Comparative Proteomic and Physiological Analyses of Two Divergent Maize Inbred Lines Provide More Insights into Drought-Stress Tolerance Mechanisms

 ,
,
Abstract
:
1. Introduction
2. Results
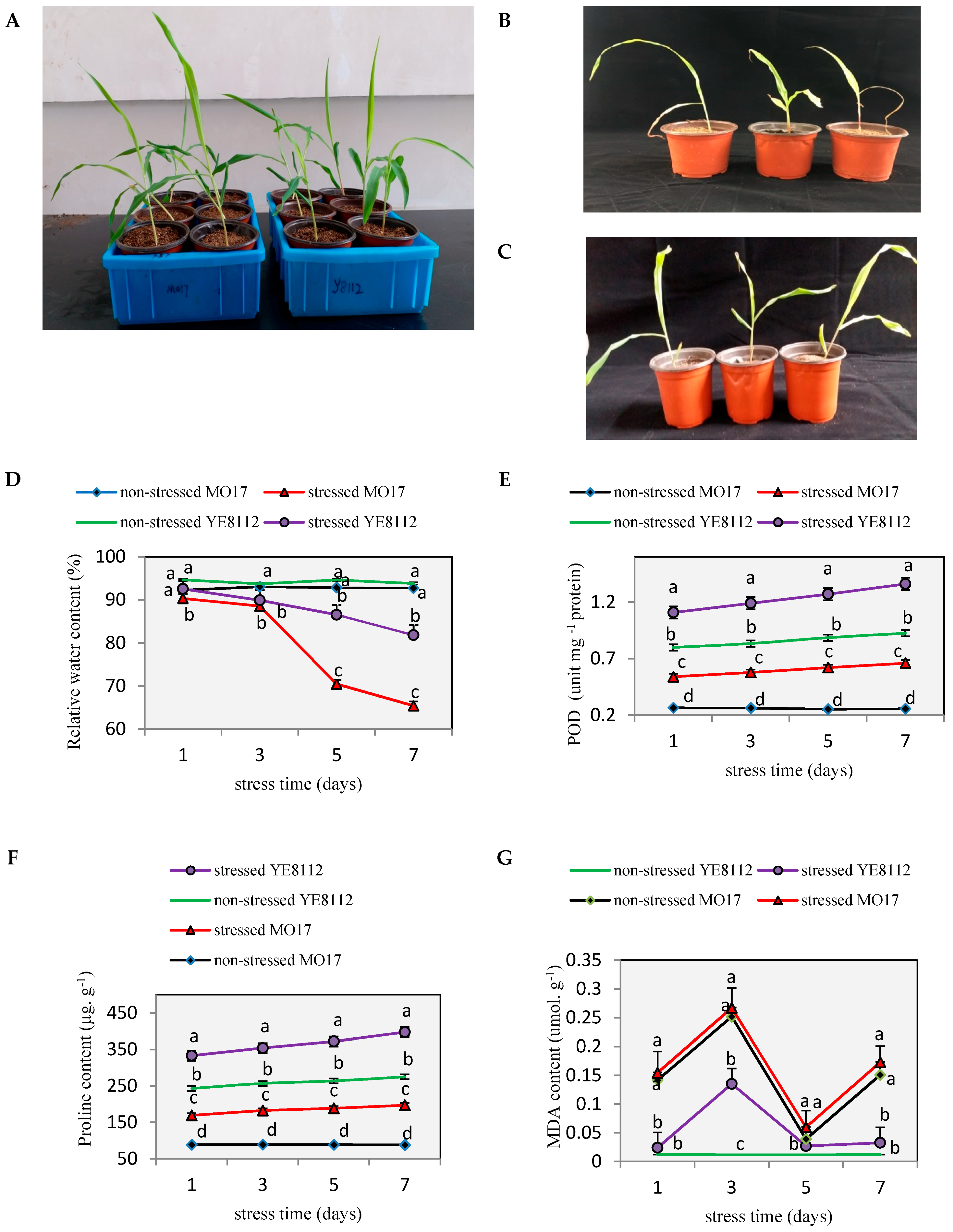
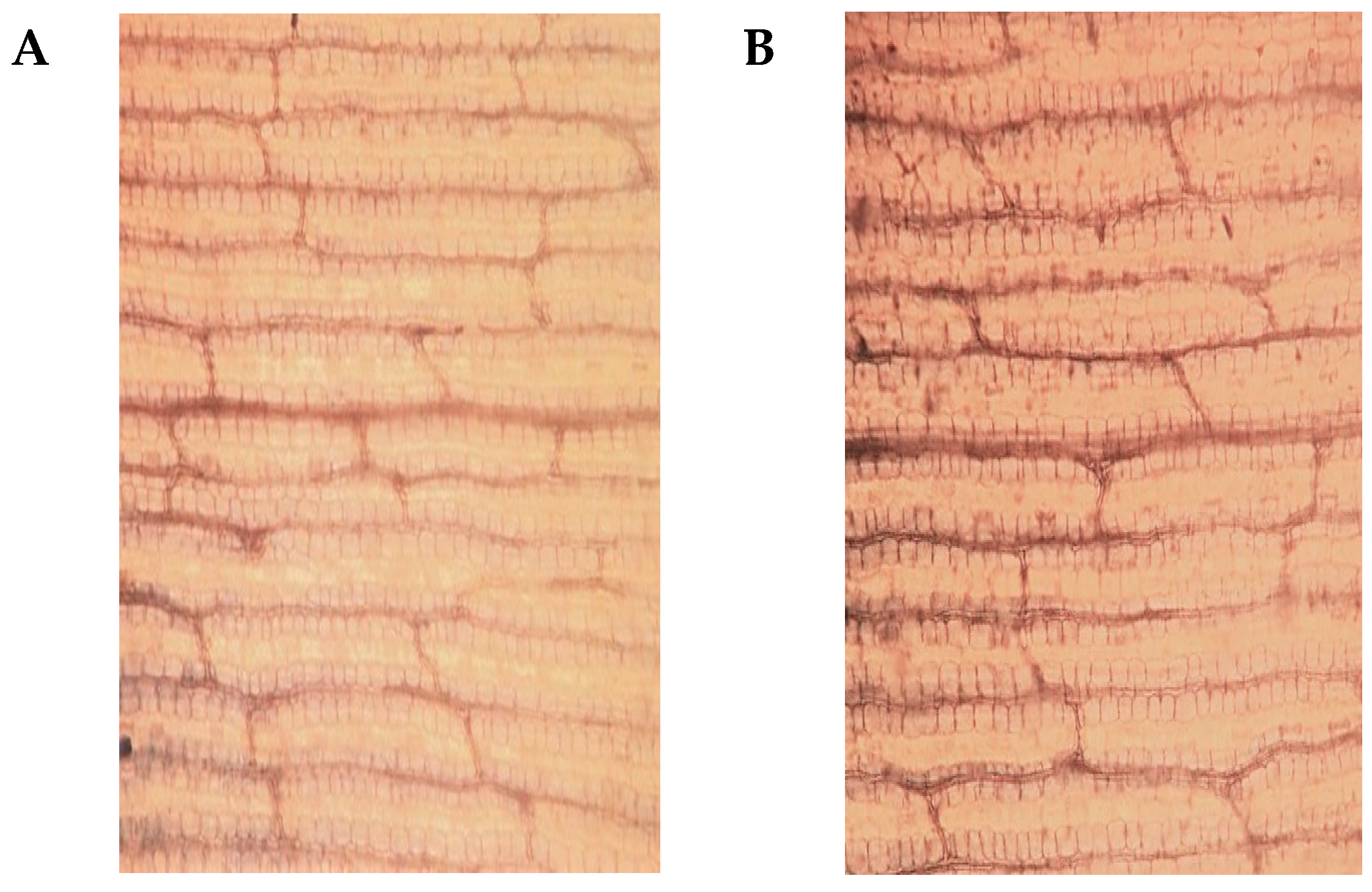
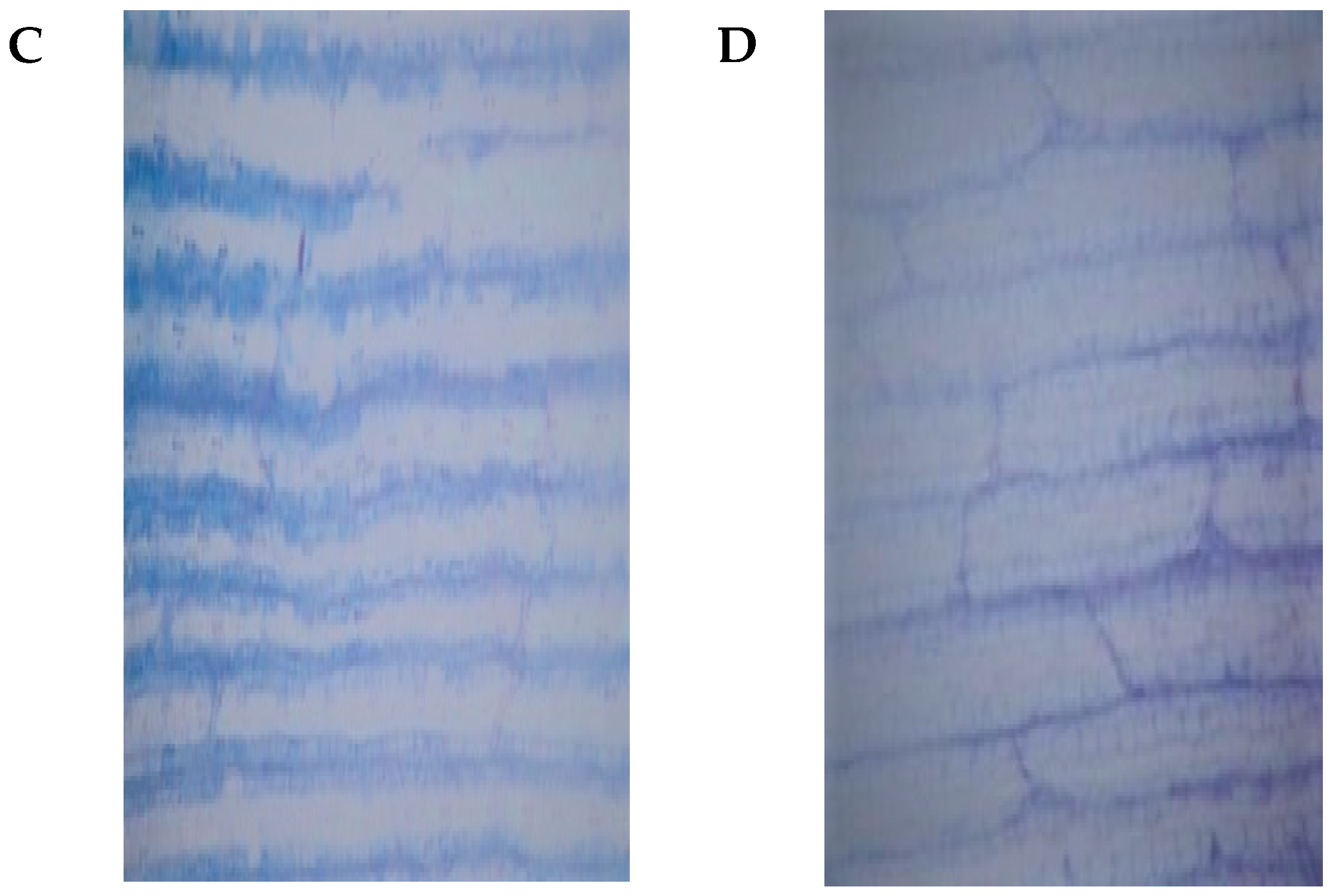
2.1. Phenotypic and Physiological Differences between YE8112 and MO17 in Response to Drought Stress
2.2. Inventory of Maize Seedling Leaf Proteins Identified by iTRAQ
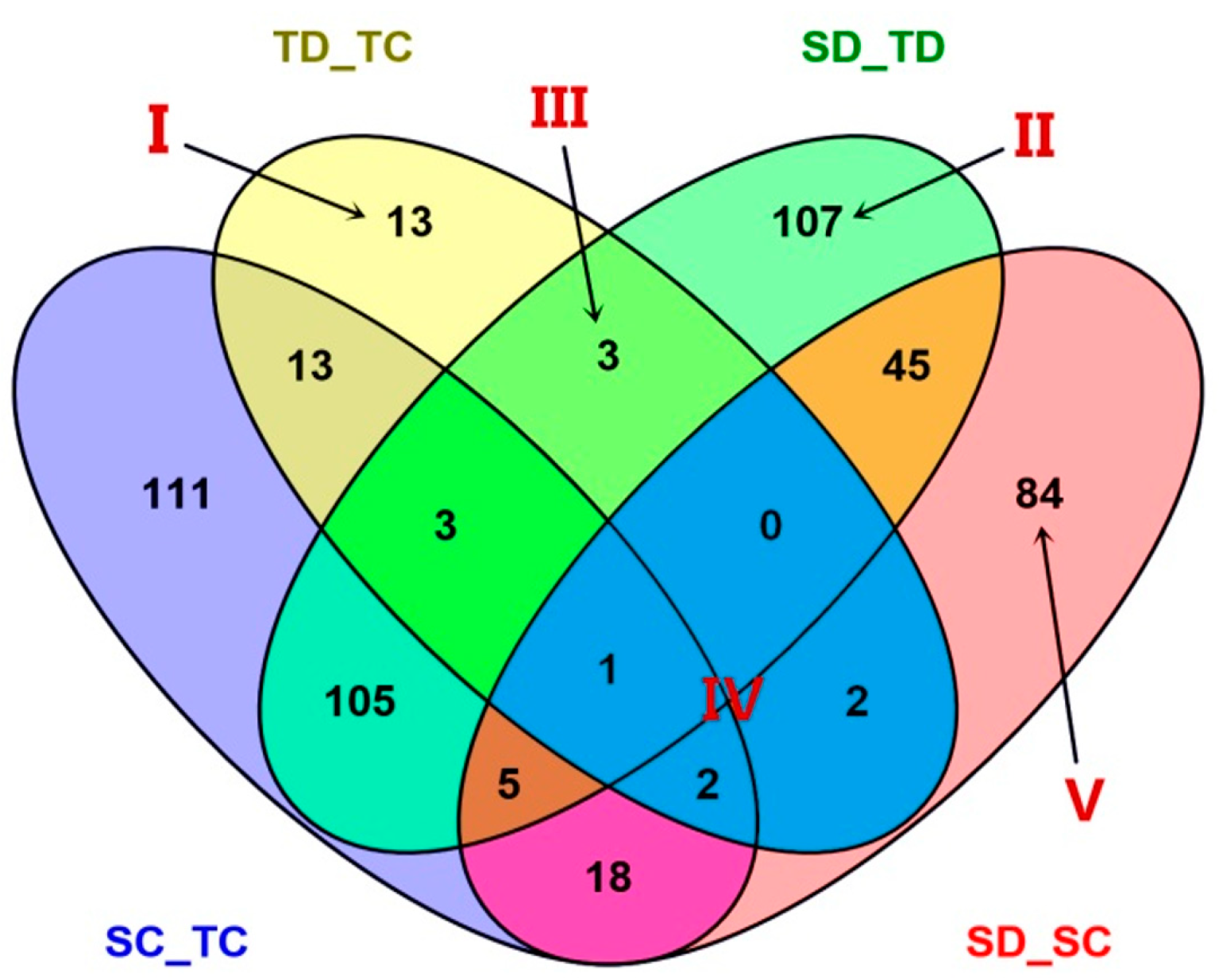
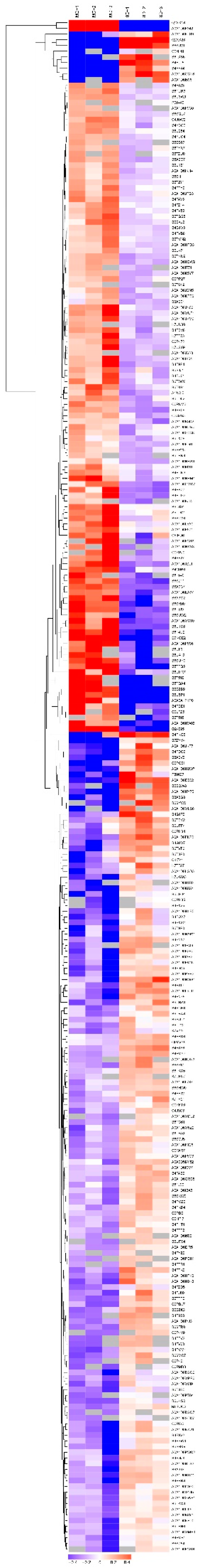
2.3. Analysis of Diffentially Abundant Proteins (DAPs) Observed in Different Experimental Comparisons
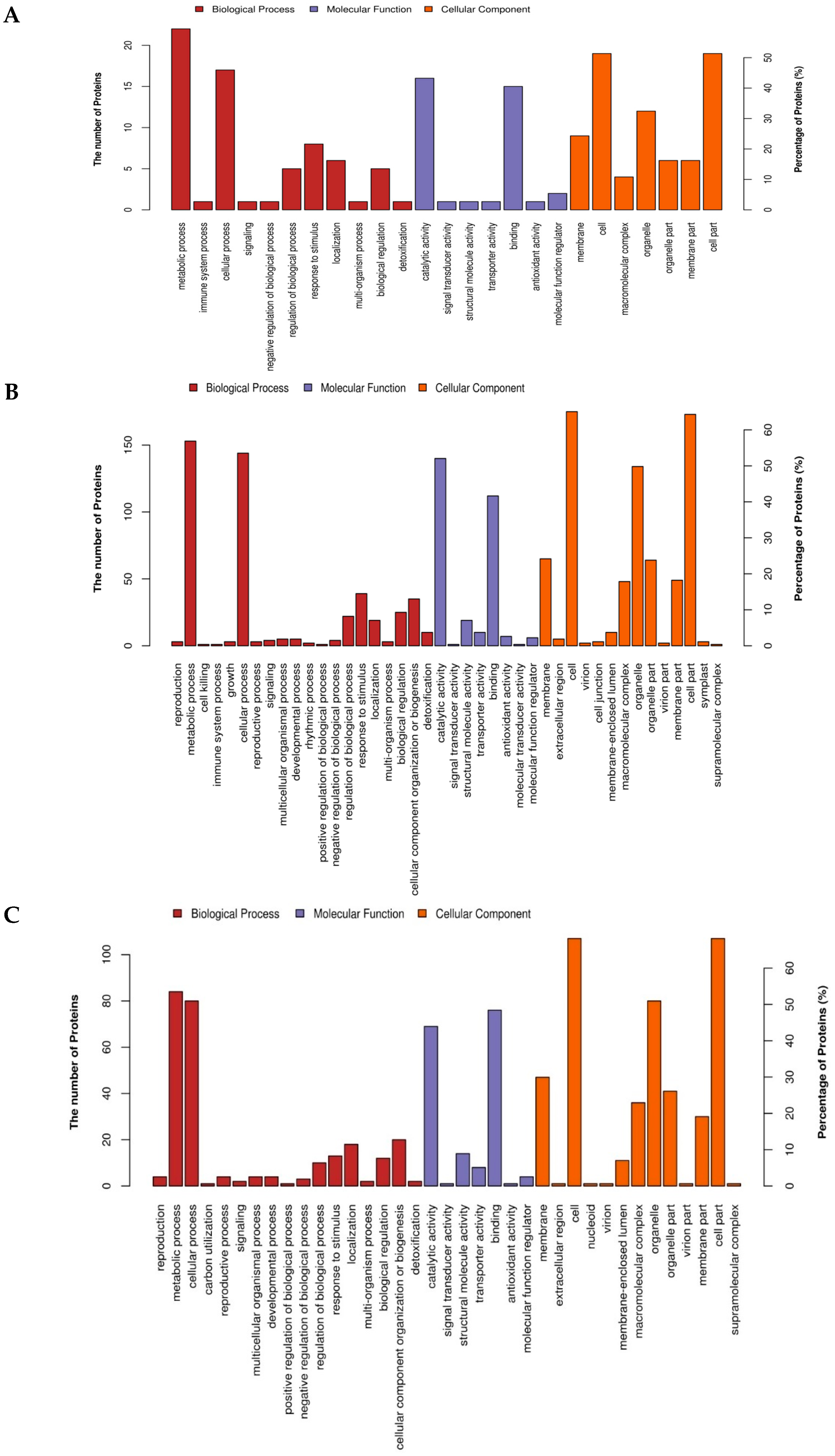
2.4. Gene Ontology (GO) Annotation and Functional Classification of the Drought Responsive DAPs
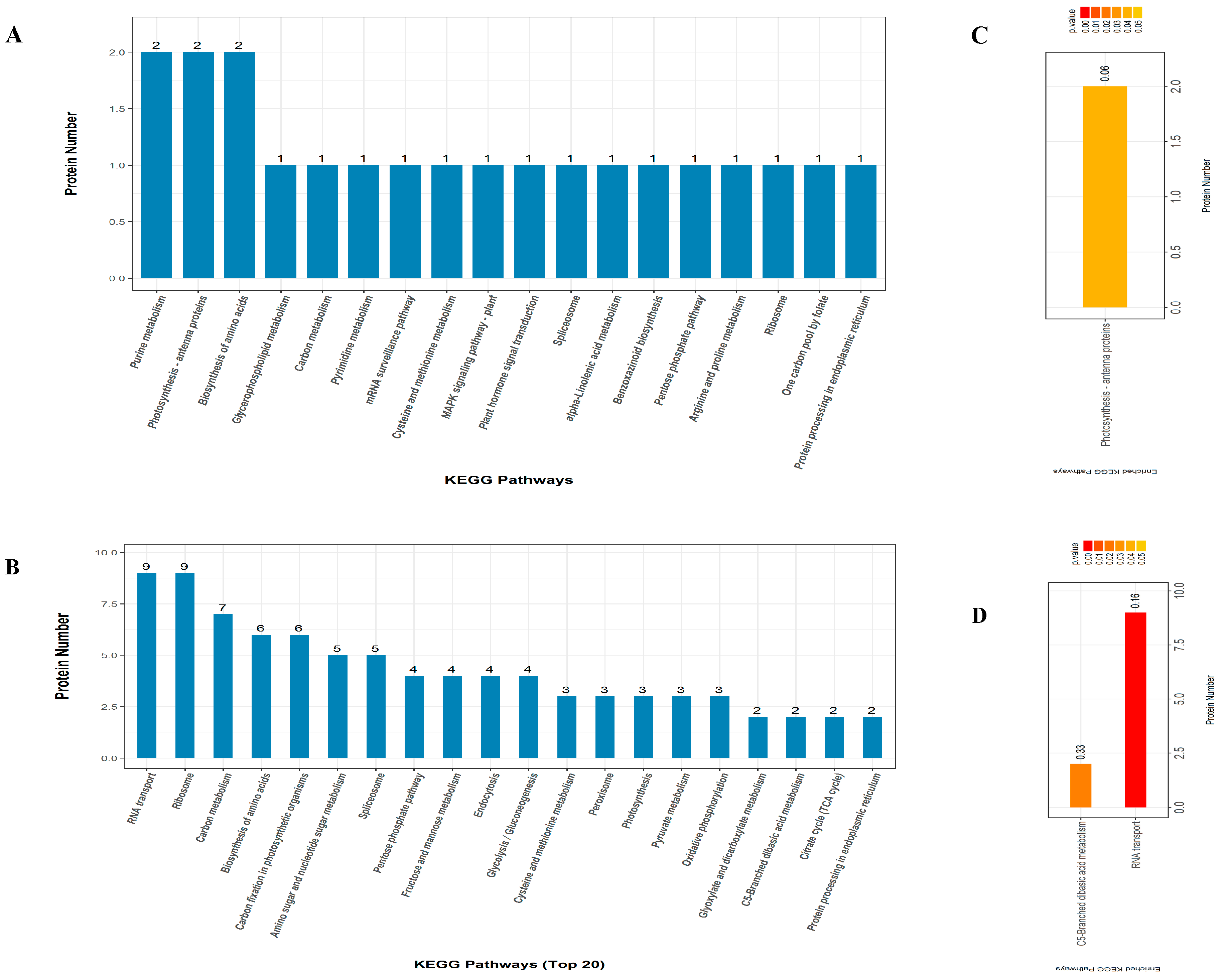
2.5. KEGG Pathway Enrichment Analysis
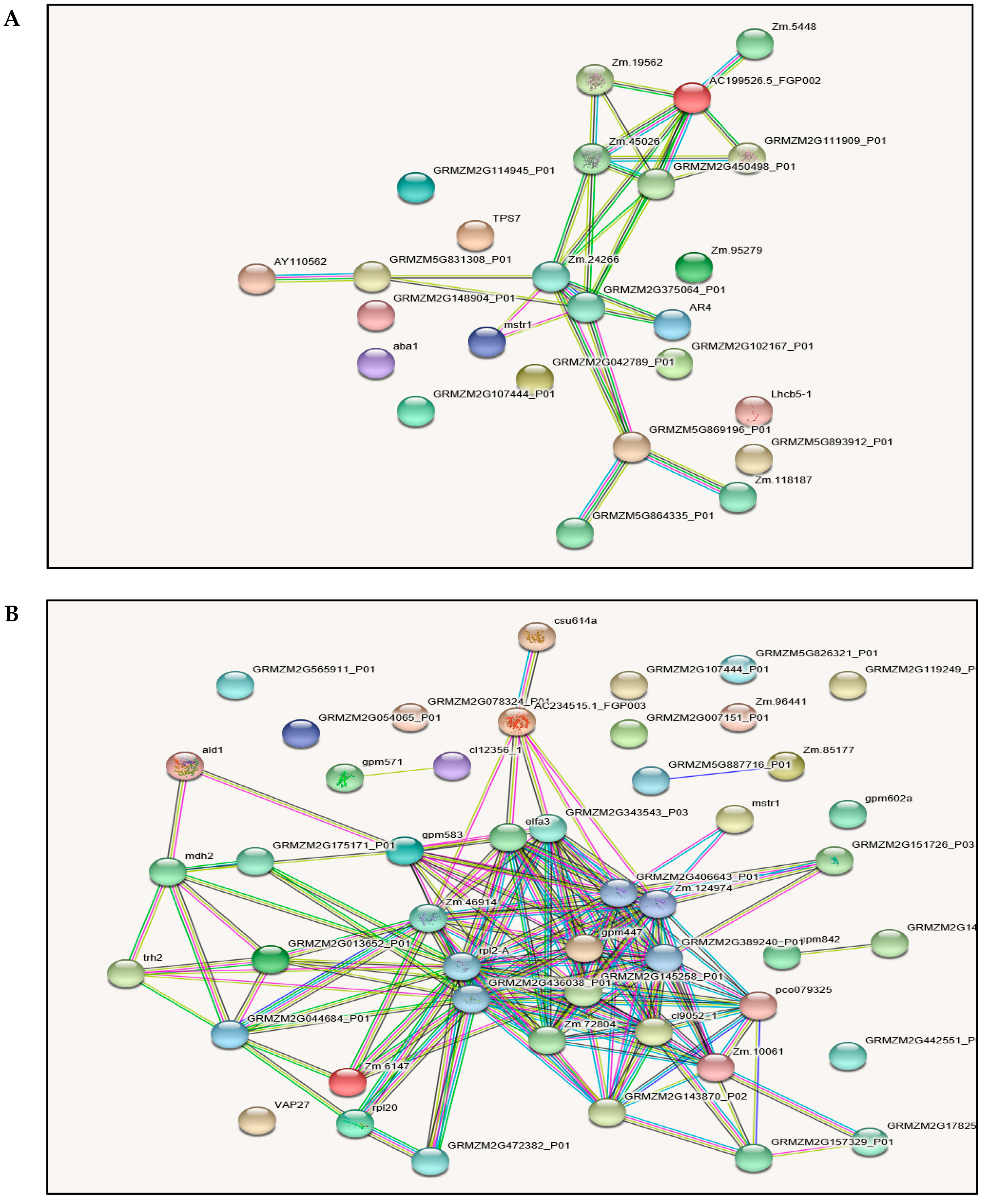
2.6. Protein-Protein Interactions (PPI)
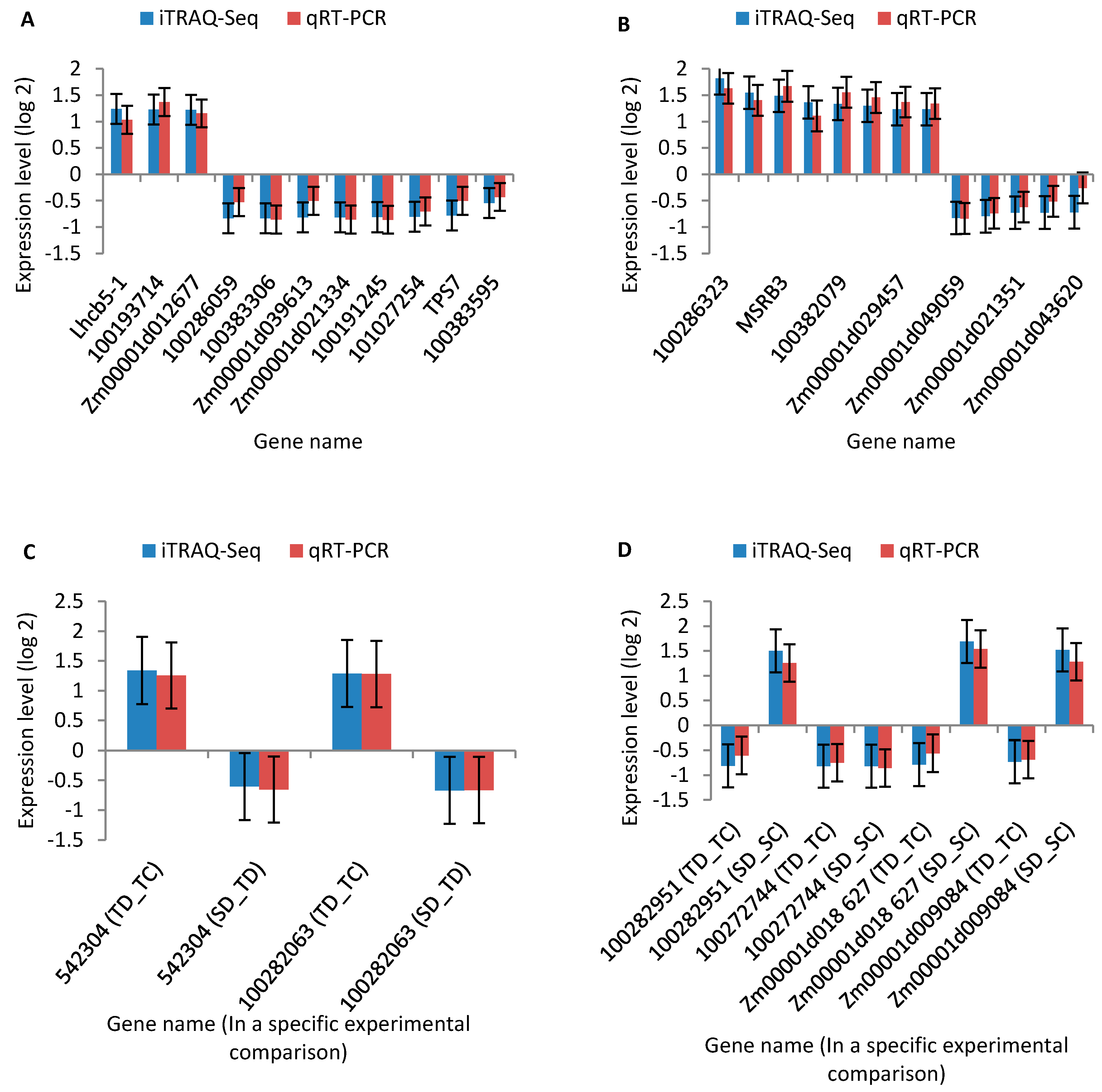
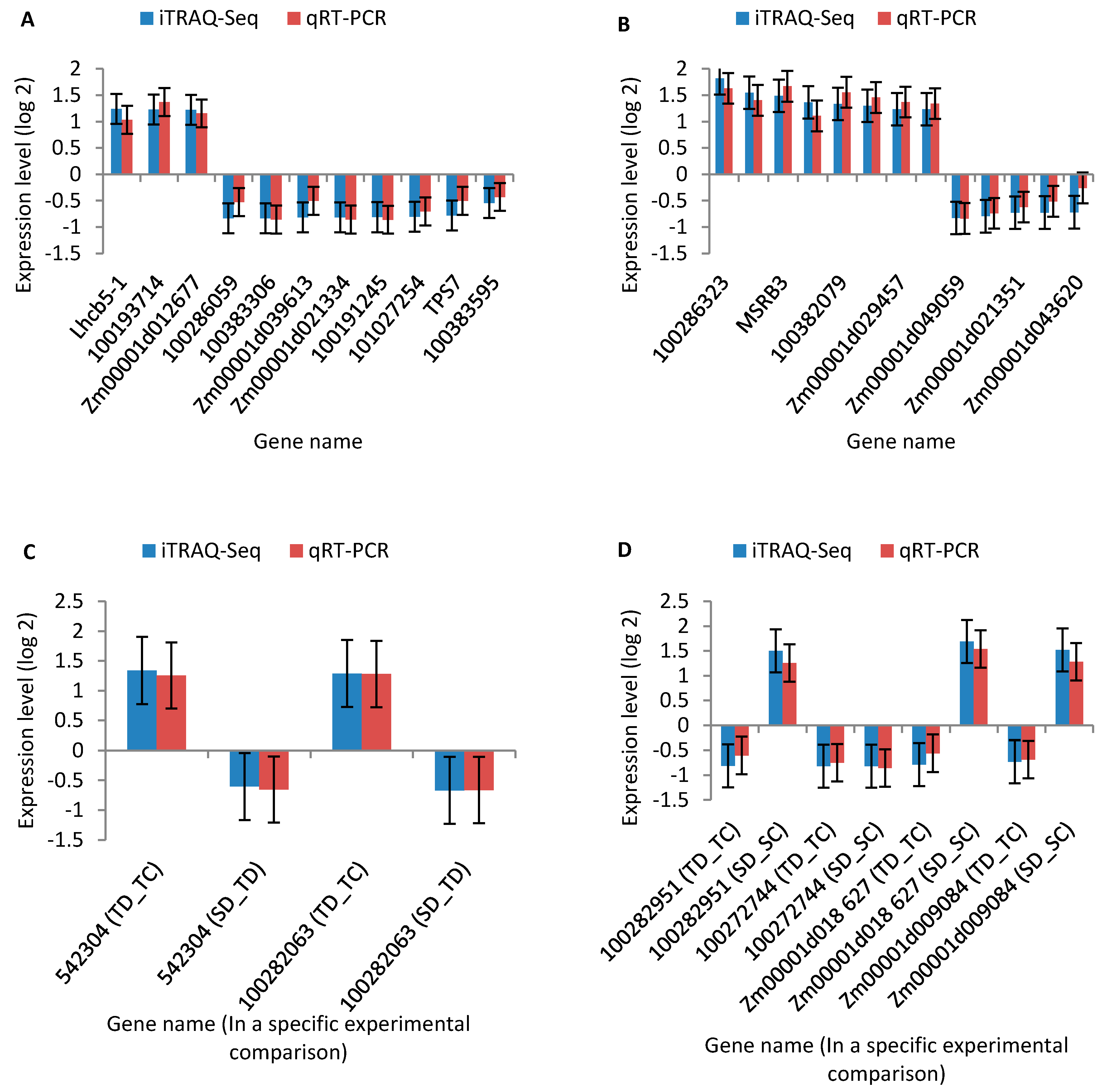
2.7. Quantitative Real-Time RT-PCR (qRT-PCR) Analysis
3. Discussion
3.1. Inbred Lines YE8112 and MO17 Showed Significant Differences in Physiological Response to Drought Stress
3.2. Drought Responsive DAPs Observed in the Tolerant Inbred Line YE8112
3.2.1. Photosynthesis (Photosystem II) Related Proteins Are the Major Drought Tolerance Signature in YE8112
3.2.2. Up-Regulation of Lipid-Metabolism Related Proteins Could Contribute to Increased Signaling and Water Conservation in the Cell
3.2.3. Enhancement of Molecular Chaperons Is a Vital Strategy for Drought Stress Tolerance in YE8112
3.2.4. Proteins/Enzymes Involved in Cellular Detoxification under Drought Stress
3.2.5. Proteins Related to ‘Response to Stimuli’ under Drought Stress
3.2.6. Key Epigenetic Regulation Mechanisms of the Tolerant Line YE8112
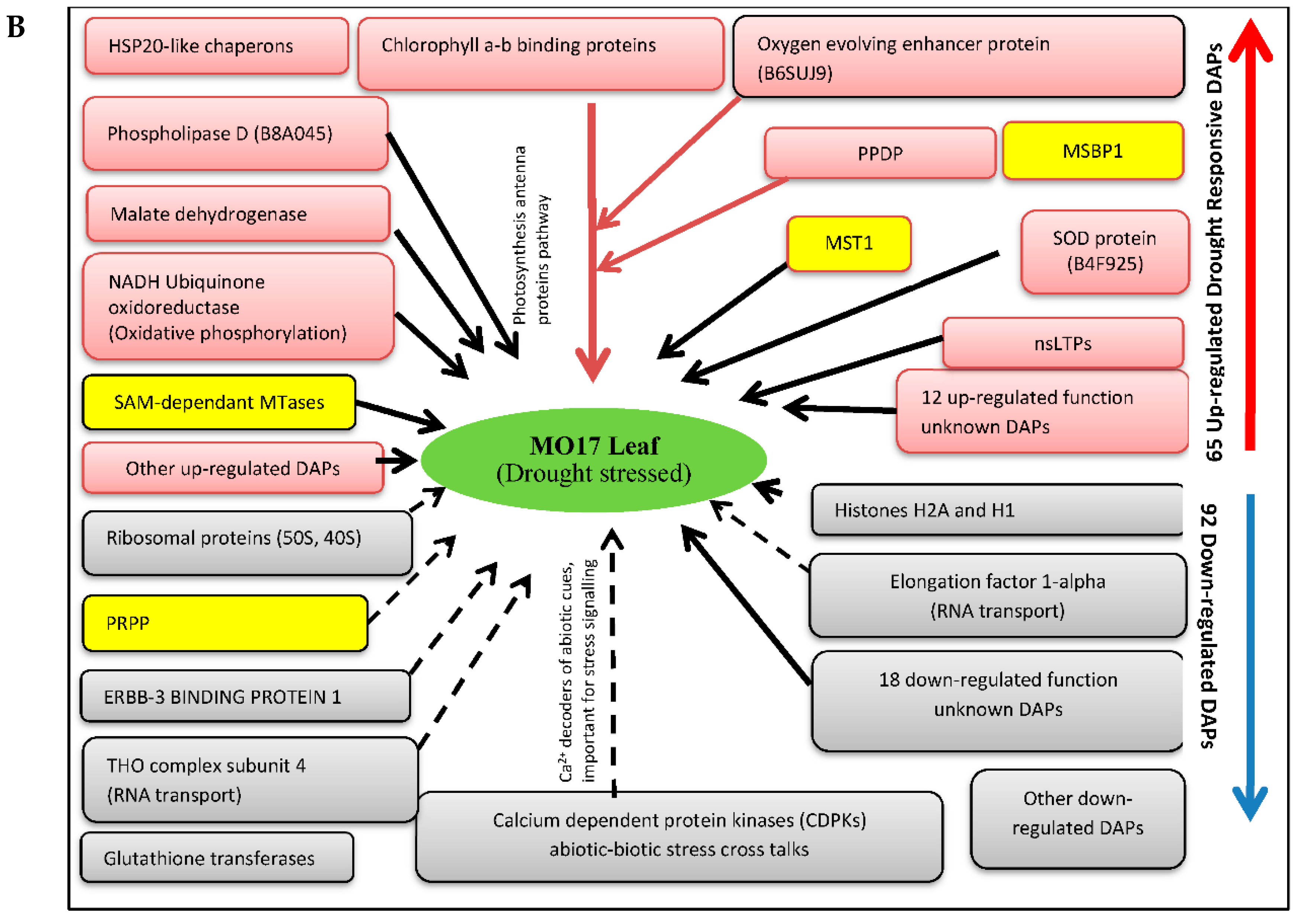
3.3. Drought Responsive DAPs Observed in Sensitive Inbred-Line MO17
3.3.1. Enhanced Expression of Heat Shock Proteins (HSP20-Like Chaperons) and 50S Ribosomal Proteins Constitutes a Critical Defensive Response in MO17
3.3.2. Up-Regulation of Cell Detoxification and Photosynthesis Related Proteins May Contribute to Enhanced Drought Stress Tolerance in MO17
3.3.3. Glutathione Transferases and Ca2+-Dependent Kinases Negatively Influenced by Short Term Drought Stress
3.3.4. Key Epigenetic Regulation Mechanisms of the Sensitive Line MO17
3.4. Overlapping Drought Responsive Proteins Between YE8112 and MO17 under Drought Conditions
3.5. Significantly Enriched Metabolic Pathways of DAPs under Drought Stress
3.6. Function-Unknown Proteins Identified Under Drought Stress Conditions
3.7. Protein-Protein Interaction (PPI) Analysis
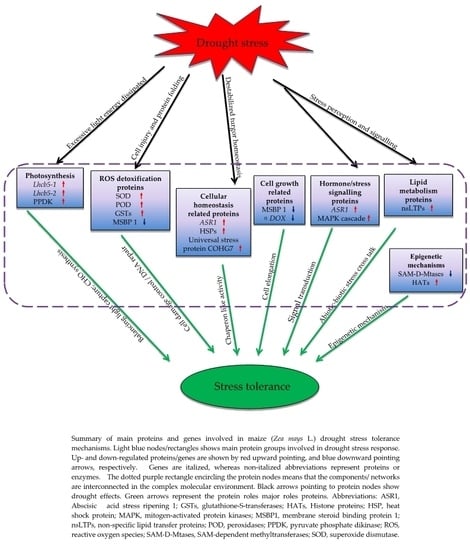
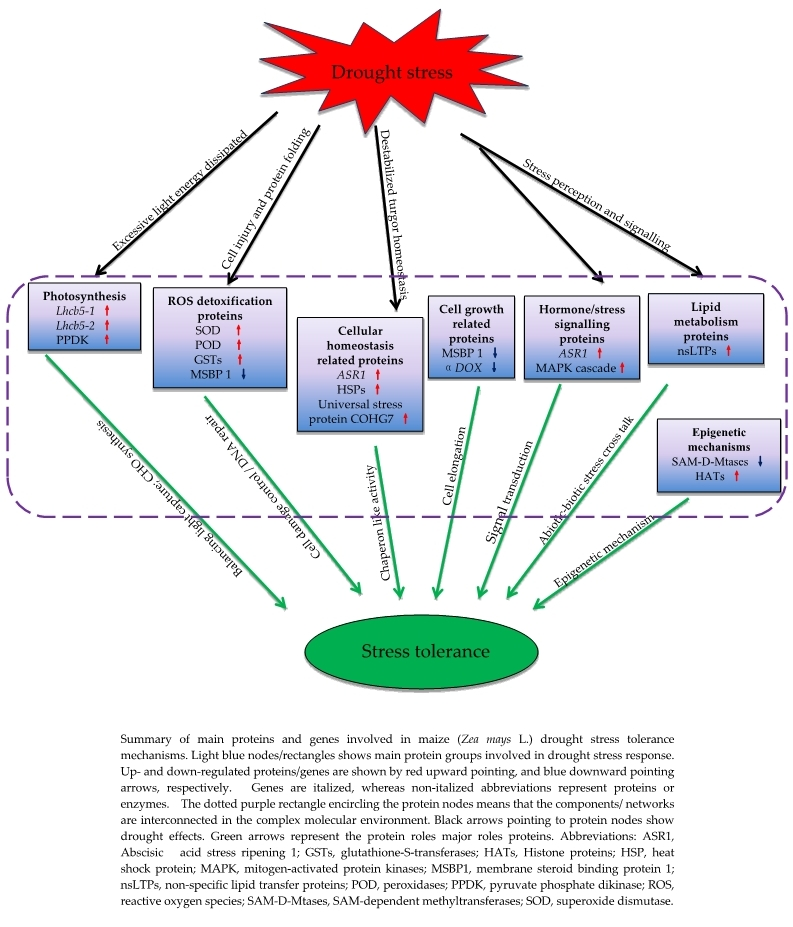
3.8. Proposed Models of Drought Stress Tolerance in Maize Seedlings
4. Materials and Methods
4.1. Plant Materials and Drought Stress Treatment
4.2. Phenotypic and Physiological Characterizations
4.3. Protein Extraction
4.4. Protein Digestion and iTRAQ (Isobaric Tags for Relative and Absolute Quantification) Labeling
4.5. Strong Cation Exchange (SCX) and LC-MS/MS Analysis
4.6. Protein Identification and Quantification
4.7. DAPs Functional Classification, Pathway Enrichment, and Hierarchal Clustering Analysis
4.8. RNA Extraction, cDNA Synthesis, and RT-qPCR Analysis
4.9. Statistical Data Analysis of Physiological Changes
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ASR1 | Abscisic acid stress ripening 1 |
| CDPKs | Calcium dependent protein kinases |
| DAPs | Differentially abundant proteins |
| GDPD | Glycerophosphodiester phosphodiesterase |
| GO | Gene ontology |
| GST | Glutathione-S-transferase |
| HSPs | Heat shock proteins |
| iTRAQ | Isobaric tags for relative and absolute quantification |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| LC-MS/MS | Liquid chromatography-tandem mass spectrometry |
| MAPK | Mitogen-activated protein kinases |
| MDA | Malondialdehyde |
| MSBP1 | Membrane steroid binding protein 1 |
| MSTs | Monosaccharide transporters |
| nsLTPs | Non-specific lipid transfer proteins |
| PPI | Protein-protein interaction |
| POD | Peroxidases |
| PRPP | Ribose-phosphate pyrophosphokinase |
| qRT-PCR | Quantitative real-time polymerase chain reaction |
| RBPs | RNA binding proteins |
| ROS | Reactive oxygen species |
| SAM | S-adenosyl-methionine |
| SAM-d-Mtases | SAM-dependent methyltransferases |
| SOD | Superoxide dismutase |
| TRX | Thioredoxin |
References
- Campos, H.; Cooper, M.; Habben, J.E.; Edmeades, G.O.; Schussler, J.R. Improving drought tolerance in maize: A view from industry. Field Crops Res. 2004, 90, 19–34. [Google Scholar] [CrossRef]
- Li, G.K.; Gao, J.; Peng, H.; Shen, Y.O.; Ding, H.P.; Zhang, Z.M.; Pan, G.T.; Lin, H.J. Proteomic changes in maize as a response to heavy metal (lead) stress revealed by iTRAQ quantitative proteomics. Genet. Mol. Res. 2016, 15, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Min, H.; Chen, C.; Wei, S.; Shang, X.; Sun, M.; Xia, R.; Liu, X.; Hao, D.; Chen, H.; Xie, Q. Identification of drought tolerant mechanisms in maize seedlings based on transcriptome analysis of recombination inbred lines. Front Plant Sci. 2016, 7, 1080. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Fu, J.; Gou, M.; Huai, J.; Liu, Y.; Jian, M.; Huang, Q.; Guo, X.; Dong, Z.; Wang, H.; et al. Genome-wide transcriptome analysis of two maize inbred lines under drought stress. Plant Mol. Biol. 2010, 72, 407–421. [Google Scholar] [CrossRef] [PubMed]
- Cui, D.; Wu, D.; Liu, J.; Li, D.; Xu, C.; Li, S.; Li, P.; Zhang, H.; Liu, X.; Jiang, C.; et al. Proteomic analysis of seedling roots of two maize inbred lines that differ significantly in the salt stress response. PLoS ONE 2015, 10, e0116697. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Shan, X.; Wu, Y.; Su, S.; Li, S.; Liu, H.; Han, J.; Xue, C.; Yuan, Y. iTRAQ-based quantitative proteomic analysis reveals new metabolic pathways responding to chilling stress in maize seedlings. J. Proteom. 2016, 146, 14–24. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Xu, W.; Velten, J.; Xin, Z.; Stout, J. Characterization of maize inbred lines for drought and heat tolerance. J. Soil Water Conserv. 2012, 67, 354–364. [Google Scholar] [CrossRef] [Green Version]
- Kaul, J.; Kumar, R.; Kumar, R.S.; Dass, S.; Bhat, B.; Kamboj, O.P.; Nara, S.; Yadav, A.K. Response of maize (Zea mays L.) hybrids to excess soil moisture stress at different growth stages. Res. Crops 2013, 4, 439–443. [Google Scholar]
- Farooq, M.; Wahid, A.; Kobayashi, N.; Fujita, D.; Basra, S.M.A. Plant drought stress: Effects, mechanisms and management. Agron. Sustain. Dev. 2009, 29, 185–212. [Google Scholar] [CrossRef]
- Lei, L.; Shi, J.; Chen, J.; Zhang, M.; Sun, S.; Xie, S.; Li, X.; Zeng, B.; Peng, L.; Hauck, A.; et al. Ribosome profiling reveals dynamic translational landscape in maize seedlings under drought stress. Plant J. 2015, 84, 1206–1218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raos, G.J.N.; Reddy, J.N.; Variar, M.; Mahender, A. Molecular breeding to improve plant breeding to improve plant resistance to abiotic stresses. Adv. Plant Breed. Strateg. 2016, 2, 283–326. [Google Scholar] [CrossRef]
- Ahmadi, A.; Emam, Y.; Pessarakli, M. Biochemical changes in maize seedlings exposed to drought stress conditions at different nitrogen levels. J. Plant Nutr. 2010, 33, 541–556. [Google Scholar] [CrossRef]
- Yin, X.G.; Jørgen, E.O.; Wang, M.; Kersebaum, K.-C.; Chen, H.; Baby, S.; Öztürk, I.; Chen, F. Adapting maize production to drought in the northeast farming region of China. Eur. J. Agron. 2016, 77, 47–58. [Google Scholar] [CrossRef]
- Maiti, R.K.; Maiti, L.E.; Sonia, M.; Maiti, A.M.; Maiti, M.; Maiti, H. Genotypic variability in maize (Zea mays L.) for resistance to drought and salinity at the seedling stage. J. Plant Physiol. 1996, 148, 741–744. [Google Scholar] [CrossRef]
- Cao, L.Z.X.; Wj, B.X.P. Discuss on evaluating method to Drought-resistance of Maize in seedling stage. J. Maize Sci. 2004, 12, 73–75. [Google Scholar]
- Miao, Z.; Han, Z.; Zhang, T.; Chen, S.; Ma, C. A systems approach to spatio-temporal understanding of the drought stress response in maize. Sci. Rep. 2017, 7, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Li, X.; Li, W.; Li, M.; Liu, X. Analysis on difference for drought responses of maize inbred lines at seedling stage. J. Maize Sci. 2004, 12, 63–65. [Google Scholar]
- Wu, B.; Li, X.; Xiao, M. Genetic variation in fifty-three maize inbred lines in relation to drought tolerance at seedling stage. Sci. Agric. Sin. 2007, 40, 665–676. [Google Scholar]
- Oliver, S.N.; Dennis, E.S.; Dolferus, R. ABA regulates apoplastic sugar transport and is a potential signal for cold-induced pollen sterility in rice. Plant Cell Physiol. 2007, 48, 1319–1330. [Google Scholar] [CrossRef] [PubMed]
- Jogaiah, S.; Govind, S.R.; Tran, L.S. Systems biology-based approaches toward understanding drought tolerance in food crops. Crit. Rev. Biotechnol. 2013, 33, 23–39. [Google Scholar] [CrossRef] [PubMed]
- Luan, M.; Xu, M.; Lu, Y.; Zhang, L.; Fan, Y.; Wang, L. Expression of zma-miR169 miRNAs and their target ZmNF-YA genes in response to abiotic stress in maize leaves. Gene 2015, 555, 178–185. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Zhu, Q.G.; Meng, Q.W.; Wu, C.A. Transcript profiling during salt stress of young cotton (Gossypium hirsutum) seedlings via Solexa sequencing. Acta Physiol. Plant. 2012, 34, 107–115. [Google Scholar] [CrossRef]
- Chen, Y.; Liu, Z.H.; Feng, L.; Zheng, Y.; Li, D.D.; Li, X.B. Genome-wide functional analysis of cotton (Gossypium hirsutum) in response to drought. PLoS ONE 2013, 8, e80879. [Google Scholar] [CrossRef] [PubMed]
- Chan, Z.L. Expression profiling of ABA pathway transcripts indicates crosstalk between abiotic and biotic stress responses in Arabidopsis. Genomics 2012, 100, 110–115. [Google Scholar] [CrossRef] [PubMed]
- Canovas, F.M.; Dumas-Gaudot, E.; Recorbet, G.; Jorrin, J.; Mock, H.P.; Rossignol, M. Plant proteome analysis. Proteomics 2004, 4, 285–298. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Zhang, H.; Wang, T.; Chen, S.; Dai, S. Proteomics-based investigation of salt-responsive mechanisms in plant roots. J. Proteom. 2013, 82, 230–253. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, S.; Hiraga, S.; Yanagawa, Y. Proteomics techniques for the development of flood tolerant crops. J. Proteome Res. 2012, 11, 68–78. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Ning, F.; Zhang, Q.; Wu, X.; Wang, W. Enhancing omics research of crop responses to drought under field conditions. Front. Plant Sci. 2017, 8, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Kosova, K.; Vitamvas, P.; Prasil, I.T.; Renaut, J. Plant proteome changes under abiotic stress-contribution of proteomics studies to understanding plant stress response. J. Proteom. 2011, 74, 1301–1322. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Jiang, T.; Fountain, J.C.; Scully, B.T.; Lee, R.D.; Kemerait, R.C.; Chen, S.; Guo, B. Protein profiles reveal diverse responsive signaling pathways in kernels of two maize inbred lines with contrasting drought sensitivity. Int. J. Mol. Sci. 2014, 15, 18892–18918. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Wu, L.; Zhao, F.; Zhang, D.; Li, N.; Zhu, G.; Li, C.; Wang, W. Phosphoproteomic analysis of the response of maize leaves to drought, heat and their combination stress. Front. Plant Sci. 2015, 6, 298. [Google Scholar] [CrossRef] [PubMed]
- Benešová, M.; Holá, D.; Fischer, L.; Jedelský, P.L.; Hnilička, F.; Wilhelmová, N.; Rothová, O.; Kočová, M.; Procházková, D.; Honnerová, J.; et al. The physiology and proteomics of drought tolerance in maize: Early stomatal closure as a cause of lower tolerance to short-term dehydration? PLoS ONE 2012, 7, e38017. [Google Scholar] [CrossRef] [PubMed]
- Gong, F.; Hu, X.; Wang, W. Proteomic analysis of crop plants under abiotic stress conditions: Where to focus our research? Front. Plant Sci. 2015, 6, 418. [Google Scholar] [CrossRef] [PubMed]
- Dong, M.; Gu, J.; Zhang, L.; Chen, P.; Liu, T.; Deng, J.; Lu, H.; Han, L.; Zhao, B. Comparative proteomics analysis of superior and inferior spikelets in hybrid rice during grain filling and response of inferior spikelets to drought stress using isobaric tags for relative and absolute quantification. J. Proteom. 2014, 109, 382–399. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Du, H.; Wang, Z.; Huang, B. Identification of proteins associated with water-deficit tolerance in C4 perennial grass species, Cynodon dactylonxCynodon transvaalensis and Cynodon dactylon. Physiol. Plant. 2011, 141, 40–55. [Google Scholar] [CrossRef] [PubMed]
- Ross, P.L.; Huang, Y.N.; Marchese, J.N.; Williamson, B.; Parker, K.; Hattan, S.; Khainovski, N.; Pillai, S.; Dey, S.; Daniels, S.; et al. Multiplexed protein quantitation in Saccharomyces cerevisiae using amine-reactive isobaric tagging reagents. Mol. Cell. Proteom. 2004, 3, 1154–1169. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Wang, W. Increasing confidence of proteomics data regarding the identification of stress-responsive proteins in crop plants. Front. Plant Sci. 2016, 7, 702. [Google Scholar] [CrossRef] [PubMed]
- Qi, S.; Li, X.; Zhang, D.; Li, X.; Hao, Z.; Weng, J.; Li, M.; Zhang, S. Trends in drought tolerance in Chinese maize cultivars from the 1950s to the 2000s. Field Crops Res. 2017, 201, 175–183. [Google Scholar] [CrossRef]
- Zhang, Z.; Tang, W.; Tao, Y.; Zheng, Y. cDNA microarray analysis of early response to submerging stress in Zea mays roots. Russ. J. Plant Physiol. 2005, 52, 43–49. [Google Scholar] [CrossRef]
- Edmeades, G.O. Progress in Achieving and Delivering Drought Tolerance in Maize—An Update; ISAA: Ithaca, NY, USA, 2013; pp. 1–39. [Google Scholar]
- Zhu, J.K. Abiotic stress signaling and responses in plants. Cell 2016, 167, 313–324. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Komatsu, S. Plant subcellular proteomics: Application for exploring optimal cell function in soybean. J. Proteom. 2016, 143, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Gill, S.S.; Tuteja, N. Reactive oxygen species and antioxidant machinery in abiotic stress tolerance in crop plants. Plant Physiol. Biochem. 2010, 48, 909–930. [Google Scholar] [CrossRef] [PubMed]
- Islam, M.; Begum, M.C.; Kabir, A.H.; Alam, M.F. Molecular and biochemical mechanisms associated with differential responses to drought tolerance in wheat (Triticum aestivum L.). J. Plant Interact. 2015, 10, 195–201. [Google Scholar] [CrossRef]
- Kamies, R.; Farrant, J.M.; Tadele, Z.; Cannarozzi, G.; Rafudeen, M.S. A Proteomic Approach to Investigate the Drought Response in the Orphan Crop Eragrostis tef. Proteome 2017, 5, 32. [Google Scholar] [CrossRef] [PubMed]
- Moussa, H.R.; Abdel-Aziz, S.M. Comparative response of drought tolerant and drought sensitive maize genotypes to water stress. Aust. J. Crop Sci. 2008, 1, 31–36. [Google Scholar]
- Miller, G.; Suzuki, N.; Ciftciyilmaz, S.; Mittler, R. Reactive oxygen species homeostasis and signalling during drought and salinity stresses. Plant Cell Environ. 2010, 33, 453–467. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Jha, A.B.; Dubey, R.S.; Pessarakli, M. Reactive Oxygen Species, Oxidative Damage, and Antioxidative Defense Mechanism in Plants under Stressful Conditions. J. Botany 2012, 10, 26. [Google Scholar] [CrossRef]
- Mattioni, C.; Lacerenza, N.G.; Troccoli, A.; De Leonardis, A.M.; Di Fonzo, N. Water and salt stress-induced alterations in proline metabolism of Triticum durum seedlings. Physiol. Plant. 1997, 101, 787–792. [Google Scholar] [CrossRef]
- Kumar, S.G.; Matta Reddy, A.; Sudhakar, C. NaCl effects on proline metabolism in two high yielding genotypes of mulberry (Morus alba L.) with contrasting salt tolerance. Plant Sci. 2003, 165, 1245–1251. [Google Scholar] [CrossRef]
- Sairam, R.K.; Roa, K.V.; Srivastava, G.C. Differential response of wheat cultivar genotypes to long term salinity stress in relation oxidative stress, antioxidant activity, and osmolyte concentration. Plant Sci. 2000, 163, 1037–1048. [Google Scholar] [CrossRef]
- Foyer, C.H.; Harbinson, J. Oxygen metabolism and the regulation of photosynthetic electron transport. In Causes of Photooxidative Stresses and Amelioration of Defense Systems in Plants; Foyer, C.H., Mullineaux, P., Eds.; CRC Press: Boca Raton, FL, USA, 1994; pp. 1–42. [Google Scholar]
- Luca, D.; Stefano, C.; Mauro, B.; David, P.; Karekl, Ž.; Niyogi, K.K.; Fleming, G.R.; Zigmantas, D.; Bassi, R. Two mechanisms for dissipation of excess light in monomeric and trimeric light-harvesting complexes. Nat. Plants 2010, 3, 17033. [Google Scholar] [CrossRef]
- Tai, F.J.; Yuan, Z.L.; Wu, X.L.; Zhao, P.F.; Hu, X.L.; Wang, W. Identification of membrane proteins in maize leaves, altered in expression under drought stress through polyethylene glycol treatment. Plant Omics J. 2011, 4, 250–256. [Google Scholar]
- Edstam, M.M.; Viitanen, L.; Salminen, T.A.; Edqvist, J. Evolutionary history of the non-specific lipid transfer proteins. Mol. Plant 2011, 4, 947–964. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.B.; Go, Y.S.; Bae, H.J.; Park, J.H.; Cho, S.H.; Cho, H.J.; Lee, D.S.; Park, O.K.; Hwang, I.; Suh, M.C. Disruption of glycosylphosphatidylinositol-anchored lipid transfer protein gene altered cuticular lipid composition, increased plastoglobules and enhanced susceptibility to infection by the fungal pathogen. Alternaria brassicicola. Plant Physiol. 2009, 150, 42–54. [Google Scholar] [CrossRef] [PubMed]
- Molina, A.; Segura, A.; García-Olmedo, F. Lipid transfer proteins (nsLTPs) from barley and maize leaves are potent inhibitors of bacterial and fungal plant pathogens. FEBS Lett. 1993, 316, 119–122. [Google Scholar] [CrossRef] [Green Version]
- Tuteja, N. Mechanisms of high salinity tolerance in plants. Methods Enzymol. 2007, 428, 419–438. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, Y.; Koizumi, R.; Shui, G.; Shimojima, M.; Wenk, M.R.; Ito, T.; Ohta, H. Arabidopsis lipins mediate eukaryotic pathway of lipid metabolism and cope critically with phosphate starvation. Proc. Natl. Acad. Sci. USA 2009, 106, 20978–20983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, Y.; Zhou, W.; El Sheery, N.I.; Peters, C.; Li, M.; Wang, X.; Huang, J. Characterization of the Arabidopsis glycerophosphodiester phosphodiesterase (GDPD) family reveals a role of the plastid-localized AtGDPD1 in maintaining cellular phosphate homeostasis under phosphate starvation. Plant J. 2011, 66, 781–795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rom, S.; Gilad, A.; Kalifa, Y.; Konrad, Z.; Karpasas, M.M.; Goldgur, Y.; Bar-Zvi, D. Mapping the DNA- and zinc-binding domains of ASR1 (abscisic acid stress ripening), an abiotic-stress regulated plant specific protein. Biochimie 2006, 88, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Konrad, Z.; Bar-Zvi, D. Synergism between the chaperone-like activity of the stress regulated ASR1 protein and the osmolyte glycine-betaine. Planta 2008, 227, 1213–1219. [Google Scholar] [CrossRef] [PubMed]
- Kalifa, Y.; Perlson, E.; Gilad, A.; Konrad, Z.; Scolnik, P.A.; Bar-Zvi, D. Over expression of the water and salt stress-regulated Asr1 gene confers an increased salt tolerance. Plant Cell Environ. 2004, 27, 1459–1468. [Google Scholar] [CrossRef]
- Kalifa, Y.; Gilad, A.; Konrad, Z.; Zaccai, M.; Scolnik, P.A.; Bar-Zvi, D. The water- and salt-stress-regulated Asr1 (abscisic acid stress ripening) gene encodes a zinc-dependent DNA-binding protein. Biochem. J. 2004, 381 Pt 2, 373–378. [Google Scholar] [CrossRef]
- Isokpehi, R.D.; Simmons, S.S.; Cohly, H.H.P.; Ekunwe, S.I.; Begonia, G.B.; Ayensu, W.K. Identification of drought-responsive universal stress proteins in Viridiplantae. Bioinform. Biol. Insights 2011, 5, 41–58. [Google Scholar] [CrossRef] [PubMed]
- Tkaczuk, K.L.; Shumilin, I.A.; Chruszcz, M.; Evdokimova, E.; Savchenko, A.; Minor, W. Structural and functional insight into the universal stress protein family. Evol. Appl. 2013, 6, 434–449. [Google Scholar] [CrossRef] [PubMed]
- Mindrebo, J.T.; Nartey, C.M.; Seto, Y.; Burkart, M.D.; Noel, J.P. Unveiling the functional diversity of the alpha/beta hydrolase superfamily in the plant kingdom. Curr. Opin. Struct. Biol. 2016, 41, 233–246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, U.; Rioflorido, I.; Hong, S.W.; Larkindale, J.; Waters, E.R.; Vierling, E. The Arabidopsis ClpB/Hsp100 family of proteins: Chaperones for stress and chloroplast development. Plant J. 2007, 49, 115–127. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.Y.; Brown, R.L.; Damann, K.E.; Cleveland, T.E. Identification of unique or elevated levels of kernel prpteins in aflatoxin-resistant maize genotypes through proteome analysis. Phytopathology 2002, 92, 1084–1094. [Google Scholar] [CrossRef] [PubMed]
- Cruz de Carvalho, M.H. Drought stress and reactive oxygen species:Production, scavenging and signaling. Plant Signal. Behav. 2008, 3, 156–165. [Google Scholar] [CrossRef] [PubMed]
- Wan, X.Y.; Liu, J.Y. Comparative proteomics analysis reveals an intimate protein network provoked by hydrogen peroxide stress in rice seedling leaves. Mol. Cell. Proteom. 2008, 7, 1469–1488. [Google Scholar] [CrossRef] [PubMed]
- Gong, H.; Jiao, Y.; Hu, W.W.; Pua, E.C. Expression of glutathione-S-transferase and its role in plant growth and development in vivo and shoot morphogenesis in vitro. Plant Mol. Biol. 2005, 57, 53–66. [Google Scholar] [CrossRef] [PubMed]
- Sytykiewicz, H.; Chrzanowski, G.; Czerniewicz, P.; Sprawka, I.; Łukasik, I.; Goławska, S.; Sempruch, C. Expression profiling of selected glutathione transferase genes in Zea mays (L.) seedlings infested with cereal aphids. PLoS ONE 2014, 9, e111863. [Google Scholar] [CrossRef] [PubMed]
- Cummins, I.; Wortley, D.J.; Sabbadin, F.; He, Z.; Coxon, C.R.; Straker, H.E.; Sellars, J.D.; Knight, K.; Edwards, L.; Hughes, D.; et al. Key role for a glutathione transferase in multiple-herbicide resistance in grass weeds. Proc. Natl. Acad. Sci. USA 2013, 110, 5812–5817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lan, T.; Yang, Z.L.; Yang, X.; Liu, Y.J.; Wang, X.R.; Zeng, Q.Y. Extensive functional diversification of the Populus glutathione S-transferase supergene family. Plant Cell 2009, 21, 3749–3766. [Google Scholar] [CrossRef] [PubMed]
- Cançado, G.M.A.; De Rosa, V.E.; Fernandez, J.H.; Maron, L.G.; Jorge, R.A.; Menossi, M. Glutathione S-transferase and aluminum toxicity in maize. Funct. Plant Biol. 2005, 32, 1045–1055. [Google Scholar] [CrossRef]
- Buchanan, B.; Schurmann, P.; Wolosiuk, R.; Jacquot, J. The ferredoxin/thioredoxin system: From discovery to molecular structures and beyond. Discov. Photosynth. 2002, 73, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Balmer, Y.; Vensel, W.H.; Cai, N.; Manieri, W.; Schürmann, P.; Hurkman, W.J.; Buchanan, B.B. A complete ferredoxin_thioredoxin system regulates fundamental processes in amyloplasts. Proc. Natl. Acad. Sci. USA 2006, 103, 2988–2993. [Google Scholar] [CrossRef] [PubMed]
- Broin, M.; Rey, P. Potato plants lacking the CDSP32 plastidic thioredoxin exhibit overoxidation of the BAS1 2-cysteine peroxiredoxin and increased lipid peroxidation in thylakoids under photooxidative stress. Plant Physiol. 2003, 132, 1335–1343. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Shi, S. Physiological and Proteomic Responses of Contrasting Alfalfa (Medicago sativa L.) Varieties to PEG-Induced Osmotic Stress. Front. Plant Sci. 2018, 9, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Wahid, A.; Gelani, S.; Ashraf, M.; Foolad, M.R. Heat tolerance in plants: An overview. Environ. Exp. Bot. 2007, 61, 199–223. [Google Scholar] [CrossRef]
- Kuroha, T.; Tokunaga, H.; Kojima, M.; Ueda, N.; Ishida, T.; Nagawa, S.; Fukuda, H.; Sugimoto, K.; Sakakibara, H. Functional analyses of LONELY GUY cytokinin-activating enzymes reveal the importance of the direct activation pathway in Arabidopsis. Plant Cell 2009, 21, 3152–3169. [Google Scholar] [CrossRef] [PubMed]
- Argueso, C.T.; Ferreira, F.J.; Kieber, J.J. Environmental perception avenues: The interaction of cytokinin and environmental response pathways. Plant Cell Environ. 2009, 32, 1147–1160. [Google Scholar] [CrossRef] [PubMed]
- Pavlů, J.; Novák, J.; Koukalová, V.; Luklová, M.; Brzobohatý, B.; Černý, M. Cytokinin at the crossroads of abiotic stress signalling pathways. Int. J. Mol. Sci. 2018, 19, 2450. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Wang, Y.; Yang, H.; Wang, W.; Wu, J.; Hu, X. Quantitative proteomic analyses identify aba-related proteins and signal pathways in maize leaves under drought conditions. Front. Plant Sci. 2016, 7, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Li, J.; Liu, H.; Chong, K.; Xu, Y. Roles of ubiquitination-mediated protein degradation in plant responses to abiotic stresses. Environ. Exp. Bot. 2015, 114, 92–103. [Google Scholar] [CrossRef]
- Bannenberg, G.; Martı’nez, M.; Rodrı’guez, M.J.; Lo’pez, M.A.; Ponce de León, I.; Hamberg, M.; Castresana, C. Functional analysis of a-DOX2, an active a-Dioxygenase critical for normal development in tomato plants. Plant Physiol. 2009, 151, 1421–1432. [Google Scholar] [CrossRef] [PubMed]
- Mazid, M.; Khan, T.A.; Mohammad, F. Role of secondary metabolites in defense mechanisms of plants. Biol. Med. 2011, 3, 232–249. [Google Scholar]
- Falara, V.; Akhtar, T.A.; Nguyen, T.T.; Spyropoulou, E.A.; Bleeker, P.M.; Schauvinhold, I.; Matsuba, Y.; Bonini, M.E.; Schilmiller, A.L.; Last, R.L.; et al. The tomato terpene synthase gene family. Plant Physiol. 2011, 157, 770–789. [Google Scholar] [CrossRef] [PubMed]
- Jia, Q.; Köllner, T.G.; Gershenzon, J.; Chen, F. MTPSLs: New terpene synthases in nonseed plants. Trends Plant Sci. 2018, 23, 121–128. [Google Scholar] [CrossRef] [PubMed]
- Jonak, C.; Heberle-Bors, E.; Hirt, H. MAP kinases: Universal multipurpose signalling tools. Plant Mol. Biol. 1994, 24, 407–416. [Google Scholar] [CrossRef] [PubMed]
- Morris, P.C. MAP kinase signaltransduction pathways in plants. New Phytol. 2001, 151, 67–89. [Google Scholar] [CrossRef]
- Xu, J.; Li, Y.; Wang, Y.; Liu, H.; Lei, L.; Yang, H.; Liu, G.; Ren, D. Activation of MAPK kinase 9 induces ethylene and camalexin biosynthesis and enhances sensitivity to salt stress in Arabidopsis. J. Biol. Chem. 2008, 283, 26996–27006. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, M.W.; Milena, R.; Morten, P.; John, M. MAP kinase cascades in arabidopsis innate immunity. Front. Plant Sci. 2012, 3, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.B.; Brendel, V. Molecular characterization and phylogeny of U2AF homologs in plants. Plant Physiol. 2006, 140, 624–636. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Singh, A. Epigenetic regulation of abiotic stress tolerance in plants. Adv. Plants Agric. Res. 2016, 5, 15406. [Google Scholar] [CrossRef]
- Yaish, M.W. DNA methylation-associated epigenetic regulation changes in stress tolerance of plants. In Molecular Stress Physiology of Plants; Rout, G.R., Das, A.B., Eds.; Springer: New Dehli, India, 2013; pp. 427–439. [Google Scholar]
- Chinnusamy, V.; Zhu, J.K. Epigenetic regulation of stress responses in plants. Curr. Opin. Plant Biol. 2009, 12, 133–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Labra, M.; Ghiani, A.; Citterio, S.; Sgorbati, S.; Sala, F.; Vannini, C.; Ruffini-Castiglione, M.; Bracale, M. Analysis of cytosine methylation pattern in response to water deficit in pea root tips. Plant Biol. 2002, 4, 694–699. [Google Scholar] [CrossRef]
- Dyachenko, O.V.; Zakharchenko, N.S.; Shevchuk, T.V.; Bohnert, H.J.; Cushman, J.C.; Buryanov, Y.I. Effect of hypermethylation of CCWGG sequences in DNA of Mesembryanthemum crystallinum plants on their adaptation to salt stress. Biochemistry 2006, 71, 461–465. [Google Scholar] [CrossRef] [PubMed]
- Kozbial, P.Z.; Mushegian, A.R. Natural history of S-adenosylmethionine-binding proteins. BMC Struct. Biol. 2005, 5, 19. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Luo, M.; Zhang, W.; Zhao, J.; Zhang, J.; Wu, K.; Tian, L.; Duan, J. Histone acetyltransferases in rice (Oryza sativa L.): Phylogenetic analysis, subcellular localization and expression. BMC Plant Biol. 2012, 12, 145. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Vinocur, B.; Shoseyov, O.; Altman, A. Role of plant heat-shock proteins and molecular chaperons in the abiotic stress response. Trends Plant Sci. 2004, 9, 244–252. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q.-L.; Kang, J.-M.; Long, R.-C.; Cui, Y.-J.; Zhang, T.-J.; Xiong, J.-B.; Yang, Q.-C.; Sun, Y. Proteomic analysis of salt and osmotic-drought stress in alfalfa seedlings. J. Integr. Agric. 2016, 15, 2266–2278. [Google Scholar] [CrossRef]
- De la Cruz, J.; Karbstein, K.; Woolford, J.L. Functions of ribosomal proteins in assembly of eukaryotic ribosomes in vivo. Annu. Rev. Biochem. 2015, 84, 93–129. [Google Scholar] [CrossRef] [PubMed]
- Ziogas, V.; Tanou, G.; Belghazi, M.; Filippou, P.; Fotopoulos, V.; Grigorios, D.; Molassiotis, A.A. Roles of sodium hydrosulfide and sodium nitroprusside as priming molecules during drought acclimation in citrus plants. Plant Mol. Biol. 2015, 89, 433–450. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y. Identification and roles of Photosystem II assembly, stability, and repair factors in Arabidopsis. Front. Plant Sci. 2016, 7, 1–27. [Google Scholar] [CrossRef] [PubMed]
- Theis, J.; Schroda, M. Revisiting the photosystem II repair cycle. Plant Signal. Behav. 2016, 11, e1218587. [Google Scholar] [CrossRef] [PubMed]
- Edwards, G.E.; Nakamoto, H.; Burnell, J.N.; Hatch, M.D. Pyruvate, Pi dikinase and NADP-malate dehydrogenase in C4 photosynthesis: Properties and mechanism of light/dark regulation. Annu. Rev. Plant Physiol. 1985, 36, 255–286. [Google Scholar] [CrossRef]
- Jiang, H.-X.; Yang, L.-T.; Qi, Y.-P.; Lu, Y.-B.; Huang, Z.-R.; Chen, L.-S. Root iTRAQ protein profile analysis of two citrus species differing in aluminium-tolerance in response to long-term aluminium-toxicity. BMC Genom. 2015, 16, 949. [Google Scholar] [CrossRef] [PubMed]
- Liese, A.; Romeis, T. Biochemical regulation of in vivo function of plant calcium-dependent protein kinases (CDPK). Biochim. Biophys. Acta 2013, 1833, 1582–1589. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Li, W.; He, Q.L.; Daud, M.K.; Chen, J.; Zhu, S. Genome-wide survey and expression analysis of calcium-dependent protein kinase in Gossypium raimondii. PLoS ONE 2014, 9, e98189. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.H.; Xu, Z.H.; Xue, H.W. Arabidopsis membrane steroid binding protein 1 is involved in inhibition of cell elongation. Plant Cell 2005, 17, 116–131. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Song, L.; Xue, H.W. Membrane steroid binding protein 1 (MSBP1) stimulates tropism by regulating vesicle trafficking and auxin redistribution. Mol. Plant 2008, 1, 1077–1078. [Google Scholar] [CrossRef] [PubMed]
- Ashihara, H. Biosynthesis of 5-phosphoribosyl-1-pyrophosphate in plants: A review. Eur. Chem. Bull. 2016, 5, 314–323. [Google Scholar]
- Büttner, M.; Sauer, N. Monosaccharide transporters in plants: Structure, function and physiology. Biochim. Biophys. Acta 2000, 1465, 263–274. [Google Scholar] [CrossRef]
- Truernit, E.; Schmid, J.; Epple, P.; Illig, J.; Sauer, N. The sink-specific and stress-regulated Arabidopsis STP4 gene: Enhanced expression of a gene encoding a monosaccharide transporter by wounding, elicitors, and pathogen challenge. Plant Cell 1996, 8, 2169–2182. [Google Scholar] [CrossRef] [PubMed]
- Büttner, M.; Truernit, E.; Baier, K.; Scholz-Starke, J.; Sontheim, M.; Lauterbach, C.; Huss, V.A.R.; Sauer, N. AtSTP3, a green leaf-specific, low affinity monosaccharide-H+ symporter of Arabidopsis thaliana. Plant Cell Environ. 2000, 23, 175–184. [Google Scholar] [CrossRef] [Green Version]
- Nouri, M.Z.; Toorchi, M.; Komatsu, S. Proteomics approach for identifying abiotic stress responsive proteins in soybean. Soybean Aleksandra Sudarić. IntechOpen 2011, 276, 514. [Google Scholar] [CrossRef]
- Alam, I.; Sharmin, S.A.; Kim, K.H.; Yang, J.K.; Choi, M.S.; Lee, B.H. Proteome analysis of soybean roots subjected to short-term drought stress. Plant Soil 2010, 333, 491–505. [Google Scholar] [CrossRef]
- Ghannoum, O. C4 photosynthesis and water stress. Ann. Bot. 2009, 103, 635–644. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.B.; Lei, L.; Lai, J.S.; Zhao, H.M.; Song, W.B. Effects of drought stress and water recovery on physiological responses and gene expression in maize seedlings. BMC Plant Biol. 2018, 18, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Zhao, F.; Zhang, D.; Zhao, Y.; Wang, W.; Yang, H.; Tai, F.; Li, C.; Hu, X. The difference of physiological and proteomic changes in maize leaves adaptation to drought, heat, and combined both stresses. Front. Plant Sci. 2016, 7, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Murata, N.; Allakhverdiev, S.I.; Nishiyama, Y. The mechanism of photoinhibition in vivo: Re-evaluation of the roles of catalase, α-tocopherol, non-photochemical quenching, and electron transport. Biochim. Biophys. Acta 2012, 1817, 1127–1133. [Google Scholar] [CrossRef] [PubMed]
- Melkozernov, A.N.; Blankenship, R.E. Photosynthetic functions of chlorophylls. In Chlorophylls and Bacteriochlorophylls; Grimm, B., Porra, R.J., Rüdiger, W., Scheer, H., Eds.; Springer: Dordrecht, The Netherlands, 2006; pp. 397–412. [Google Scholar]
- Dudhate, A.; Shinde, H.; Tsugama, D.; Liu, S.; Takano, T. Transcriptomic analysis reveals the differentially expressed genes and pathways involved in drought tolerance in pearl millet [Pennisetum glaucum (L.) R. Br]. PLoS ONE 2018, 13, e0195908. [Google Scholar] [CrossRef] [PubMed]
- Xiao, L.; Zhang, L.; Yang, G.; Zhu, H.; He, Y. Transcriptome of Protoplasts Reprogrammed into Stem Cells in Physcomitrella patens. PLoS ONE 2012, 7, e35961. [Google Scholar] [CrossRef] [PubMed]
- Izaurralde, E.; Mattaj, I.W. Transport of RNA between nucleus and cytoplasm. Semin. Cell Biol. 1992, 3, 279–288. [Google Scholar] [CrossRef]
- Galmés, J.; Flexas, J.; Savé, R.; Medrano, H. Water relations and stomatal characteristics of Mediterranean plants with different growth forms and leaf habits: Responses to water stress and recovery. Plant Soil 2007, 290, 139–155. [Google Scholar] [CrossRef]
- Keogh, R.C.; Deverall, B.J.; Mcleod, S. Comparison of histological and physiological responses to phakopspora pachyrhizi in pesistant and susceptible soybean. Great Br. 1980, 74, 329–333. [Google Scholar] [CrossRef]
- Lade, H.; Kadam, A.; Paul, D.; Govindwar, S. A low-cost wheat bran medium for biodegradation of the benzidine-based carainogenic dye trypan blue using a micronial consortium. Environ. Res. Public Health 2015, 12, 3480–3505. [Google Scholar] [CrossRef] [PubMed]
- Han, L.B.; Song, G.L.; Zhang, X. Preliminary observation of physiological responses of three turfgrass species to traffic stress. HortTechnology 2008, 18, 139–143. [Google Scholar]
- Dhindsa, R.S.; Plumb-Dhindsa, P.; Thorpe, T.A. Leaf senescence: Correlated with increased leaves of membrane permeability and lipid peroxidation, and decreased levels of superoxide dismutase and catalase. J. Exp. Bot. 1981, 32, 93–101. [Google Scholar] [CrossRef]
- Bates, T.S.; Waldren, R.P.; Teare, I.D. Rapid determination of free proline for water-stress studies. Plant Soil 1973, 39, 205–207. [Google Scholar] [CrossRef]
- Wisniewski, J.R.; Zougman, A.; Nagaraj, N.; Mann, M. Universal sample preparation method for proteome analysis. Nat. Methods 2009, 6, 359–362. [Google Scholar] [CrossRef] [PubMed]
- Swägger, H. Tricine-SDS-PAGE. Nat. Protoc. 2006, 1, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.; Zhao, Y.; Wang, Y.; Shi, Z.; Zhang, P.; Zhang, Y.; Song, W.; Zhao, J. Comparative proteomics of contrasting maize genotypes provides insights into salt-stress tolerance mechanisms. J. Proteome Res. 2018, 17, 141–153. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Wang, H.Y.; Zhang, T. Comprehensive and quantitative proteomic analysis of metamorphosis-related proteins in the veined rapa whelk. Int. J. Mol. Sci. 2016, 17, 924. [Google Scholar] [CrossRef] [PubMed]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Gotz, S.; García-Gómez, J.M.; Terol, J.; Williams, T.D.; Nagaraj, S.H.; Nueda, M.J.; Robles, M.; Talón, M.; Dopazo, J.; Conesa, A. High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res. 2008, 36, 3420–3435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Comparisons 1 | Up-Regulated 2 | Down-Regulated 3 | Total 4 |
|---|---|---|---|
| SD_SC | 65 | 92 | 157 |
| TD_TC | 11 | 26 | 37 |
| SD_TD | 116 | 153 | 269 |
| SC_TC | 119 | 139 | 258 |
| No. | Protein ID 1 | Gene Name/ID 2 | Description 3 | Coverage (%) 4 | Peptide Fragments 5 | Fold Change 6 | p Value 7 | Pathways 8 |
|---|---|---|---|---|---|---|---|---|
| 1 | C0HJ06 | 541618 | Uncharacterized protein 9 | 22.4 | 1 | 1.37 | 0.0109 | MAPK signaling pathway/Plant hormone signaling |
| 2 | Q41746 | Lhcb5-1 | Chlorophyll a-b binding protein, chloroplastic | 55.8 | 10 | 1.24 | 0.0131 | X3 |
| 3 | C0HGH7 | 100193714 | Universal stress family protein | 20.4 | 3 | 1.23 | 0.0430 | |
| 4 | A0A1D6GAZ6 | ZEAMMB73_Zm00001d012677 | Glycerophosphodiester phosphodiesterase GDPD5 | 16.8 | 5 | 1.22 | 0.0136 | Glycerophospholipid metabolism |
| 5 | C0P948 | Zm00001d024886 | Uncharacterized protein | 55.9 | 20 | 1.21 | 0.0350 | |
| 6 | A0A1D6PQ00 | 100286059 | U2 snRNP auxiliary factor large subunit | 9.4 | 2 | 0.83 | 0.0171 | Spliceosome |
| 7 | A0A1D6IUI1 | 100383306 | Ubiquitin carboxyl-terminal hydrolase 13 | 2.7 | 3 | 0.83 | 0.0217 | |
| 8 | A0A1D6MJP2 | ZEAMMB73_Zm00001d039613 | Uncharacterized protein | 19.9 | 4 | 0.82 | 0.0111 | |
| 9 | B4FTP2 | ZEAMMB73_Zm00001d021334 | Thioredoxin-like protein CDSP32 chloroplastic | 23.7 | 6 | 0.81 | 0.0246 | |
| 10 | B4F845 | 100191245 | Uncharacterized protein | 3.0 | 1 | 0.81 | 0.0027 | |
| 11 | H9BG22 | 101027254 | Alpha-dioxygenase | 4.4 | 3 | 0.80 | 0.0162 | alpha-linolenic acid metabolism |
| 12 | Q5GJ59 | TPS7 | Terpene synthase 7 | 14.8 | 5 | 0.78 | 0.0179 | |
| 13 | C0PHF6 | 100383595 | AAA-ATPase ASD mitochondrial | 10.6 | 5 | 0.55 | 0.0487 |
| No. | Protein ID 1 | Gene Name/ID 2 | Description 3 | Coverage (%) 4 | Peptide Fragments 5 | Fold Change 6 | p Value 7 | Pathways 8 |
|---|---|---|---|---|---|---|---|---|
| 1 | B4FV94 | Zm00001d032197 | Chlorophyll a-b binding protein, chloroplastic | 49.8 | 7 | 1.66 | 0.0326 | Photosynthesis-antenna proteins |
| 2 | B4FCG6 | Zm00001d004386 | 9 Uncharacterized protein | 9.0 | 1 | 1.48 | 0.0036 | |
| 3 | B4FTN5 | 100273215 | Metal-dependent protein hydrolase | 5.7 | 1 | 1.45 | 0.0459 | |
| 4 | B8A3B7 | Zm00001d043059 | Uncharacterized protein | 20.8 | 3 | 1.33 | 0.0278 | |
| 5 | C0P6L9 | Zm00001d053377 | Uncharacterized protein | 40.2 | 7 | 1.33 | 0.0011 | Ribosome |
| 6 | B4FLE3 | 100282216 | HSP20-like chaperones superfamily protein | 33.0 | 4 | 1.32 | 0.0484 | |
| 7 | B6U3Z0 | Zm00001d053377 | 50S ribosomal protein L21 | 42.5 | 7 | 1.31 | 0.0149 | Ribosome |
| 8 | K7TP80 | Zm00001d024014 | Zinc finger (C3HC4-type RING finger) family protein | 36.0 | 14 | 1.31 | 0.0028 | |
| 9 | A0A1D6JW44 | Zm00001d028428 | Calcium-binding EF-hand family protein | 9.0 | 1 | 1.30 | 0.0014 | |
| 10 | A0A097PND9 | Zm00001d015195 | AT5G11810-like protein (Fragment) | 6.9 | 1 | 1.29 | 0.0358 | |
| 11 | B4FE30 | 100193174 | 10 kDa chaperonin | 45.9 | 5 | 1.29 | 0.0024 | |
| 12 | B4FZU8 | 100274264 | Malate dehydrogenase | 56.8 | 12 | 1.28 | 0.0150 | Carbon metabolism, Pyruvate metabolism, Cysteine and methionine metabolism |
| 13 | Q4A1J8 | cc3 | Cysteine proteinase inhibitor | 11.3 | 1 | 1.28 | 0.0293 | |
| 14 | A0A1X7YHJ3 | Zm00001d000282 | Photosystem II CP47 reaction center protein | 46.9 | 16 | 1.28 | 0.0062 | Photosynthesis |
| 15 | B4FWP6 | Zm00001d039452 | Uncharacterized protein | 9.9 | 4 | 1.27 | 0.0374 | Spliceosome |
| 16 | B4FTL2 | Zm00001d044931 | Protein TIC 22 chloroplastic | 9.3 | 2 | 1.27 | 0.0001 | |
| 17 | C0P8X5 | 100284068 | Electron transfer flavoprotein subunit beta mitochondrial | 14.9 | 1 | 1.25 | 0.0020 | |
| 18 | A0A1D6HE45 | ZEAMMB73_Zm00001d017330 | ATP-dependent Clp protease proteolytic subunit | 33.7 | 5 | 1.25 | 0.0218 | |
| 19 | Q2XX37 | plt2 | Non-specific lipid-transfer protein | 46.2 | 4 | 1.25 | 0.0435 | |
| 20 | A0A1D6JYF7 | 103634473 | Kinesin-like protein | 3.1 | 1 | 1.24 | 0.0409 | |
| 21 | A0A1D6E501 | ZEAMMB73_Zm00001d002880 | 3-isopropylmalate dehydrogenase | 50.1 | 12 | 1.24 | 0.0449 | Oxocarboxylic acid metabolism, C5-Branched dibasic acid metabolism, Biosynthesis of amino acids |
| 22 | A0A1D6L0Y0 | ZEAMMB73_Zm00001d033634 | Uncharacterized protein | 7.6 | 1 | 1.24 | 0.0111 | |
| 23 | A0A096PRE6 | 100282938 | Fibrillin1 | 31.4 | 9 | 1.23 | 0.0421 | |
| 24 | K7UWX4 | ZEAMMB73_Zm00001d051062 | GrpE protein homolog | 44.2 | 11 | 1.23 | 0.0083 | |
| 25 | B4FMA5 | 100217267 | Chaperone DnaJ-domain superfamily protein | 14.6 | 2 | 1.23 | 0.0378 | |
| 26 | B7ZZT1 | Zm00001d027326 | Uncharacterized protein | 6.5 | 1 | 1.22 | 0.0039 | |
| 27 | B8A045 | 100279815 | Phospholipase D | 2.9 | 2 | 1.22 | 0.0211 | Endocytosis, Ether lipid metabolism, Glycero phospholipid metabolism |
| 28 | B6TGF1 | Zm00001d009640 | Malate dehydrogenase 2 mitochond. | 72.4 | 14 | 1.22 | 0.0092 | Carbon metabolism, Pyruvate metabolism, Cysteine and methionine metabolism, Carbon fixation in photosynthetic organisms |
| 29 | A0A1D6FI49 | ZEAMMB73_Zm00001d009189 | TPR repeat | 6.0 | 1 | 1.22 | 0.0283 | |
| 30 | B6UHD9 | Zm00001d021715 | Peptide chain release factor 2 | 8.0 | 2 | 1.22 | 0.0374 | |
| 31 | B6TDF7 | 100282980 | Plastid-specific 30S ribosomal protein 2 | 45.4 | 9 | 1.21 | 0.0014 | RNA transport, RNA degradation, mRNA surveillance pathway |
| 32 | Q1KKB7 | nad1 | NADH-ubiquinone oxidoreductase chain 1 | 5.9 | 1 | 1.21 | 0.0355 | Oxidative phosphorylation |
| 33 | A0A059Q7D4 | psbD | Photosystem II D2 protein | 25.2 | 7 | 1.20 | 0.0058 | Photosynthesis |
| 34 | C4J3Q4 | 100277436 | YCF37-like protein | 17.7 | 2 | 1.20 | 0.0017 | |
| 35 | B4FTK9 | 100282281 | Alpha/beta-Hydrolases superfamily protein | 33.6 | 6 | 1.20 | 0.0421 | |
| 36 | B6TBW4 | 100282838 | ERBB-3 BINDING PROTEIN 1 | 30.5 | 10 | 0.83 | 0.0174 | |
| 37 | A0A1D6DVJ8 | ZEAMMB73_Zm00001d002006 | H(+)-ATPase 5 | 34.6 | 18 | 0.83 | 0.0322 | Oxidative phosphorylation |
| 38 | A0A1D6DYT2 | 100383868 | Signal recognition particle 14 kDa protein | 11.3 | 1 | 0.83 | 0.0172 | Protein export |
| 39 | B6T346 | 100279524 | THO complex subunit 4 | 14.2 | 3 | 0.83 | 0.0390 | mRNA surveillance pathway, RNA transport |
| 40 | A0A1D6GKY6 | 100192032 | Uncharacterized protein | 4.9 | 1 | 0.83 | 0.0411 | |
| 41 | B6SJ21 | 100280585 | Guanine nucleotide-binding protein beta subunit-like protein | 59.3 | 13 | 0.83 | 0.0232 | |
| 42 | C0PI72 | Zm00001d017459 | Uncharacterized protein | 8.3 | 1 | 0.82 | 0.0201 | Valine, leucine and isoleucine degradation |
| 43 | C0HI59 | 100381692 | Uncharacterized protein | 13.3 | 5 | 0.82 | 0.0181 | |
| 44 | A0A1D6M4E1 | ZEAMMB73_Zm00001d038192 | Glutathione transferase41 | 8.6 | 1 | 0.82 | 0.0025 | Glutathione metabolism |
| 45 | A0A1D6GES6 | 103625778 | DNA gyrase subunit A chloroplastic/mitochondrial | 1.9 | 1 | 0.81 | 0.0372 | |
| 46 | B6TIL4 | Zm00001d048954 | GDP-mannose 3,5-epimerase 2 | 20.5 | 6 | 0.81 | 0.0265 | Amino sugar and nucleotide sugar metabolism, Ascorbate and aldarate metabolism |
| 47 | B6T3J2 | 100282096 | Eukaryotic translation initiation factor 2 beta subunit | 12.9 | 3 | 0.81 | 0.0206 | RNA transport |
| 48 | A0A1D6F8L4 | 100194138 | Coatomer subunit gamma | 7.2 | 4 | 0.81 | 0.0316 | |
| 49 | C0PI69 | Zm00001d040286 | Uncharacterized protein | 18.5 | 2 | 0.81 | 0.0092 | |
| 50 | A0A0B4J3C2 | ZEAMMB73_Zm00001d037873 | Elongation factor 1-alpha | 42.1 | 15 | 0.81 | 0.0463 | RNA transport |
| 51 | B4FEV5 | Zm00001d031689 | Uncharacterized protein | 13.8 | 1 | 0.81 | 0.0400 | Plant-pathogen interaction |
| 52 | P26566 | rpl20 | 50S ribosomal protein L20, chloroplastic | 20.2 | 3 | 0.81 | 0.0476 | Ribosome |
| 53 | A0A1D6KBW7 | ZEAMMB73_Zm00001d030317 | Hsp20/alpha crystallin family protein | 17.8 | 2 | 0.81 | 0.0098 | |
| 54 | A0A1D6ICZ3 | 542526 | Calcium dependent protein kinase8 | 7.0 | 3 | 0.80 | 0.0465 | Plant-pathogen interaction |
| 55 | B4FAJ4 | Zm00001d008739 | Uncharacterized protein | 2.8 | 1 | 0.80 | 0.0260 | Peroxisome |
| 56 | B6T9T5 | N/A | Uncharacterized protein | 4.3 | 1 | 0.80 | 0.0002 | |
| 57 | Q9M7E2 | Zm00001d036904 | Elongation factor 1-alpha | 30.7 | 10 | 0.80 | 0.0134 | RNA transport |
| 58 | B7ZZ42 | 103650526 | Heat shock 70 kDa protein 3 | 58.6 | 30 | 0.80 | 0.0076 | Spliceosome, Endocytosis, Protein processing in endoplasmic reticulum |
| 59 | A0A1D6N9X4 | 103651144 | Insulin-degrading enzyme-like 1 peroxisomal | 3.5 | 3 | 0.79 | 0.0149 | |
| 60 | A0A1D6IHP2 | 103633334 | ARM repeat superfamily protein | 6.5 | 5 | 0.79 | 0.0161 | |
| 61 | B4FLV6 | 100286322 | Protein translation factor SUI1 | 20.0 | 3 | 0.79 | 0.0269 | RNA transport |
| 62 | B4FQM2 | 100282190 | Pyrophosphate--fructose 6-phosphate 1-phosphotransferase subunit beta | 6.7 | 2 | 0.79 | 0.0123 | Fructose and mannose metabolism, Pentose phosphate pathway, Glycolysis/Gluconeogenesis |
| 63 | B6TP02 | Zm00001d017866 | Aspartic proteinase nepenthesin-1 | 5.6 | 2 | 0.78 | 0.0276 | |
| 64 | A0A1D6PW61 | 100191474 | DNA topoisomerase 1 beta | 3.1 | 1 | 0.78 | 0.0189 | |
| 65 | B6SR37 | Zm00001d011799 | Uncharacterized protein | 17.3 | 2 | 0.78 | 0.0070 | |
| 66 | A0A1D6JQY8 | 100192907 | Uroporphyrinogen-III synthase | 2.8 | 1 | 0.78 | 0.0294 | Porphyrin and chlorophyll metabolism |
| 67 | A0A1D6IIC2 | ZEAMMB73_Zm00001d021999 | Nuclear transport factor 2 (NTF2) family protein | 5.6 | 1 | 0.77 | 0.0092 | |
| 68 | B6U4J6 | Zm00001d045774 | Embryogenesis transmembrane protein | 4.5 | 1 | 0.77 | 0.0258 | |
| 69 | C0P626 | Zm00001d011454 | Carbonic anhydrase | 74.3 | 13 | 0.77 | 0.0272 | Nitrogen metabolism |
| 70 | Q9M7E3 | Zm00001d009868 | Elongation factor 1-alpha | 37.8 | 13 | 0.76 | 0.0045 | RNA transport |
| 71 | B6SI29 | 100501869 | Histone H2A | 29.3 | 4 | 0.76 | 0.0326 | |
| 72 | B4FIA6 | 100194327 | Histone H2A | 28.9 | 3 | 0.76 | 0.0406 | |
| 73 | A0A1D6JVL9 | ZEAMMB73_Zm00001d028377 | Small nuclear ribonucleoprotein Sm D3 | 21.7 | 2 | 0.75 | 0.0384 | Spliceosome |
| 74 | B6SLI1 | 100282946 | 40S ribosomal protein S30 | 16.1 | 1 | 0.74 | 0.0110 | Ribosome |
| 75 | A0A1D6LBT4 | 100279572 | Protein prenyltransferase superfamily protein | 7.0 | 1 | 0.72 | 0.0475 | |
| 76 | A0A1D6P0E7 | ZEAMMB73_Zm00001d046001 | Triose phosphate/phosphate translocator TPT chloroplastic | 22.1 | 2 | 0.72 | 0.0111 | |
| 77 | B4FFS7 | Zm00001d036233 | Uncharacterized protein | 7.8 | 1 | 0.71 | 0.0347 | |
| 78 | A0A1D6FPL0 | 100382596 | Fructose-16-bisphosphatase cytosolic | 21.2 | 8 | 0.70 | 0.0179 | Fructose and mannose metabolism, Pentose phosphate pathway |
| 79 | Q8LLS4 | Pgk-1 | Phosphoglycerate kinase (Fragment) | 32.2 | 9 | 0.69 | 0.0440 | Carbon metabolism, Glycolysis/Gluconeogenesis. |
| 80 | A0A1D6K8W1 | ZEAMMB73_Zm00001d030005 | Dynamin-related protein 1E | 2.7 | 1 | 0.68 | 0.0411 | |
| 81 | A0A1D6QSH1 | 100383873 | Cullin-associated NEDD8-dissociated protein 1 | 3.6 | 3 | 0.65 | 0.0139 | |
| 82 | B6TNP4 | Zm00001d034479 | Histone H1 | 41.0 | 11 | 0.65 | 0.0485 | |
| 83 | A0A1D6MEZ2 | ZEAMMB73_Zm00001d039282 | Serine/threonine-protein kinase AGC1-5 | 1.4 | 1 | 0.55 | 0.0120 | |
| 84 | E7DDW6 | Zm00001d026630 | Clathrin light chain 2 | 23.0 | 4 | 0.52 | 0.0203 |
| No. | Protein ID 1 | Gene Name/ID 2 | Description 3 | Coverage (%) 4 | Peptide Fragments 5 | YE8112 Fold Change 6 | p Value 7 | SD_TD Fold Change 8 | p Value 7 | Pathways 9 |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | B6SQW8 | Zm00001d024893 | Uncharacterized protein | 27.2 | 3 | 1.59 | 0.0155 | 0.53 | 0.0093 | No significant enrichment |
| 2 | B4FKG5 | 542304 | Abscisic acid stress ripening 1 | 47.1 | 4 | 1.34 | 0.0096 | 0.60 | 0.0325 | No significant enrichment |
| 3 | A0A1D6HWS1 | 100282063 | Dirigent protein | 34.3 | 4 | 1.29 | 0.0207 | 0.67 | 0.0118 | Not significant enrichment |
| No. | Protein ID | Gene Name/ID | Description | Coverage (%) | Peptide Fragments | YE8112 | MO17 | Pathways | ||
|---|---|---|---|---|---|---|---|---|---|---|
| Fold Change | p Value | Fold Change | p Value | |||||||
| 1 | B6TD62 | 100282951 | Membrane steroid-binding protein 1 | 35.8 | 5 | 0.81 | 0.0223 | 1.50 | 0.0142 | |
| 2 | A0A1D6GZE2 | 100272744 | Ribose-phosphate pyrophosphokinase | 5.4 | 1 | 0.82 | 0.0078 | 0.82 | 0.0068 | Purine metabolism/Carbon metabolism/Pentose phosphate pathway |
| 3 | C4J0F8 | Zm00001d038865 | Uncharacterized protein | 32.5 | 4 | 0.80 | 0.0090 | 0.81 | 0.0465 | Ribosome |
| 4 | C0PHL2 | Zm00001d018627 | Monosaccharide transporter1 | 3.8 | 1 | 0.79 | 0.0051 | 1.69 | 0.0495 | |
| 5 | C0HDZ4 | Zm00001d009084 | SAM-dependent methyltransferase superfamily protein | 14.1 | 2 | 0.73 | 0.0218 | 1.52 | 0.0245 | |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zenda, T.; Liu, S.; Wang, X.; Jin, H.; Liu, G.; Duan, H. Comparative Proteomic and Physiological Analyses of Two Divergent Maize Inbred Lines Provide More Insights into Drought-Stress Tolerance Mechanisms. Int. J. Mol. Sci. 2018, 19, 3225. https://doi.org/10.3390/ijms19103225
Zenda T, Liu S, Wang X, Jin H, Liu G, Duan H. Comparative Proteomic and Physiological Analyses of Two Divergent Maize Inbred Lines Provide More Insights into Drought-Stress Tolerance Mechanisms. International Journal of Molecular Sciences. 2018; 19(10):3225. https://doi.org/10.3390/ijms19103225
Chicago/Turabian StyleZenda, Tinashe, Songtao Liu, Xuan Wang, Hongyu Jin, Guo Liu, and Huijun Duan. 2018. "Comparative Proteomic and Physiological Analyses of Two Divergent Maize Inbred Lines Provide More Insights into Drought-Stress Tolerance Mechanisms" International Journal of Molecular Sciences 19, no. 10: 3225. https://doi.org/10.3390/ijms19103225