iTRAQ-Based Proteomic Analysis of Ogura-CMS Cabbage and Its Maintainer Line




Abstract
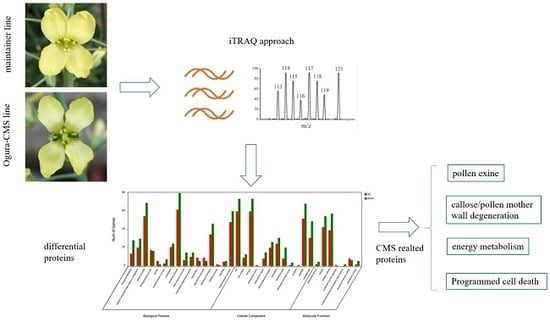
:
1. Introduction
2. Results
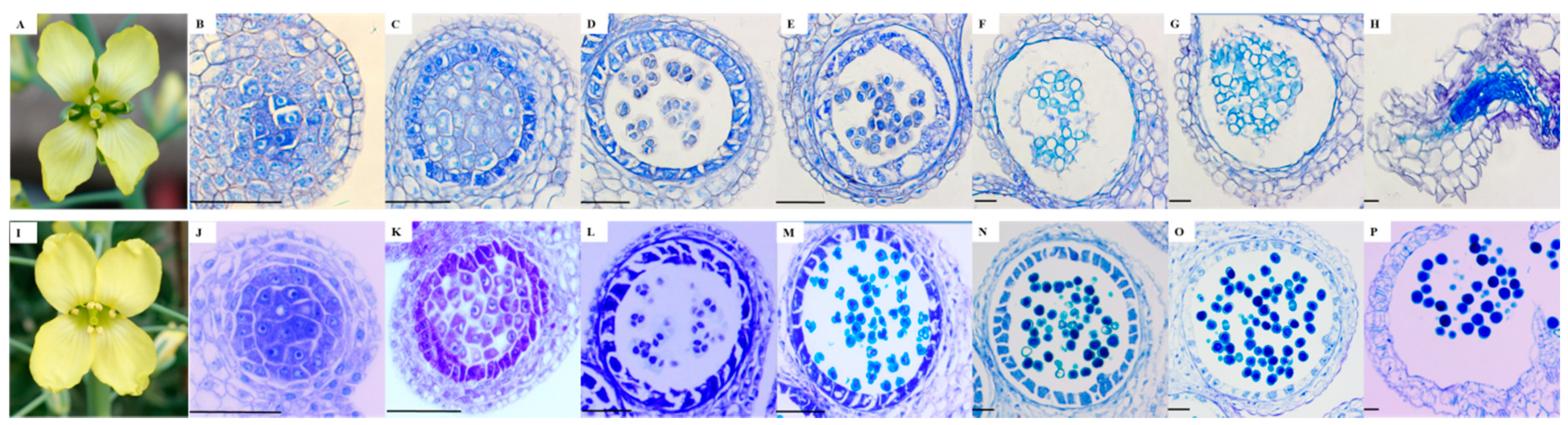
2.1. Morphological and Microscopic Examination
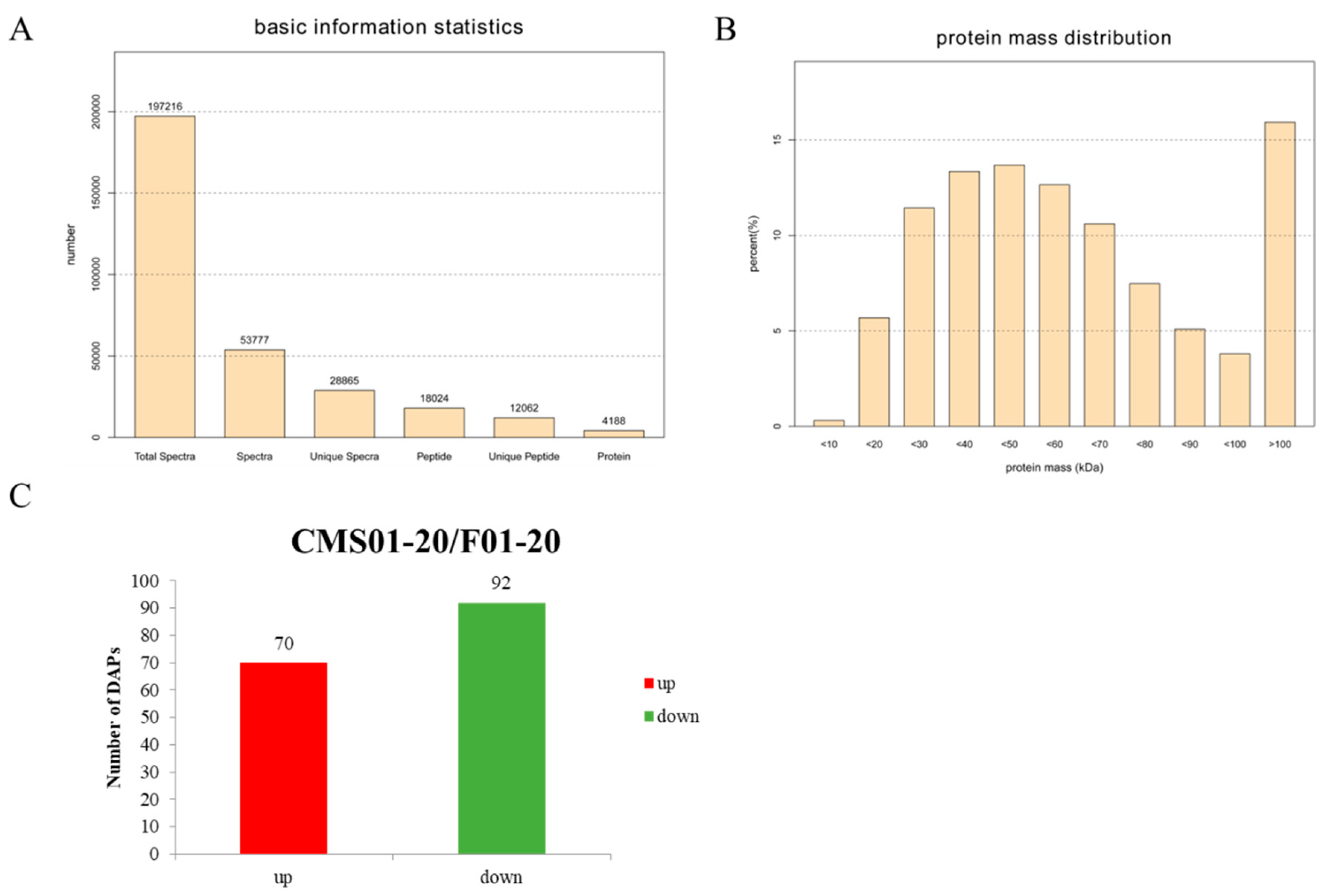
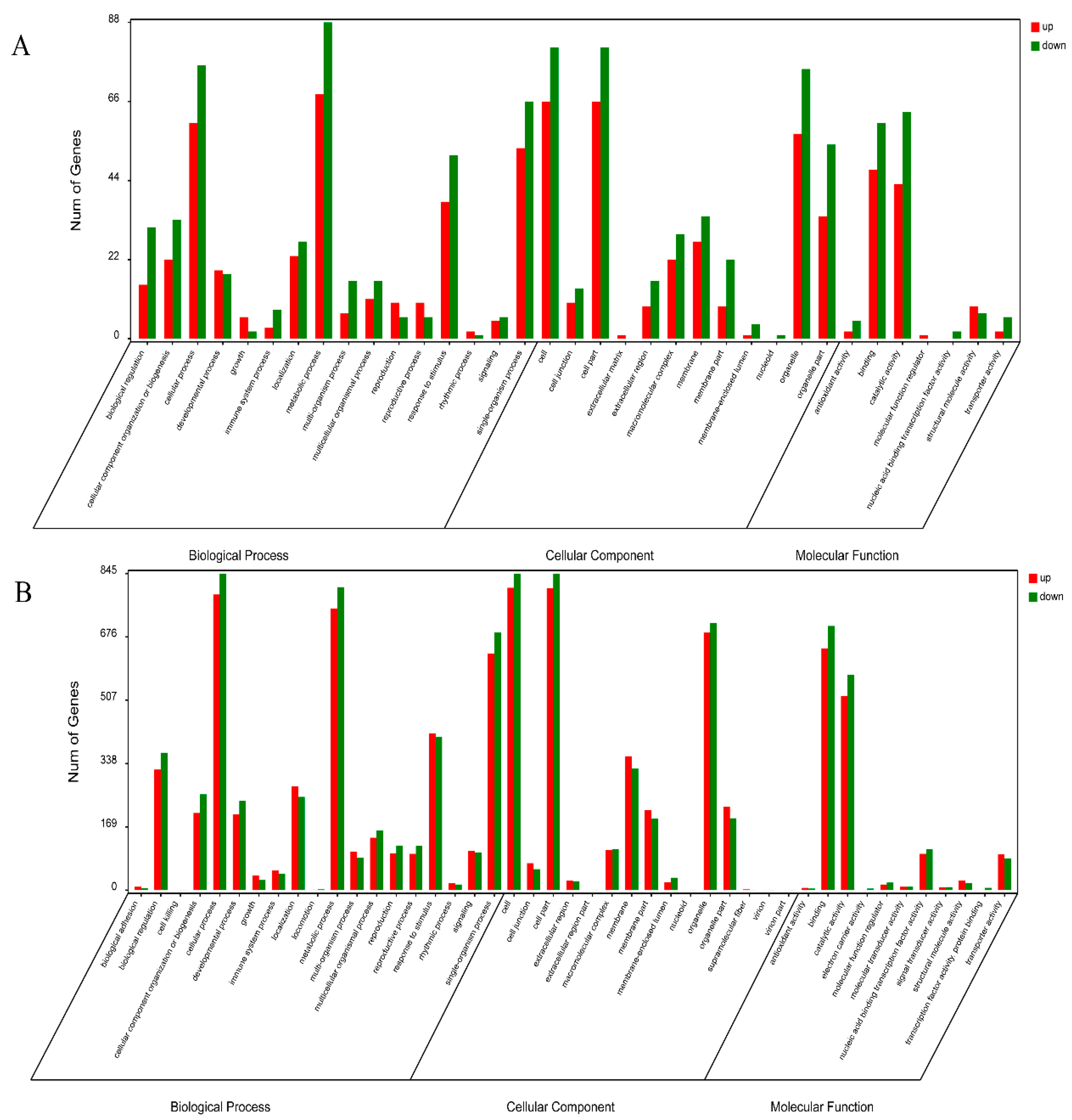
2.2. Overview of the Protein Species Identified Using iTRAQ Data
2.3. Overview of the DAPs between CMS01-20 and F01-20
2.4. DAPs Involved in Oxidative Phosphorylation and TCA Cycle
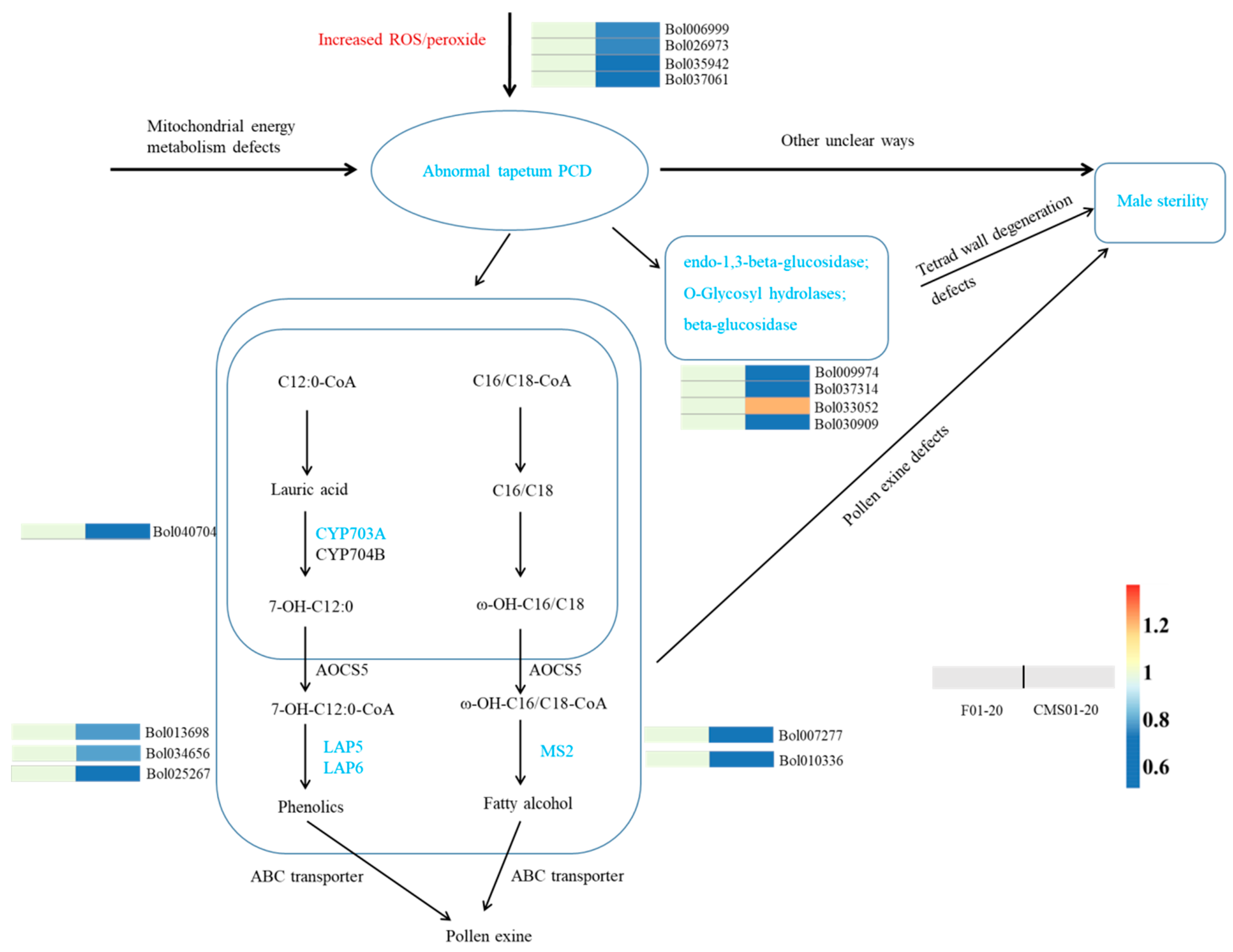
2.5. Other Ogura-CMS Related DAPs and Pathways
2.6. Joint Proteome–Transcriptome Analysis
3. Materials and Methods
3.1. Plant Materials and Sample Preparation
3.2. Microscopy
3.3. iTRAQ Analysis and Protein Species Annotation
3.4. RNA-Seq Analysis and Conjoint Analysis with Proteome Data
3.5. Quantitative RT-PCR Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Fang, Z.Y.; Liu, Y.M.; Yang, L.M.; Wang, X.W.; Zhuang, M.; Zhang, Y.Y.; Sun, P.T. A survey of research in genetic breedings of cabbage in China. Acta Hort. Sin. 2002, 29, S657–S663. [Google Scholar]
- Prakash, C.; Verma, T. Heterosis in cytoplasmic male sterile lines of cabbage. Crucif. Newslett. 2004, 25, 49–50. [Google Scholar]
- Chen, L.; Liu, Y.G. Male sterility and fertility restoration in crops. Annu. Rev. Plant Biol. 2014, 65, 579–606. [Google Scholar] [CrossRef] [PubMed]
- Fang, Z.Y.; Sun, P.T.; Liu, Y.M.; Yang, L.M.; Wang, X.W.; Hou, A.F.; Bian, C.S. A male sterile line with dominant gene (MS) in cabbage (Brassica oleracea var. capitata) and its utilization for hybrid seed production. Euphytica 1997, 97, 265–268. [Google Scholar]
- Fang, Z.Y.; Sun, P.T.; Liu, Y.M. Investigation of different types of male sterility and application of dominant male sterility in cabbage. China Veg. 2001, 1, 6–10. [Google Scholar]
- Ogura, H. Studies of a new male-sterility in Japanese radish, with special reference to the utilization of this sterility towards the practical raising of hybrid seeds. Mem. Fac. Agric. Kagoshima Univ. 1968, 6, 39–78. [Google Scholar]
- Bannerrot, H.; Boulidard, L.; Couderon, Y.; Temple, J. Transfer of cytoplasmic male sterility from Raphanus sativus to Brassica oleracea. Proc. Eucarpia Meet. Crucif. 1974, 25, 52–54. [Google Scholar]
- Sigareva, M.; Earle, E. Direct transfer of a cold-tolerant Ogura male-sterile cytoplasm into cabbage (Brassica oleracea ssp. capitata) via protoplast fusion. Theor. Appl. Genet. 1997, 94, 213–220. [Google Scholar] [CrossRef]
- Walters, W.T.; Mutschler, A.M.; Earle, D.E. Protoplast fusion-derived Ogura male-sterile cauliflower with cold tolerance. Plant Cell Rep. 1992, 10, 624–628. [Google Scholar] [CrossRef] [PubMed]
- Pelletier, G.; Primard, C.; Vedel, F.; Chetrit, P.; Remy, R.; Renard, M. Intergeneric cytoplasmic hybridization in Cruciferae by protoplast fusion. Mol. Genet. Genomics 1983, 191, 244–250. [Google Scholar] [CrossRef]
- Wang, Q.B.; Zhang, Y.Y.; Fang, Z.Y.; Liu, Y.M.; Yang, L.M.; Zhuang, M. Chloroplast and mitochondrial SSR help to distinguish allo-cytoplasmic male sterile types in cabbage (Brassica oleracea L. var. capitata). Mol. Breed. 2012, 30, 709–716. [Google Scholar] [CrossRef]
- Chase, C.D. Cytoplasmic male sterility: A window to the world of plant mitochondrial-nuclear interactions. Trends Genet. 2007, 23, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Huang, W.; Huang, Q.; Qin, X.; Yu, C.; Wang, L.; Li, S.; Zhu, R.; Zhu, Y. Mitochondria and cytoplasmic male sterility in plants. Mitochondrion 2014, 19, 282–288. [Google Scholar] [CrossRef] [PubMed]
- Tuteja, R.; Saxena, R.K.; Davila, J.; Shah, T.; Chen, W.; Xiao, Y.; Fan, G.; Saxena, K.B.; Alverson, A.J.; Spillane, C.; et al. Cytoplasmic male sterility-associated chimeric open reading frames identified by mitochondrial genome sequencing of four Cajanus genotypes. DNA Res. 2013, 20, 485–495. [Google Scholar] [CrossRef] [PubMed]
- Carlsson, J.; Glimelius, K. Cytoplasmic male-sterility and nuclear encoded fertility restoration. Plant Mitochond. 2011, 1, 469–491. [Google Scholar]
- Woodson, J.D.; Chory, J. Coordination of gene expression between organellar and nuclear genomes. Nat. Rev. Genet. 2008, 9, 383–395. [Google Scholar] [CrossRef] [PubMed]
- Duroc, Y.; Hiard, S.; Vrielynck, N.; Ragu, S.; Budar, F. The Ogura sterility-inducing protein forms a large complex without interfering with the oxidative phosphorylation components in rapeseed mitochondria. Plant Mol. Biol. 2009, 70, 123–137. [Google Scholar] [CrossRef] [PubMed]
- Bonhomme, S.; Budar, F.; Lancelin, D.; Small, I.; Defrance, M.C.; Pelletier, G. Sequence and transcript analysis of the Nco2.5 Ogura specific fragment correlated with cytoplasmic male sterility in Brassica cybrids. Mol. Gen. Genet. 1992, 235, 340–348. [Google Scholar] [CrossRef] [PubMed]
- Grelon, M.; Budar, F.; Bonhomme, S.; Pelletier, G. Ogura cytoplasmic male-sterility (CMS)-associated orf138 is translated into a mitochondrial membrane polypeptide in male-sterile Brassica cybrids. Mol. Gen. Genet. 1994, 243, 540–547. [Google Scholar] [CrossRef] [PubMed]
- Krishnasamy, S.; Makaroff, C.A. Characterization of the radish mitochondrial orfB locus: Possible relationship with male sterility in Ogura radish. Curr. Genet. 1993, 24, 156–163. [Google Scholar] [CrossRef] [PubMed]
- Duroc, Y.; Gaillard, C.; Hiard, S.; Defrance, M.C.; Pelletier, G.; Budar, F. Biochemical and functional characterization of ORF138, a mitochondrial protein responsible for Ogura cytoplasmic male sterility in Brassiceae. Biochimie 2005, 87, 1089–1100. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.; Kim, W.K.; Lim, Y.P.; Kim, Y.K.; Hur, Y. Ogura-CMS in Chinese cabbage (Brassica rapa ssp. pekinensis) causes delayed expression of many nuclear genes. Plant Sci. 2013, 199, 7–17. [Google Scholar] [PubMed]
- Wang, S.; Wang, C.; Zhang, X.X.; Chen, X.; Liu, J.J.; Jia, X.F.; Jia, S.Q. Transcriptome de novo assembly and analysis of differentially expressed genes related to cytoplasmic male sterility in cabbage. Plant Physiol. Biochem. 2016, 105, 224–232. [Google Scholar] [CrossRef] [PubMed]
- Shu, J.; Zhang, L.; Liu, Y.; Li, Z.; Fang, Z.; Yang, L.; Zhuang, M.; Zhang, Y.; Lv, H. Normal and abortive buds transcriptomic profiling of broccoli ogu cytoplasmic male sterile line and its maintainer. Int. J. Mol. Sci. 2018, 19, 2501. [Google Scholar] [CrossRef] [PubMed]
- Xing, M.; Sun, C.; Li, H.; Hu, S.; Lei, L.; Kang, J. Integrated analysis of transcriptome and proteome changes related to the Ogura cytoplasmic male sterility in cabbage. PLoS ONE 2018, 13, e0193462. [Google Scholar] [CrossRef] [PubMed]
- Mihr, C.; Baumgärtner, M.; Dieterich, J.H.; Schmitz, U.K.; Braun, H.P. Proteomic approach for investigation of cytoplasmic male sterility (CMS) in Brassica. J. Plant Physiol. 2001, 158, 787–794. [Google Scholar] [CrossRef]
- Sheoran, I.S.; Sawhney, V.K. Proteome analysis of the normal and Ogura (ogu) CMS anthers of Brassica napus to identify proteins associated with male sterility. Botany 2010, 88, 217–230. [Google Scholar] [CrossRef]
- Liu, X.; Han, F.; Kong, C.; Fang, Z.; Yang, L.; Zhang, Y.; Zhuang, M.; Liu, Y.; Li, Z.; Lv, H. Rapid introgression of the Fusarium wilt resistance gene into an elite cabbage line through the combined application of a microspore culture, genome background analysis, and disease resistance-specific marker assisted foreground selection. Front. Plant Sci. 2017, 8, 354. [Google Scholar] [CrossRef] [PubMed]
- González-Melendi, P.; Uyttewaal, M.; Morcillo, C.N.; Mora, J.R.H.; Fajardo, S.; Budar, F.; Lucas, M.M. A light and electron microscopy analysis of the events leading to male sterility in Ogu-INRA CMS of rapeseed (Brassica napus). J. Exp. Bot. 2008, 59, 827–838. [Google Scholar] [CrossRef] [PubMed]
- Tratt, J. Identifying the Wall-Degrading Enzymes Responsible for Microspore Release from the Pollen Tetrad. Ph.D. Thesis, University of Bath, Bath, UK, 2016. [Google Scholar]
- Balk, J.; Leaver, C.J. The PET1-CMS mitochondrial mutation in sunflower is associated with premature programmed cell death and cytochrome c release. Plant Cell 2001, 13, 1803–1818. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Wan, C.; Kong, J.; Zhang, Z.; Li, Y.; Zhu, Y. Programmed cell death during microgenesis in a Honglian CMS line of rice is correlated with oxidative stress in mitochondria. Funct. Plant Biol. 2004, 31, 369–376. [Google Scholar] [CrossRef]
- Luo, D.P.; Xu, H.; Liu, Z.L.; Guo, J.X.; Li, H.Y.; Chen, L.T.; Fang, C.; Zhang, Q.Y.; Bai, M.; Yao, N.; et al. A detrimental mitochondrial-nuclear interaction causes cytoplasmic male sterility in rice. Nat. Genet. 2013, 45, 573–577. [Google Scholar] [CrossRef] [PubMed]
- Yi, J.; Moon, S.; Lee, Y.S.; Zhu, L.; Liang, W.; Zhang, D.; Jung, K.H.; An, G. Defective Tapetum Cell Death 1 (DTC1) regulates ROS levels by binding to metallothionein during tapetum degeneration. Plant Physiol. 2016, 170, 1611–1623. [Google Scholar] [CrossRef] [PubMed]
- Min, L.; Zhu, L.; Tu, L.; Deng, F.; Yuan, D.; Zhang, X. Cotton GhCKI disrupts normal male reproduction by delaying tapetum programmed cell death via inactivating starch synthase. Plant J. 2013, 75, 823–835. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Ding, X.; Han, S.; He, T.; Zhang, H.; Yang, L.; Yang, S.; Gai, J. Differential proteomics analysis to identify proteins and pathways associated with male sterility of soybean using iTRAQ-based strategy. J. Proteomics 2016, 138, 72–82. [Google Scholar] [CrossRef] [PubMed]
- Ji, J.; Yang, L.; Fang, Z.; Zhuang, M.; Zhang, Y.; Lv, H.; Liu, Y.; Li, Z. Complementary transcriptome and proteome profiling in cabbage buds of a recessive male sterile mutant provides new insights into male reproductive development. J. Proteomics 2018, 179, 80–91. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Pang, C.; Wei, H.; Song, M.; Meng, Y.; Ma, J.; Fan, S.; Yu, S. iTRAQ-facilitated proteomic profiling of anthers from a photosensitive male sterile mutant and wild-type cotton (Gossypium hirsutum L.). J. Proteomics 2015, 126, 68–81. [Google Scholar] [CrossRef] [PubMed]
- Pan, Z.; Zeng, Y.; An, J.; Ye, J.; Xu, Q.; Deng, X. An integrative analysis of transcriptome and proteome provides new insights into carotenoid biosynthesis and regulation in sweet orange fruits. J. Proteomics 2012, 75, 2670–2684. [Google Scholar] [CrossRef] [PubMed]
- Lou, P.; Kang, J.; Zhang, G.; Bonnema, G.; Fang, Z.; Wang, X. Transcript profiling of a dominant male sterile mutant (Ms-cd1) in cabbage during flower bud development. Plant Sci. 2007, 172, 111–119. [Google Scholar] [CrossRef]
- Chu, P.; Yan, G.; Yang, Q.; Zhai, L.; Zhang, C.; Zhang, F.; Guan, R. iTRAQ-based quantitative proteomics analysis of Brassica napus, leaves reveals pathways associated with chlorophyll deficiency. J. Proteomics 2015, 113, 244–259. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Liu, Y.; Yang, X.; Tong, C.; Edwards, D.; Parkin, I.A.; Zhao, M.; Ma, J.; Yu, J.; Huang, S.; et al. The Brassica oleracea genome reveals the asymmetrical evolution of polyploid genomes. Nat. Commun. 2014, 5, 3930. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.; Pertea, G.; Trapnell, C.; Pimentel, H.; Kelley, R.; Salzberg, S. TopHat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013, 14, R36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCt method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Zheng, B.B.; Fang, Y.N.; Pan, Z.Y.; Sun, L.; Deng, X.X.; Grosser, J.W.; Guo, W.W. iTRAQ-based quantitative proteomics analysis revealed alterations of carbohydrate metabolism pathways and mitochondrial proteins in a male sterile Cybrid pummelo. J. Proteome Res. 2014, 13, 2998–3015. [Google Scholar] [CrossRef] [PubMed]
- Sabar, M.; Gagliardi, D.; Balk, J.; Leaver, C.J. ORFB is a subunit of F1F(O)-ATP synthase: Insight into the basis of cytoplasmic male sterility in sunflower. EMBO Rep. 2003, 4, 381–386. [Google Scholar] [CrossRef] [PubMed]
- Bergman, P.; Edqvist, J.; Farbos, I.; Glimelius, K. Male-sterile tobacco displays abnormal mitochondrial atp1 transcript accumulation and reduced floral ATP/ADP ratio. Plant Mol. Biol. 2000, 42, 531–544. [Google Scholar] [CrossRef] [PubMed]
- Ducos, E.; Touzet, P.; Boutry, M. The male sterile G, cytoplasm of wild beet displays modified mitochondrial respiratory complexes. Plant J. 2001, 26, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Li, S.; Yi, P.; Wan, C.; Chen, Z.; Zhu, Y. A Honglian CMS line of rice displays aberrant F0 of F0 F1-ATPase. Plant Cell Rep. 2007, 26, 1065–1071. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Gao, F.; Ji, Y.; Liu, Y.; Dan, Z.; Yang, P.; Zhu, Y.; Li, S. ORFH79 impairs mitochondrial function via interaction with a subunit of electron transport chain complex III in Honglian cytoplasmic male sterile rice. New Phytol. 2013, 198, 408–418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ariizumi, T.; Toriyama, K. Genetic regulation of sporopollenin synthesis and pollen exine development. Annu. Rev. Plant Biol. 2011, 62, 437–460. [Google Scholar] [CrossRef] [PubMed]
- Shirley, B.W.; Kubasek, W.L.; Storz, G.; Bruggemann, E.; Koornneef, M.; Ausubel, F.M.; Goodman, H.M. Analysis of Arabidopsis mutants deficient in flavonoid biosynthesis. Plant J. 1995, 8, 659–671. [Google Scholar] [CrossRef] [PubMed]
- Dobritsa, A.A.; Lei, Z.; Nishikawa, S.I.; Urbanczyk-Wochniak, E.; Huhman, D.V.; Preuss, D.; Sumner, L.W. LAP5 and LAP6 encode anther-specific proteins with similarity to chalcone synthase essential for pollen exine development in Arabidopsis thaliana. Plant Physiol. 2010, 110, 157446. [Google Scholar] [CrossRef]
- Morant, M.; Jørgensen, K.; Schaller, H.; Pinot, F.; Møller, B.L.; Werck-Reichhart, D.; Bak, S. CYP703 is an ancient cytochrome P450 in land plants catalyzing in-chain hydroxylation of lauric acid to provide building blocks for sporopollenin synthesis in pollen. Plant Cell 2007, 19, 1473–1487. [Google Scholar] [CrossRef] [PubMed]
- Aarts, M.G.M.; Hodge, R.; Kalantidis, K.; Florack, D.; Wilson, Z.A.; Mulligan, B.J.; Stiekema, W.J.; Scott, R.; Pereira, A. The Arabidopsis MALE STERILITY 2 protein shares similarity with reductases in elongation/condensation complexes. Plant J. 1997, 12, 615–623. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Yu, X.H.; Zhang, K.; Shi, J.; De Oliveira, S.; Schreiber, L.; Shanklin, J.; Zhang, D. Male Sterile2 encodes a plastid-localized fatty acyl carrier protein reductase required for pollen exine development in Arabidopsis. Plant Physiol. 2011, 157, 842–853. [Google Scholar] [CrossRef] [PubMed]
- Wallace, S.; Chater, C.C.; Kamisugi, Y.; Cuming, A.C.; Wellman, C.H.; Beerling, D.J.; Fleming, A.J. Conservation of Male Sterility 2 function during spore and pollen wall development supports an evolutionarily early recruitment of a core component in the sporopollenin biosynthetic pathway. New Phytol. 2015, 205, 390–401. [Google Scholar] [CrossRef] [PubMed]
- Piffanelli, P.; Ross, J.H.E.; Murphy, D.J. Biogenesis and function of the lipidic structures of pollen grains. Sexual Plant Reprod. 1998, 11, 65–80. [Google Scholar] [CrossRef]
- Preuss, D.; Rhee, S.Y.; Davis, R.W. Tetrad analysis possible in Arabidopsis with mutation of the QUARTET (QRT) genes. Science 1994, 264, 1458–1460. [Google Scholar] [CrossRef] [PubMed]
- Rhee, S.Y.; Osborne, E.; Poindexter, P.D.; Somerville, C.R. Microspore separation in the quartet 3 mutants of Arabidopsis is impaired by a defect in a developmentally regulated polygalacturonase required for pollen mother cell wall degradation. Plant Physiol. 2003, 133, 1170–1180. [Google Scholar] [CrossRef] [PubMed]
- Jiang, P.; Zhang, X.; Zhu, Y.; Zhu, W.; Xie, H.; Wang, X. Metabolism of reactive oxygen species in cotton cytoplasmic male sterility and its restoration. Plant Cell Rep. 2007, 26, 1627–1634. [Google Scholar] [CrossRef] [PubMed]
- Gechev, T.S.; Hille, J. Hydrogen peroxide as a signal controlling plant programmed cell death. J. Cell Biol. 2005, 168, 17–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Von Malek, B.; van der Graaff, E.; Schneitz, K.; Keller, B. The Arabidopsis male-sterile mutant dde2-2 is defective in the ALLENE OXIDE SYNTHASE gene encoding one of the key enzymes of the jasmonic acid biosynthesis pathway. Planta 2002, 216, 187–192. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID | Description | Up/Down in CMS Line | |
|---|---|---|---|
| Oxidative phosphorylation | Bol015119 | NADH-ubiquinone oxidoreductase B18 subunit | down |
| Bol009135 | ATP synthase subunit d, mitochondrial-like | down | |
| Bol025034 | ATP synthase subunit d, mitochondrial-like | down | |
| Bol017288 | mitochondrial ATP synthase 6 KD subunit | down | |
| Bol025922 | mitochondrial ATP synthase 6 KD subunit | down | |
| Bol015469 | ATP synthase 6 kDa subunit | down | |
| Bol012326 | cytochrome c | down | |
| Bol010838 | cytochrome c oxidase subunit Vc | down | |
| TCA cycle | Bol022522 | pyruvate dehydrogenase E1 component subunit beta-2 | up |
| Bol008536 | pyruvate dehydrogenase E1 component subunit beta-2 | up | |
| Bol008657 | 2-oxoglutarate dehydrogenase | up | |
| Bol029048 | aconitate hydratase 1 | up | |
| Bol029509 | citrate synthase 4 | up | |
| pollen wall | Bol013698 | LAP5; Chalcone and stilbene synthase family protein | down |
| Bol025267 | LAP6; Chalcone and stilbene synthase family protein | down | |
| Bol007277 | MS2; fatty acyl-CoA reductase | down | |
| Bol034656 | LAP5; Chalcone and stilbene synthase family protein | down | |
| Bol040704 | cytochrome P450 703A2 | down | |
| Bol010336 | MS2; fatty acyl-CoA reductase | down | |
| tetrad wall | Bol009974 | probable glucan endo-1,3-beta-glucosidase A6 | down |
| Bol037314 | O-Glycosyl hydrolases family 17 protein; | down | |
| Bol033052 | beta-d-xylosidase 1 | up | |
| Bol030909 | beta-glucosidase 43 isoform X2 | down | |
| PCD | Bol006999 | catalase-3 | down |
| Bol026973 | catalase-3 | down | |
| Bol035942 | allene oxide synthase | down | |
| Bol037061 | peroxisomal | down | |
| Bol005496 | stromal ascorbate peroxidase | down | |
| Bol004624 | glutathione S-transferase F9 | down | |
| Bol033376 | glutathione S-transferase F9 | down |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Han, F.; Zhang, X.; Yang, L.; Zhuang, M.; Zhang, Y.; Li, Z.; Fang, Z.; Lv, H. iTRAQ-Based Proteomic Analysis of Ogura-CMS Cabbage and Its Maintainer Line. Int. J. Mol. Sci. 2018, 19, 3180. https://doi.org/10.3390/ijms19103180
Han F, Zhang X, Yang L, Zhuang M, Zhang Y, Li Z, Fang Z, Lv H. iTRAQ-Based Proteomic Analysis of Ogura-CMS Cabbage and Its Maintainer Line. International Journal of Molecular Sciences. 2018; 19(10):3180. https://doi.org/10.3390/ijms19103180
Chicago/Turabian StyleHan, Fengqing, Xiaoli Zhang, Limei Yang, Mu Zhuang, Yangyong Zhang, Zhansheng Li, Zhiyuan Fang, and Honghao Lv. 2018. "iTRAQ-Based Proteomic Analysis of Ogura-CMS Cabbage and Its Maintainer Line" International Journal of Molecular Sciences 19, no. 10: 3180. https://doi.org/10.3390/ijms19103180
APA StyleHan, F., Zhang, X., Yang, L., Zhuang, M., Zhang, Y., Li, Z., Fang, Z., & Lv, H. (2018). iTRAQ-Based Proteomic Analysis of Ogura-CMS Cabbage and Its Maintainer Line. International Journal of Molecular Sciences, 19(10), 3180. https://doi.org/10.3390/ijms19103180