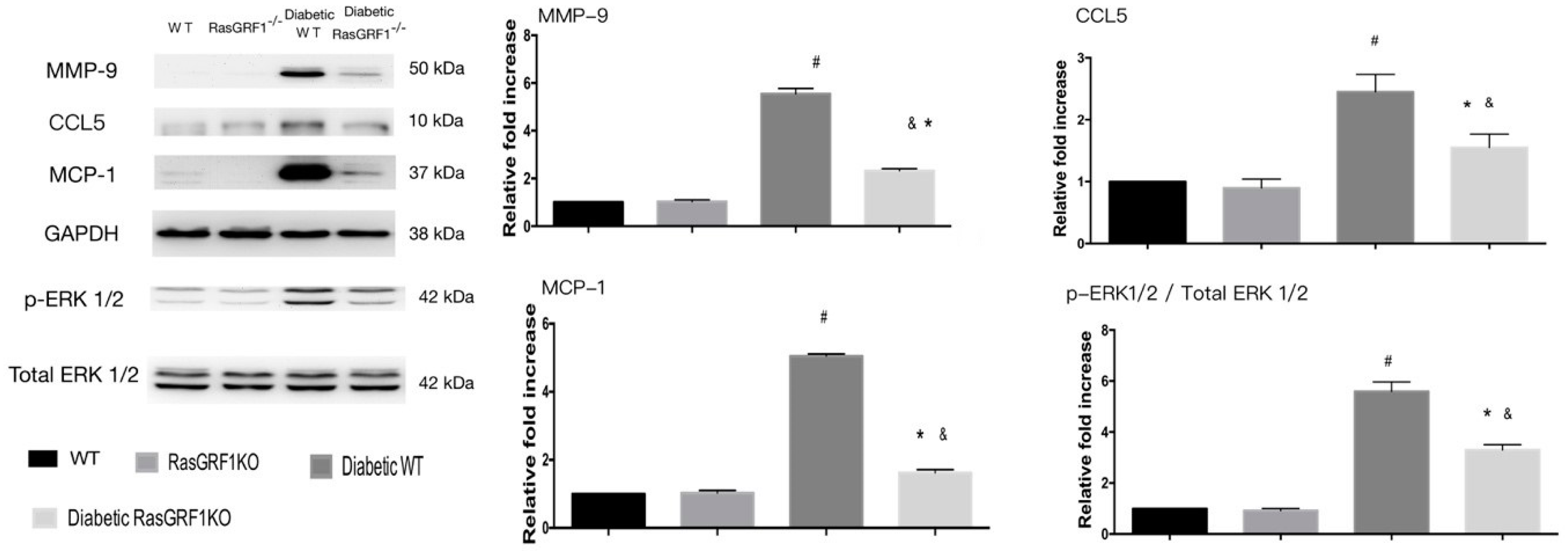
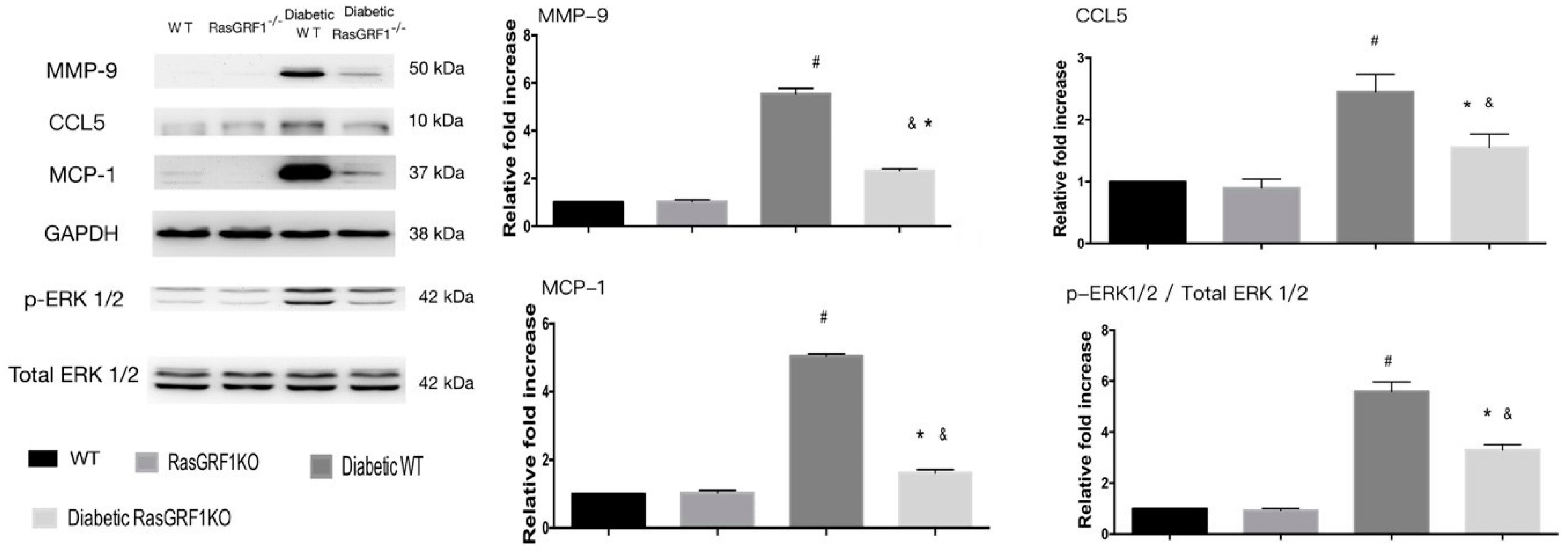
2.7. Effects of Deficiency of RasGRF1 on Protein Expressions of Oxidative Stress and Extracellular Matrix in Diabetic Mice
2.8. Effects of Deficiency of RasGRF1 on Protein Expressions of Inflammatory Cytokine and Chemo-Attractive Cytokines in Diabetic Mice
3. Discussion
3.1. Inflammatory Process and Oxidative Stress Are Major Causes of DCM
3.2. RasGRF1-Regulated Inflammatory Cytokines Secrete an Endoplasmic Reticulum-Dependent Reactive Oxidant Species Production
3.3. The Role of RasGRF1 in DCM and Clinical Impact
4. Materials and Methods
4.1. Animals
4.2. Diabetic Mice Model Induction and Study Protocol
4.3. ELISA for Measure of Serum IL-6 Level
4.3.1. Histology and Immunofluorescent Staining
4.3.2. Quantitative Realtime qPCR
4.3.3. Western Blotting
5. Statistics
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Boudina, S.; Abel, E.D. Diabetic cardiomyopathy revisited. Circulation 2007, 115, 3213–3223. [Google Scholar] [CrossRef] [PubMed]
- From, A.M.; Scott, C.G.; Chen, H.H. The development of heart failure in patients with diabetes mellitus and pre-clinical diastolic dysfunction a population-based study. J. Am. Coll. Cardiol. 2010, 55, 300–305. [Google Scholar] [CrossRef] [PubMed]
- Kamalesh, M. Diabetes and prognosis: Are systolic and diastolic heart failure different? Heart 2009, 95, 178–179. [Google Scholar] [CrossRef] [PubMed]
- Nasir, S.; Aguilar, D. Congestive heart failure and diabetes mellitus: Balancing glycemic control with heart failure improvement. Am. J. Cardiol. 2012, 110, 50B–57B. [Google Scholar] [CrossRef] [PubMed]
- Desai, A.; Fang, J.C. Heart failure with preserved ejection fraction: Hypertension, diabetes, obesity/sleep apnea, and hypertrophic and infiltrative cardiomyopathy. Heart Fail. Clin. 2008, 4, 87–97. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.H.; Young, J.B. Cardiomyopathy and heart failure in diabetes. Endocrinol. Metab. Clin. N. Am. 2001, 30, 1031–1046. [Google Scholar] [CrossRef]
- Ding, W.Y.; Liu, L.; Wang, Z.H.; Tang, M.X.; Ti, Y.; Han, L.; Zhang, L.; Zhang, Y.; Zhong, M.; Zhang, W. FP-receptor gene silencing ameliorates myocardial fibrosis and protects from diabetic cardiomyopathy. J. Mol. Med. 2014, 92, 629–640. [Google Scholar] [CrossRef] [PubMed]
- Jellis, C.; Wright, J.; Kennedy, D.; Sacre, J.; Jenkins, C.; Haluska, B.; Martin, J.; Fenwick, J.; Marwick, T.H. Association of imaging markers of myocardial fibrosis with metabolic and functional disturbances in early diabetic cardiomyopathy. Circ. Cardiovasc. Imaging 2011, 4, 693–702. [Google Scholar] [CrossRef] [PubMed]
- Huynh, K.; McMullen, J.R.; Julius, T.L.; Tan, J.W.; Love, J.E.; Cemerlang, N.; Kiriazis, H.; Du, X.J.; Ritchie, R.H. Cardiac-specific IGF-1 receptor transgenic expression protects against cardiac fibrosis and diastolic dysfunction in a mouse model of diabetic cardiomyopathy. Diabetes 2010, 59, 1512–1520. [Google Scholar] [CrossRef] [PubMed]
- Lim, G.B. Heart failure: Macrophages promote cardiac fibrosis and diastolic dysfunction. Nat. Rev. Cardiol. 2018, 15, 196–197. [Google Scholar] [CrossRef] [PubMed]
- Nevers, T.; Salvador, A.M.; Velazquez, F.; Ngwenyama, N.; Carrillo-Salinas, F.J.; Aronovitz, M.; Blanton, R.M.; Alcaide, P. Th1 effector T cells selectively orchestrate cardiac fibrosis in nonischemic heart failure. J. Exp. Med. 2017, 214, 3311–3329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Savvatis, K.; Kang, J.S.; Fan, P.; Zhong, H.; Schwartz, K.; Barry, V.; Mikels-Vigdal, A.; Karpinski, S.; Kornyeyev, D.; et al. Targeting LOXL2 for cardiac interstitial fibrosis and heart failure treatment. Nat. Commun. 2016, 7, 13710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weber, K.T.; Diez, J. Targeting the Cardiac Myofibroblast Secretome to Treat Myocardial Fibrosis in Heart Failure. Circ. Heart fail. 2016, 9, e003315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Linthout, S.; Seeland, U.; Riad, A.; Eckhardt, O.; Hohl, M.; Dhayat, N.; Richter, U.; Fischer, J.W.; Bohm, M.; Pauschinger, M.; et al. Reduced MMP-2 activity contributes to cardiac fibrosis in experimental diabetic cardiomyopathy. Basic Res. Cardiol. 2008, 103, 319–327. [Google Scholar] [CrossRef] [PubMed]
- Shiomi, T.; Tsutsui, H.; Ikeuchi, M.; Matsusaka, H.; Hayashidani, S.; Suematsu, N.; Wen, J.; Kubota, T.; Takeshita, A. Streptozotocin-induced hyperglycemia exacerbates left ventricular remodeling and failure after experimental myocardial infarction. J. Am. Coll. Cardiol. 2003, 42, 165–172. [Google Scholar] [CrossRef] [Green Version]
- Wu, R.F.; Liao, C.; Hatoum, H.; Fu, G.; Ochoa, C.D.; Terada, L.S. RasGRF Couples Nox4-Dependent Endoplasmic Reticulum Signaling to Ras. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 98–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abreu, J.R.; de Launay, D.; Sanders, M.E.; Grabiec, A.M.; van de Sande, M.G.; Tak, P.P.; Reedquist, K.A. The Ras guanine nucleotide exchange factor RasGRF1 promotes matrix metalloproteinase-3 production in rheumatoid arthritis synovial tissue. Arthritis Res. Ther. 2009, 11, R121. [Google Scholar] [CrossRef] [PubMed]
- Kowluru, R.A.; Zhong, Q.; Santos, J.M. Matrix metalloproteinases in diabetic retinopathy: Potential role of MMP-9. Expert Opin. Investig. Drugs 2012, 21, 797–805. [Google Scholar] [CrossRef] [PubMed]
- Clotet, S.; Riera, M.; Pascual, J.; Soler, M.J. RAS and sex differences in diabetic nephropathy. Am. J. Physiol. Ren. Physiol. 2016, 310, F945–F957. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Fernandez, B.; Ortiz, A.; Gomez-Guerrero, C.; Egido, J. Therapeutic approaches to diabetic nephropathy–beyond the RAS. Nat. Rev. Nephrol. 2014, 10, 325–346. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Li, H.; Liu, J.; Han, P.; Zhang, C.; Bai, H.; Yuan, X.; Wang, X.; Li, L.; Ma, H.; et al. MicroRNA-23b Targets Ras GTPase-Activating Protein SH3 Domain-Binding Protein 2 to Alleviate Fibrosis and Albuminuria in Diabetic Nephropathy. J. Am. Soc. Nephrol. 2016, 27, 2597–2608. [Google Scholar] [CrossRef] [PubMed]
- Cieslik, K.A.; Trial, J.; Entman, M.L. Mesenchymal stem cell-derived inflammatory fibroblasts promote monocyte transition into myeloid fibroblasts via an IL-6-dependent mechanism in the aging mouse heart. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2015, 29, 3160–3170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Z.; Tong, Q.; Zhang, Z.; Wang, S.; Zheng, Y.; Liu, Q.; Qian, L.B.; Chen, S.Y.; Sun, J.; Cai, L. Inhibition of HDAC3 prevents diabetic cardiomyopathy in OVE26 mice via epigenetic regulation of DUSP5-ERK1/2 pathway. Clin. Sci. 2017, 131, 1841–1857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Purvis, G.S.D.; Chiazza, F.; Chen, J.; Azevedo-Loiola, R.; Martin, L.; Kusters, D.H.M.; Reutelingsperger, C.; Fountoulakis, N.; Gnudi, L.; Yaqoob, M.M.; et al. Annexin A1 attenuates microvascular complications through restoration of Akt signalling in a murine model of type 1 diabetes. Diabetologia 2018, 61, 482–495. [Google Scholar] [CrossRef] [PubMed]
- Vulesevic, B.; McNeill, B.; Giacco, F.; Maeda, K.; Blackburn, N.J.; Brownlee, M.; Milne, R.W.; Suuronen, E.J. Methylglyoxal-Induced Endothelial Cell Loss and Inflammation Contribute to the Development of Diabetic Cardiomyopathy. Diabetes 2016, 65, 1699–1713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, Q.; Wang, J.; Wang, L.; Zhang, Y.; Yin, H.; Li, Y.; Tong, C.; Liang, G.; Zheng, C. Attenuation of inflammatory response by a novel chalcone protects kidney and heart from hyperglycemia-induced injuries in type 1 diabetic mice. Toxicol. Appl. Pharmacol. 2015, 288, 179–191. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, J.H.; Zhang, Y.Y.; Wang, Y.Z.; Wang, J.; Zhao, Y.; Jin, X.X.; Xue, G.L.; Li, P.H.; Sun, Y.L.; et al. Deletion of interleukin-6 alleviated interstitial fibrosis in streptozotocin-induced diabetic cardiomyopathy of mice through affecting TGFbeta1 and miR-29 pathways. Sci. Rep. 2016, 6, 23010. [Google Scholar] [CrossRef] [PubMed]
- Al-Rasheed, N.M.; Al-Rasheed, N.M.; Hasan, I.H.; Al-Amin, M.A.; Al-Ajmi, H.N.; Mahmoud, A.M. Sitagliptin attenuates cardiomyopathy by modulating the JAK/STAT signaling pathway in experimental diabetic rats. Drug Des. Dev. Ther. 2016, 10, 2095–2107. [Google Scholar] [Green Version]
- Zhang, X.; Ma, S.; Zhang, R.; Li, S.; Zhu, D.; Han, D.; Li, X.; Li, C.; Yan, W.; Sun, D.; et al. Oncostatin M-induced cardiomyocyte dedifferentiation regulates the progression of diabetic cardiomyopathy through B-Raf/Mek/Erk signaling pathway. Acta Biochim. Biophys. Sin. 2016, 48, 257–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colicelli, J. Human RAS superfamily proteins and related GTPases. Sci. STKE Signal Transduct. Knowl. Environ. 2004, 2004, RE13. [Google Scholar] [CrossRef] [PubMed]
- Nishida, K.; Otsu, K. Inflammation and metabolic cardiomyopathy. Cardiovasc. Res. 2017, 113, 389–398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melendez, G.C.; McLarty, J.L.; Levick, S.P.; Du, Y.; Janicki, J.S.; Brower, G.L. Interleukin 6 mediates myocardial fibrosis, concentric hypertrophy, and diastolic dysfunction in rats. Hypertension 2010, 56, 225–231. [Google Scholar] [CrossRef] [PubMed]
- Leung, K.F.; Baron, R.; Seabra, M.C. Thematic review series: Lipid posttranslational modifications. geranylgeranylation of Rab GTPases. J. Lipid Res. 2006, 47, 467–475. [Google Scholar] [CrossRef] [PubMed]
- Wojtkowiak, J.W.; Gibbs, R.A.; Mattingly, R.R. Working together: Farnesyl transferase inhibitors and statins block protein prenylation. Mol. Cell. Pharmacol. 2009, 1, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.Y.; Sun, J.S.; Tsuang, Y.H.; Chen, M.H.; Weng, P.W.; Lin, F.H. Simvastatin promotes osteoblast viability and differentiation via Ras/Smad/Erk/BMP-2 signaling pathway. Nutr. Res. 2010, 30, 191–199. [Google Scholar] [CrossRef] [PubMed]
- Kishimoto, I.; Makino, H.; Ohata, Y.; Tamanaha, T.; Tochiya, M.; Anzai, T.; Kusano, K.; Noguchi, T.; Yasuda, S.; Ogawa, H. Intensity of statin therapy and new hospitalizations for heart failure in patients with type 2 diabetes. BMJ Open Diabetes Res. Care 2015, 3, e000137. [Google Scholar] [CrossRef] [PubMed]
- Alehagen, U.; Benson, L.; Edner, M.; Dahlstrom, U.; Lund, L.H. Association between use of statins and outcomes in heart failure with reduced ejection fraction: Prospective propensity score matched cohort study of 21 864 patients in the Swedish Heart Failure Registry. Circ. Heart Fail. 2015, 8, 252–260. [Google Scholar] [CrossRef] [PubMed]
- Callahan, L.A.; Supinski, G.S. Hyperglycemia-induced diaphragm weakness is mediated by oxidative stress. Crit. Care 2014, 18, R88. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Control (WT B6) | RasGRF1−/− | Diabetes WT | Diabetes RasGRF1−/− | |
|---|---|---|---|---|
| Blood glucose (mg/mL) | 172 ± 16 | 162 ± 12 | 542 ± 43 * | 587 ± 52 * |
| BW (g) | 32.2 ± 0.48 | 29.5 ± 0.54 & | 25.8 ± 0.42 * | 26.1 ± 0.36 * |
| HW (mg) | 162.2 ± 3.2 | 157.2 ± 3.2 | 138.9 ± 4.3 | 134.2 ± 4.1 |
| HW/BW | 4.67 ± 0.11 | 4.42 ± 0.13 | 4.51 ± 0.18 | 4.78 ± 0.17 |
| IL-6 (ng/mL) | 234 ± 21 | 198 ± 19 | 764 ± 78 * | 674 ± 56 & |
| LVSd (mm) | 0.78 ± 0.05 | 0.78 ± 0.06 | 0.71 ± 0.03 | 0.72 ± 0.05 |
| LVPWd (mm) | 0.92 ± 0.06 | 0.89 ± 0.07 | 0.87 ± 0.06 | 0.89 ± 0.05 |
| LVIDd (mm) | 2.63 ± 0.31 | 2.58 ± 0.17 | 3.12 ± 0.21 * | 2.89 ± 0.13 * |
| LVIDs (mm) | 1.42 ± 0.13 | 1.52 ± 0.18 | 1.98 ± 0.19 * | 1.73 ± 0.13 * |
| LV mass index | 101 ± 21 | 98 ± 18 | 86 ± 32 * | 88± 29 * |
| EF (%) | 67 ± 8.1 | 68 ± 7.2 | 51 ± 6.3 * | 54 ± 7.4* |
| FS | 45 ± 6.3 | 43 ± 5.4 | 37 ± 6.2 * | 39 ± 7.2 * |
| E/E′ | 17 ± 3.8 | 16 ± 4.2 | 27 ± 3.8 * | 21 ± 3.4 & |
| E/A | 1.6 ± 0.76 | 1.5 ± 0.81 | 1.1 ± 0.89 * | 1.3 ± 0.76 & |
| Gene | Specific Oligonucleotide Primers for Target Sequences |
|---|---|
| Vimentin | F5′-GCAAAGATTCCACTTTGCGT-3 R5′-GAAATTGCAGGAGGAGATGC-3 |
| Fibronectin | CGAGGTGACAGAGACCACAA-3 CTGGAGTCAAGCCAGACACA-3 |
| NOX2 | F5′-CCTCTACCAAAACCATTCGGAG-3 R5′-CTGTCCACGTACAATTCGTTCA-3 |
| NOX4 | F5′-AAGGTCCCTAGCAGGAGAACA-3 R5′-GCTACATGCACACCTGAGAAA-3 |
| MMP-9 | F5′-GCTGACTACGATAAGGACGGCA-3 R5′-TAGTGGTGCAGGCAGAGTAGGA-3 |
| MCP-1 | F5′-GCTACAAGAGGATCACCAGCAG-3 R5′-GTCTGGACCCATTCCTTCTTGG-3 |
| CCL5 | F5′-CCTGCTGCTTTGCCTACCTCTC-3 R5′-ACACACTTGGCGGTTCCTTCGA-3 |
| Beta-actin | F-5′-CCA ACC GCG AGA AGA TGA-3 R5′-CCA GAG GCG TAC AGG GAT AG-3 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tsai, T.-H.; Lin, C.-J.; Chua, S.; Chung, S.-Y.; Chen, S.-M.; Lee, C.-H.; Hang, C.-L. Deletion of RasGRF1 Attenuated Interstitial Fibrosis in Streptozotocin-Induced Diabetic Cardiomyopathy in Mice through Affecting Inflammation and Oxidative Stress. Int. J. Mol. Sci. 2018, 19, 3094. https://doi.org/10.3390/ijms19103094
Tsai T-H, Lin C-J, Chua S, Chung S-Y, Chen S-M, Lee C-H, Hang C-L. Deletion of RasGRF1 Attenuated Interstitial Fibrosis in Streptozotocin-Induced Diabetic Cardiomyopathy in Mice through Affecting Inflammation and Oxidative Stress. International Journal of Molecular Sciences. 2018; 19(10):3094. https://doi.org/10.3390/ijms19103094
Chicago/Turabian StyleTsai, Tzu-Hsien, Cheng-Jei Lin, Sarah Chua, Sheng-Ying Chung, Shyh-Ming Chen, Chien-Ho Lee, and Chi-Ling Hang. 2018. "Deletion of RasGRF1 Attenuated Interstitial Fibrosis in Streptozotocin-Induced Diabetic Cardiomyopathy in Mice through Affecting Inflammation and Oxidative Stress" International Journal of Molecular Sciences 19, no. 10: 3094. https://doi.org/10.3390/ijms19103094