Caspase-3 Mediated Cell Death in the Normal Development of the Mammalian Cerebellum


Abstract

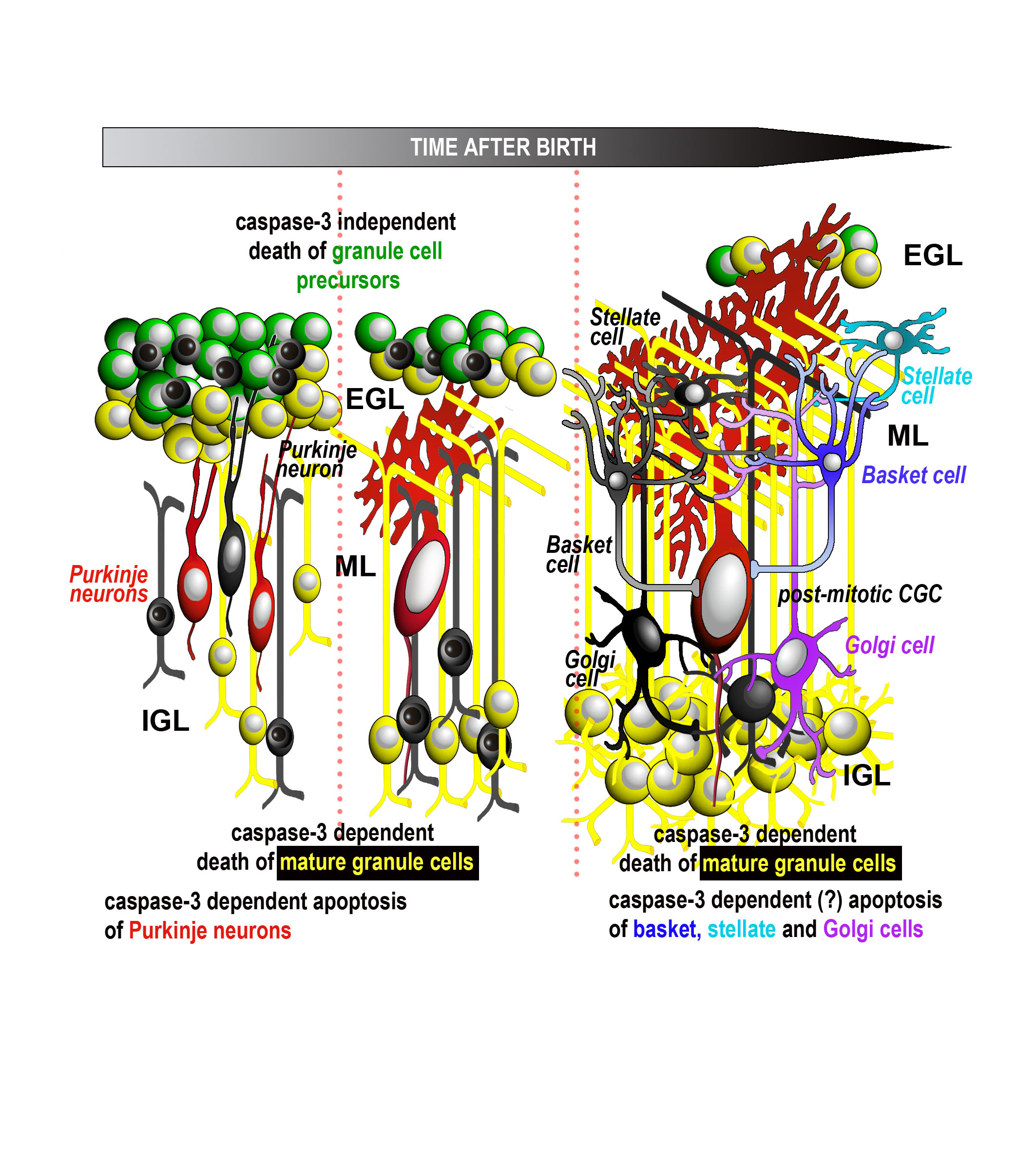{kind=link}
{kind=link}
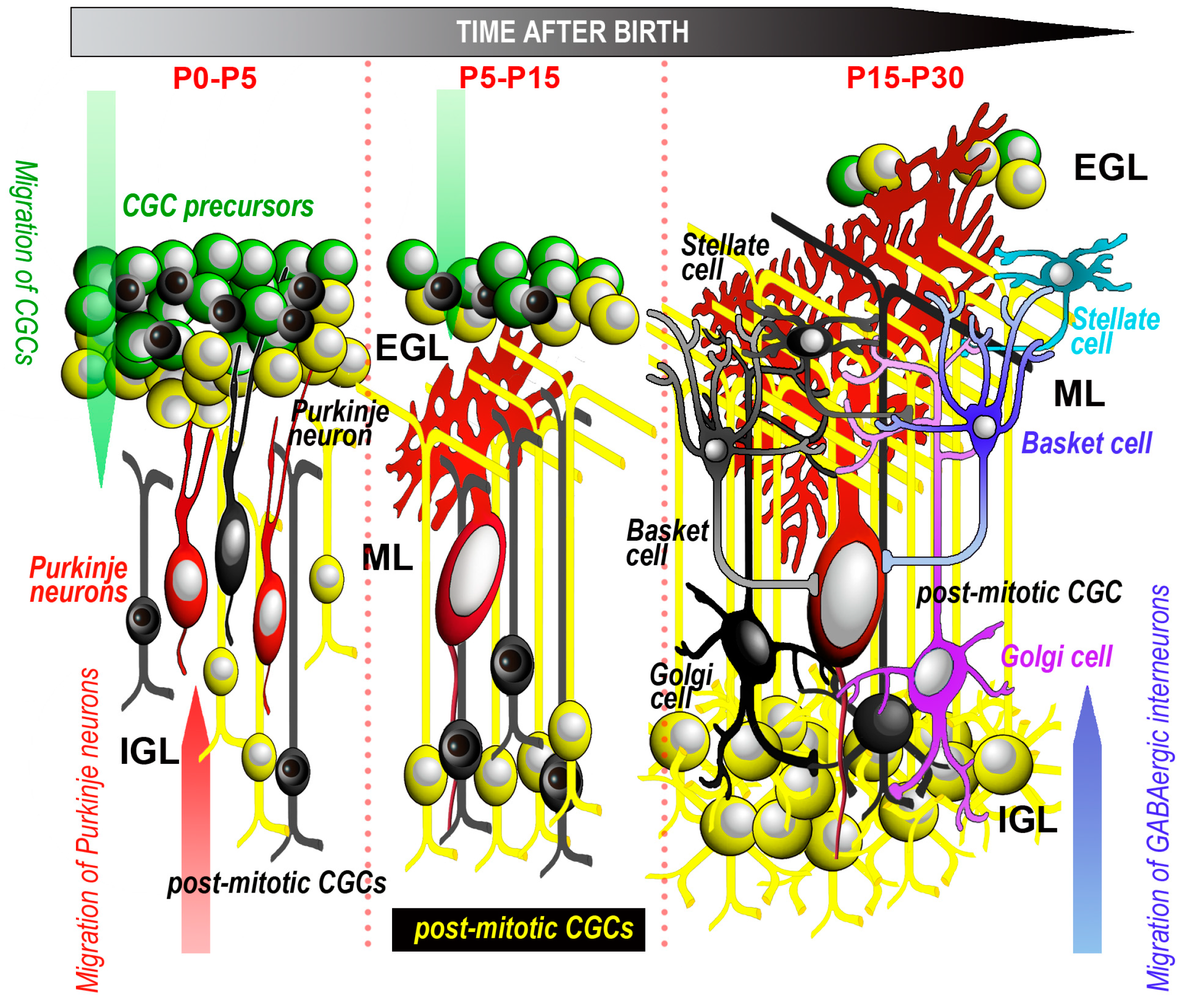{kind=link}
1. Introduction
2. The Recognition of Cell Death in Cerebellar Neurons
2.1. The Struggle for the Recognition of Cell Death of Neurons
2.2. The Discovery of PCD and NOND in Cerebellum
3. Caspase-3 in Cerebellar Development
3.1. Modes of Cell Death in Neurons and Their Relevance to Cerebellar Development
Apoptosis
3.2. Intervention of Caspase-3 in Cerebellar Development
3.2.1. Basic Features of Cerebellar Development
3.2.2. CGCs
3.2.2.1. Human Studies
3.2.2.2. Animal Studies
Relationship of cell proliferation and NOND
Rats
Mice
Rabbits
Detection of active caspase-3
Molecular pathways and regulation of NOND in CGCs
3.2.2.3. Summary of Current Knowledge
3.2.3. PNs
3.2.4. GABAergic Interneurons
3.3. Glia
4. Non-Apoptotic Role of Caspase-3 in Cerebellar Development
5. Future Perspectives
Funding
Conflicts of Interest
Abbreviations
| Aif | Apoptosis-inducing factor |
| Apaf1 | Apoptotic peptidase activating factor 1 |
| BCL-2 | B-cell lymphoma 2 |
| BDNF | Brain-derived neurotrophic factor |
| CGC(s) | Cerebellar granule cell(s) |
| CHL1 | Close homolog of L1 |
| CNS | Central nervous system |
| CPP32 | 32-kDa putative cysteine protease |
| ECFP | Enhanced cyan fluorescent protein |
| EGL | External granular layer |
| EGZ | External germinal zone |
| Fh | Flathead |
| FRET | Fluorescence resonance energy transfer |
| GAD67 | Glutamic acid decarboxylase 67 |
| GFP | Green fluorescent protein |
| Hq | Harlequin |
| IAP(s) | Inhibitors of apoptosis protein(s) |
| IGL | Internal granular layer |
| LFG | Lifeguard |
| mGluR5 | Metabotropic glutamate receptor 5 |
| MLKL | Mixed lineage kinase domain like pseudokinase |
| NMDA | N-methyl-d-aspartate |
| NOND | Naturally occurring neuronal death |
| OKA | Okadaic acid |
| p75NTR | p75 neurotrophin receptor |
| PARP-1 | Poly ADP-ribose polymerase-1 |
| PCD | Programmed cell death |
| PCNA | Proliferating cell nuclear antigen |
| PN(s) | Purkinje neuron(s) |
| proBDNF | BDNF precursor molecule |
| RGZ | Rostral germinal zone |
| RIPK1 | Serine/threonine-protein kinase 1 |
| RIPK3 | Serine/threonine-protein kinase 3 |
| RL | Rhombic lip |
| TrkB | Tropomyosin receptor kinase B |
| TUNEL | Terminal deoxynucleotidyl transferase dUTP nick end labeling |
| VZ | Ventricular zone |
References
- Yagami, T.; Yamamoto, Y.; Koma, H. Pathophysiological roles of intracellular proteases in neuronal development and neurological diseases. Mol. Neurobiol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Lossi, L.; Castagna, C.; Merighi, A. Neuronal cell death: An overview of its different forms in central and peripheral neurons. In Neuronal Cell Death; Lossi, L., Merighi, A., Eds.; Springer: New York, NY, USA, 2015; pp. 1–18. [Google Scholar]
- Castagna, C.; Merighi, A.; Lossi, L. Cell death and neurodegeneration in the postnatal development of cerebellar vermis in normal and Reeler mice. Ann. Anat. 2016, 207, 76–90. [Google Scholar] [CrossRef] [PubMed]
- Nishiyama, J.; Matsuda, K.; Kakegawa, W.; Yamada, N.; Motohashi, J.; Mizushima, N.; Yuzaki, M. Reevaluation of neurodegeneration in lurcher mice: Constitutive ion fluxes cause cell death with, not by, autophagy. J. Neurosci. 2010, 30, 2177–2187. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Yang, S.; Zhu, G. Postnatal calpain inhibition elicits cerebellar cell death and motor dysfunction. Oncotarget 2017, 8, 87997–88007. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Hersheson, J.; Lopez, D.; Hammer, M.; Liu, Y.; Lee, K.H.; Pinto, V.; Seinfeld, J.; Wiethoff, S.; Sun, J.; et al. Defects in the CAPN1 Gene result in alterations in cerebellar development and cerebellar ataxia in mice and humans. Cell Rep. 2016, 16, 79–91. [Google Scholar] [CrossRef] [PubMed]
- Cougnoux, A.; Cluzeau, C.; Mitra, S.; Li, R.; Williams, I.; Burkert, K.; Xu, X.; Wassif, C.A.; Zheng, W.; Porter, F.D. Necroptosis in Niemann-Pick disease, type C1: A potential therapeutic target. Cell Death Dis. 2016, 7, e2147. [Google Scholar] [CrossRef] [PubMed]
- Yamada, T.; Adachi, Y.; Fukaya, M.; Iijima, M.; Sesaki, H. Dynamin-related protein 1 deficiency leads to receptor-interacting protein kinase 3-mediated necroptotic neurodegeneration. Am. J. Pathol. 2016, 186, 2798–2802. [Google Scholar] [CrossRef] [PubMed]
- Lossi, L.; Merighi, A. In vivo cellular and molecular mechanisms of neuronal apoptosis in the mammalian CNS. Prog. Neurobiol. 2003, 69, 287–312. [Google Scholar] [CrossRef]
- Lossi, L.; Gambino, G.; Mioletti, S.; Merighi, A. In vivo analysis reveals different apoptotic pathways in pre- and post-migratory cerebellar granule cells of rabbit. J. Neurobiol. 2004, 60, 437–452. [Google Scholar] [CrossRef] [PubMed]
- Lossi, L.; Tamagno, I.; Merighi, A. Molecular morphology of neuronal apoptosis: Activation of caspase 3 during postnatal development of mouse cerebellar cortex. J. Mol. Histol. 2004, 35, 621–629. [Google Scholar] [CrossRef] [PubMed]
- Rao, M.S.; Jacobson, M. Developmental Neurobiology, IV ed.; Kluwer Academic/Plenum Publishers: New York, NY, USA; Boston, MA, USA; Dordrecht, The Netherlands; London, UK; Moscow, Russia, 2005. [Google Scholar]
- Beard, J. On the early development of Lepidosteus osseus—Preliminary notice. Proc. R. Soc. Lond. 1889, 46, 108–118. [Google Scholar] [CrossRef]
- Beard, J. The transient ganglion cells and their nerves in Raja batis. Anat. Anz. 1892, 7, 191–206. [Google Scholar]
- Collin, R. Histolyse de certains neuroblastes au cours du développement du tube nerveux chez le poulet. C. R. Soc. Biol. 1906, 60, 1080–1081. [Google Scholar]
- Ernst, M. Über untergang von zellen während der normalen entwicklung bei wirbeltieren. Z. Anat. Entwicklungsgesch. 1926, 79, 228–262. [Google Scholar] [CrossRef]
- Glücksmann, P.D. Cell deaths in normal vertebrate ontogeny. Biol. Rev. 1951, 26, 59–86. [Google Scholar] [CrossRef] [PubMed]
- Hamburger, V. Regression versus peripheral control of differentiation in motor hypoplasia. Am. J. Anat 1958, 102, 365–409. [Google Scholar] [CrossRef] [PubMed]
- Hughes, A. Cell degeneration in the larval ventral horn of Xenopus laevis (Daudin). J. Embryol. Exp. Morphol. 1961, 9, 269–284. [Google Scholar] [PubMed]
- Hughes, A.F.; Lewis, P.R. Effect of limb ablation on neurones in Xenopus larvae. Nature 1961, 189, 333–334. [Google Scholar] [CrossRef]
- Prestige, M.C. Cell turnover in the spinal ganglia of Xenopus laevis tadpoles. J. Embryol. Exp. Morphol. 1965, 13, 63–72. [Google Scholar]
- Levi-Montalcini, R.; Cohen, S. Effects of the extract of the mouse submaxillary salivary glands on the sympathetic system of mammals. Ann. N. Y. Acad. Sci. 1960, 85, 324–341. [Google Scholar]
- Marzban, H.; Del Bigio, M.R.; Alizadeh, J.; Ghavami, S.; Zachariah, R.M.; Rastegar, M. Cellular commitment in the developing cerebellum. Front. Cell. Neurosci. 2015, 8, 450. [Google Scholar] [CrossRef] [PubMed]
- Griffin, W.S.T.; Woodward, D.J.; Chanda, R. Quantification of cell death in developing cerebellum by a 14C tracer method. Brain Res. Bull. 1978, 3, 369–372. [Google Scholar] [CrossRef]
- Lauder, J.M. The effects of early hypo- and hyperthyroidism on the development of rat cerebellar cortex. III. Kinetics of cell proliferation in the external granular layer. Brain Res. 1977, 126, 31–51. [Google Scholar] [CrossRef]
- Rabié, A.; Favre, C.; Clavel, M.C.; Legrand, J. Effects of thyroid dysfunction on the development of the rat cerebellum, with special reference to cell death within the internal granular layer. Brain Res. 1977, 120, 521–531. [Google Scholar] [CrossRef]
- Janowsky, J.S.; Finlay, B.L. Cell degeneration in early development of the forebrain and cerebellum. Anat. Embryol. 1983, 167, 439–447. [Google Scholar] [CrossRef] [PubMed]
- Swisher, D.A.; Wilson, D.B. Cerebellar histogenesis in the lurcher (Lc) mutant mouse. J. Comp. Neurol. 1977, 173, 205–217. [Google Scholar] [CrossRef] [PubMed]
- Landis, S.C.; Mullen, R.J. The development and degeneration of Purkinje cells in pcd mutant mice. J. Comp. Neurol. 1978, 177, 125–143. [Google Scholar] [CrossRef] [PubMed]
- Landis, D.M.D.; Sidman, R.L. Electron microscopic analysis of postnatal histogenesis in the cerebellar cortex of staggerer mutant mice. J. Comp. Neurol. 1978, 179, 831–864. [Google Scholar] [CrossRef]
- Herrup, K.; Sunter, K. Numerical matching during cerebellar development: Quantitative analysis of granule cell death in staggerer mouse chimeras. J. Neurosci. 1987, 7, 829–836. [Google Scholar] [CrossRef]
- Sotelo, C.; Triller, A. Fate of presynaptic afferents to Purkinje cells in the adult nervous mutant mouse: A model to study presynaptic stabilization. Brain Res. 1979, 175, 11–36. [Google Scholar] [CrossRef]
- Smeyne, R.J.; Goldowitz, D. Development and death of external granular layer cells in the weaver mouse cerebellum: A quantitative study. J. Neurosci. 1989, 9, 1608–1620. [Google Scholar] [CrossRef] [PubMed]
- Wood, K.A.; Dipasquale, B.; Youle, R.J. In situ labeling of granule cells for apoptosis-associated DNA fragmentation reveals different mechanisms of cell loss in developing cerebellum. Neuron 1993, 11, 621–632. [Google Scholar] [CrossRef]
- Gavrieli, Y.; Sherman, Y.; Ben-Sasson, S.A. Identification of programmed cell death in situ via specific labeling of nuclear DNA fragmentation. J. Cell Biol. 1992, 119, 493–501. [Google Scholar] [CrossRef] [PubMed]
- Lossi, L.; Zagzag, D.; Greco, M.A.; Merighi, A. Apoptosis of undifferentiated progenitors and granule cell precursors in the post-natal human cerebellar cortex correlates with expression of BCL-2, ICE and CPP-32 proteins. J. Comp. Neurol. 1998, 399, 359–372. [Google Scholar] [CrossRef]
- White, L.D.; Barone, S., Jr. Qualitative and quantitative estimates of apoptosis from birth to senescence in the rat brain. Cell Death Differ. 2001, 8, 345–356. [Google Scholar] [CrossRef]
- Norman, D.J.; Feng, L.; Cheng, S.S.; Gubbay, J.; Chan, E.; Heintz, N. The lurcher gene induces apoptotic death in cerebellar Purkinje cells. Development 1995, 121, 1183–1193. [Google Scholar] [PubMed]
- Owens, G.P.; Mahalik, T.J.; Hahn, W.E. Expression of the death-associated gene RP-8 in granule cell neurons undergoing postnatal cell death in the cerebellum of weaver mice. Dev. Brain Res. 1995, 86, 35–47. [Google Scholar] [CrossRef]
- Smeyne, R.J.; Chu, T.; Lewin, A.; Bian, F.; Crisman, S.; Kunsch, C.; Lira, S.A.; Oberdick, J. Local control of granule cell generation by cerebellar Purkinje cells. Mol. Cell. Neurosci. 1995, 6, 230–251. [Google Scholar] [CrossRef]
- Krueger, B.K.; Burne, J.F.; Raff, M.C. Evidence for large-scale astrocyte death in the developing cerebellum. J. Neurosci. 1995, 15, 3366–3374. [Google Scholar] [CrossRef]
- Yamaguchi, Y.; Miura, M. Programmed cell death in neurodevelopment. Dev. Cell 2015, 32, 478–490. [Google Scholar] [CrossRef]
- Virchow, R. Cellularpathologie in Ihre Begrundung auf Physiologische und Pathologische Gewebelehre; A. Hirschwal: Berlin, Germany, 1858. [Google Scholar]
- Schweichel, J.U.; Merker, H.J. The morphology of various types of cell death in prenatal tissues. Teratology 1973, 7, 253–266. [Google Scholar] [CrossRef] [PubMed]
- Clarke, P.G.H. Developmental cell death: Morphological diversity and multiple mechanisms. Anat. Embryol. 1990, 181, 195–213. [Google Scholar] [CrossRef] [PubMed]
- Fricker, M.; Tolkovsky, A.M.; Borutaite, V.; Coleman, M.; Brown, G.C. Neuronal cell death. Physiol. Rev. 2018, 98, 813–880. [Google Scholar] [CrossRef] [PubMed]
- Kerr, J.F.; Wyllie, A.H.; Currie, A.R. Apoptosis: A basic biological phenomenon with wide-ranging implications in tissue kinetics. Br. J. Cancer 1972, 26, 239–257. [Google Scholar] [CrossRef] [PubMed]
- Lossi, L.; Mioletti, S.; Merighi, A. Synapse-independent and synapse-dependent apoptosis of cerebellar granule cells in postnatal rabbits occur at two subsequent but partly overlapping developmental stages. Neuroscience 2002, 112, 509–523. [Google Scholar] [CrossRef]
- Lossi, L.; Mioletti, S.; Aimar, P.; Bruno, R.; Merighi, A. In vivo analysis of cell proliferation and apoptosis in the CNS. In Cellular and Molecular Methods in Neuroscience Research; Merighi, A., Carmignoto, G., Eds.; Springer: New York, NY, USA, 2002; pp. 235–258. [Google Scholar]
- Julien, O.; Wells, J.A. Caspases and their substrates. Cell Death Differ. 2017, 24, 1380–1389. [Google Scholar] [CrossRef] [PubMed]
- Boland, K.; Flanagan, L.; Prehn, J.H. Paracrine control of tissue regeneration and cell proliferation by Caspase-3. Cell Death Dis. 2013, 4, e725. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Goldman, J.E. Generation of cerebellar interneurons from dividing progenitors in the white matter. Neuron 1996, 16, 47–54. [Google Scholar] [CrossRef]
- Hibi, M.; Shimizu, T. Development of the cerebellum and cerebellar neural circuits. Dev. Neurobiol. 2011, 72, 282–301. [Google Scholar] [CrossRef] [PubMed]
- Altman, J.; Bayer, S.A. Development of the Cerebellar System in Relation to Its Evolution, Structure and Functions; CRC Press: Boca Raton, IL, USA, 1997. [Google Scholar]
- Simonati, A.; Tosati, C.; Rosso, T.; Piazzola, E.; Rizzuto, N. Cell proliferation and death: Morphological evidence during corticogenesis in the developing human brain. Microsc. Res. Tech. 1999, 45, 341–352. [Google Scholar] [CrossRef]
- Ábrahám, H.; Tornóczky, T.; Kosztoláínyi, G.; Seress, L. Cell formation in the cortical layers of the developing human cerebellum. Int. J. Dev. Neurosci. 2001, 19, 53–62. [Google Scholar] [CrossRef]
- Lavezzi, A.M.; Ottaviani, G.; Terni, L.; Matturri, L. Histological and biological developmental characterization of the human cerebellar cortex. Int. J. Dev. Neurosci. 2006, 24, 365–371. [Google Scholar] [CrossRef] [PubMed]
- Nat, R.; Voiculescu, B.; Stanciu, C.; Vidulescu, C.; Cergan, R.; Badiu, C.; Popescu, L.M. Apoptosis in human embryo development: 2. Cerebellum. J. Cell. Mol. Med. 2007, 5, 179–187. [Google Scholar] [CrossRef]
- Sohma, O.; Mizuguchi, M.; Takashima, S.; Yamada, M.; Ikeda, K.; Ohta, S. High expression of Bcl-x protein in the developing human cerebellar cortex. J. Neurosci. Res. 1996, 43, 175–182. [Google Scholar] [CrossRef]
- Gagliardini, V.; Fernandez, P.-A.; Lee, R.K.K.; Drexler, H.C.A.; Rotello, R.J.; Fishman, M.C.; Yuan, J. Prevention of vertebrate neuronal death by the crmA gene. Science 1994, 263, 826–828. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Alnemri, T.; Litwack, G.; Alnemri, E.S. CPP32, a novel human apoptotic protein with homology to Caenorahbitis elegans cell death protein Ced-3 and mammalian interleukin-1-beta-converting enzyme. J. Biol. Chem. 1994, 269, 30761–30764. [Google Scholar]
- Alnemri, E.S.; Livingston, J.N.; Nicholson, D.W.; Salvesen, G.; Thornberry, N.A.; Wong, W.W.; Yuan, J. Human ICE/CED-3 protease nomenclature. Cell 1996, 87, 171–181. [Google Scholar] [CrossRef]
- Deo, K.; Bijlani, V.; Deo, M.G. “Physiological” and cytotoxic cell death in protein deficiency. A study in developing cerebellum in rats. Acta Neuropathol. 1979, 46, 221–225. [Google Scholar] [CrossRef]
- Tanaka, M.; Marunouchi, T. Immunohistochemical analysis of developmental stage of external granular layer neurons which undergo apoptosis in postnatal rat cerebellum. Neurosci. Lett. 1998, 242, 85–88. [Google Scholar] [CrossRef]
- Sinha, R.A.; Khare, P.; Rai, A.; Maurya, S.K.; Pathak, A.; Mohan, V.; Nagar, G.K.; Mudiam, M.K.R.; Godbole, M.M.; Bandyopadhyay, S. Anti-apoptotic role of omega-3-fatty acids in developing brain: Perinatal hypothyroid rat cerebellum as apoptotic model. Int. J. Dev. Neurosci. 2009, 27, 377–383. [Google Scholar] [CrossRef]
- Vidovic, M.; Chen, M.M.; Lu, Q.Y.; Kalloniatis, K.F.; Martin, B.M.; Tan, A.H.Y.; Lynch, C.; Croaker, G.D.; Cass, D.T.; Song, Z.M. Deficiency in endothelin receptor B reduces proliferation of neuronal progenitors and increases apoptosis in postnatal rat cerebellum. Cell. Mol. Neurobiol. 2008, 28, 1129–1138. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, B.D.; Gibbons, B.; Allen, L.R.; Stella, J.; D’Mello, S.R. Aberrant apoptosis in the neurological mutant Flathead is associated with defective cytokinesis of neural progenitor cells. Dev. Brain Res. 2001, 130, 53–63. [Google Scholar] [CrossRef]
- Robert, M.C.; Furlan, G.; Rosso, N.; Gambaro, S.E.; Apitsionak, F.; Vianello, E.; Tiribelli, C.; Gazzin, S. Alterations in the cell cycle in the cerebellum of hyperbilirubinemic Gunn rat: A possible link with apoptosis? PLoS ONE 2013, 8, e79073. [Google Scholar] [CrossRef] [PubMed]
- Kubera, C.; Hernandez, A.L.; Heng, V.; Bordey, A. Transient mGlu5R inhibition enhances the survival of granule cell precursors in the neonatal cerebellum. Neuroscience 2012, 219, 271–279. [Google Scholar] [CrossRef] [PubMed]
- Wüllner, U.; Loschmann, P.A.; Weller, M.; Klockgether, T. Apoptotic cell death in the cerebellum of mutant weaver and lurcher mice. Neurosci. Lett. 1995, 200, 109–112. [Google Scholar] [CrossRef]
- Cocito, C.; Merighi, A.; Giacobini, M.; Lossi, L. Alterations of cell proliferation and apoptosis in the hypoplastic Reeler cerebellum. Front. Cell. Neurosci. 2016, 10, 141. [Google Scholar] [CrossRef] [PubMed]
- Kanungo, A.K.; Liadis, N.; Robertson, J.; Woo, M.; Henderson, J.T. Excitatory tonus is required for the survival of granule cell precursors during postnatal development within the cerebellum. Neuroscience 2009, 158, 1364–1377. [Google Scholar] [CrossRef]
- Klein, J.A.; Longo-Guess, C.M.; Rossmann, M.P.; Seburn, K.L.; Hurd, R.E.; Frankel, W.N.; Bronson, R.T.; Ackerman, S.L. The harlequin mouse mutation downregulates apoptosis-inducing factor. Nature 2002, 419, 367–374. [Google Scholar] [CrossRef] [PubMed]
- Ishimura, R.; Martin, G.R.; Ackerman, S.L. Loss of apoptosis-inducing factor results in cell-type-specific neurogenesis defects. J. Neurosci. 2008, 28, 4938–4948. [Google Scholar] [CrossRef]
- Rosen, A.; Casciola-Rosen, L. Macromolecular substrates for the ICE-like proteases during apoptosis. J. Cell. Biochem. 1997, 64, 50–54. [Google Scholar] [CrossRef]
- Perez-Pouchoulen, M.; VanRyzin, J.W.; McCarthy, M.M. Morphological and phagocytic profile of microglia in the developing rat cerebellum. eNeuro 2015, 2, eN-0036. [Google Scholar] [CrossRef] [PubMed]
- Vogel, M.W.; Herrup, K. Numerical matching in the mammalian CNS: Lack of a competitive advantage of early over late-generated cerebellar granule cells. J. Comp. Neurol. 1989, 283, 118–128. [Google Scholar] [CrossRef] [PubMed]
- Lossi, L.; Cocito, C.; Alasia, S.; Merighi, A. Ex vivo imaging of active caspase 3 by a FRET-based molecular probe demonstrates the cellular dynamics and localization of the protease in cerebellar granule cells and its regulation by the apoptosis-inhibiting protein survivin. Mol. Neurodegener. 2016, 11, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Oomman, S.; Finckbone, V.; Dertien, J.; Attridge, J.; Henne, W.; Medina, M.; Mansouri, B.; Singh, H.; Strahlendorf, H.; Strahlendorf, J. Active caspase-3 expression during postnatal development of rat cerebellum is not systematically or consistently associated with apoptosis. J. Comp. Neurol. 2004, 476, 154–173. [Google Scholar] [CrossRef] [PubMed]
- Cabrera, O.; Dougherty, J.; Singh, S.; Swiney, B.S.; Farber, N.B.; Noguchi, K.K. Lithium protects against glucocorticoid induced neural progenitor cell apoptosis in the developing cerebellum. Brain Res. 2014, 1545, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.S.; Li, M.S.; Du, J.; Jiang, Q.Y.; Wang, L.; Yan, S.Y.; Yu, D.M.; Deng, J.B. Neuronal apoptosis in the developing cerebellum. Anat. Histol. Embryol. 2011, 40, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Sawada, M.; Miura, M.; Maronouchi, T. Insulin-like growth factor-I analogue prevents apoptosis mediated through an interleukin-1 beta converting enzyme (caspase-1)-like protease of cerebellar external granular layer neurons: Developmental stage-specific mechanisms of neuronal cell death. Neuroscience 1998, 84, 89–100. [Google Scholar] [CrossRef]
- Katic, J.; Loers, G.; Tosic, J.; Schachner, M.; Kleene, R. The cell adhesion molecule CHL1 interacts with patched-1 to regulate apoptosis during postnatal cerebellar development. J. Cell Sci. 2017, 130, 2606. [Google Scholar] [CrossRef]
- Vogel, M.W. Cell death, Bcl-2, Bax, and the cerebellum. Cerebellum 2002, 1, 277–287. [Google Scholar] [CrossRef]
- Jung, A.R.; Kim, T.W.; Rhyu, I.J.; Kim, H.; Lee, Y.D.; Vinsant, S.; Oppenheim, R.W.; Sun, W. Misplacement of Purkinje cells during postnatal development in bax knock-out mice: A novel role for programmed cell death in the nervous system? J. Neurosci. 2008, 28, 2941. [Google Scholar] [CrossRef]
- Bezvenyuk, Z.; Salminen, A.; Solovyan, V. Excision of DNA loop domains as a common step in caspase-dependent and -independent types of neuronal cell death. Mol. Brain Res. 2000, 81, 191–196. [Google Scholar] [CrossRef]
- Allais, A.; Burel, D.; Roy, V.; Arthaud, S.; Galas, L.; Isaac, E.R.; Desfeux, A.; Parent, B.; Fournier, A.; Chapillon, P.; et al. Balanced effect of PACAP and FasL on granule cell death during cerebellar development: A morphological, functional and behavioural characterization. J. Neurochem. 2010, 113, 329–340. [Google Scholar] [CrossRef] [PubMed]
- Hurtado de, M.T.; Perez-Garcia, C.G.; Kroll, T.T.; Hoong, N.H.; O’Leary, D.D.; Verma, I.M. Antiapoptotic protein Lifeguard is required for survival and maintenance of Purkinje and granular cells. Proc. Natl. Acad. Sci. USA 2011, 108, 17189–17194. [Google Scholar] [CrossRef] [PubMed]
- Altieri, D.C. Survivin and IAP proteins in cell-death mechanisms. Biochem. J. 2010, 430, 199–205. [Google Scholar] [CrossRef] [PubMed]
- Tamm, I.; Wang, Y.; Sausville, E.; Scudiero, D.A.; Vigna, N.; Oltersdorf, T.; Reed, J.C. IAP-family protein survivin inhibits caspase activity and apoptosis induced by Fas (CD95), Bax, caspases, and anticancer drugs. Cancer Res. 1998, 58, 5315. [Google Scholar]
- Caldas, H.; Jiang, Y.; Holloway, M.P.; Fangusaro, J.; Mahotka, C.; Conway, E.M.; Altura, R.A. Survivin splice variants regulate the balance between proliferation and cell death. Oncogene 2005, 24, 1994–2007. [Google Scholar] [CrossRef]
- Jiang, Y.; De Bruin, A.; Caldas, H.; Fangusaro, J.; Hayes, J.; Conway, E.M.; Robinson, M.L.; Altura, R.A. Essential role for survivin in early brain development. J. Neurosci. 2005, 25, 6962–6970. [Google Scholar] [CrossRef]
- Blancas, S.; Moran, J. Role for apoptosis-inducing factor in the physiological death of cerebellar neurons. Neurochem. Int. 2011, 58, 934–942. [Google Scholar] [CrossRef]
- Blancas, S.; Fado, R.; Rodriguez-Alvarez, J.; Moran, J. Endogenous XIAP, but not other members of the inhibitory apoptosis protein family modulates cerebellar granule neurons survival. Int. J. Dev. Neurosci. 2014, 37, 26–35. [Google Scholar] [CrossRef]
- Moghaddam, B.; Adams, B.; Verma, A.; Daly, D. Activation of glutamatergic neurotransmission by ketamine: A novel step in the pathway from NMDA receptor blockade to dopaminergic and cognitive disruptions associated with the prefrontal cortex. J. Neurosci. 1997, 17, 2921–2927. [Google Scholar] [CrossRef]
- Monti, B.; Contestabile, A. Blockade of the NMDA receptor increases developmental apoptotic elimination of granule neurons and activates caspases in the rat cerebellum. Eur. J. Neurosci. 2001, 12, 3117–3123. [Google Scholar] [CrossRef]
- Borghesani, P.R.; Peyrin, J.M.; Klein, R.; Rubin, J.; Carter, A.R.; Schwartz, P.M.; Luster, A.; Corfas, G.; Segal, R.A. BDNF stimulates migration of cerebellar granule cells. Development 2002, 129, 1435–1442. [Google Scholar] [PubMed]
- Menshanov, P.N.; Lanshakov, D.A.; Dygalo, N.N. proBDNF is a major product of bdnf gene expressed in the perinatal rat cortex. Physiol. Res. 2015, 64, 925–934. [Google Scholar] [PubMed]
- Ibáñez, C.F.; Simi, A. p75 neurotrophin receptor signaling in nervous system injury and degeneration: Paradox and opportunity. Trends Neurosci. 2012, 35, 431–440. [Google Scholar] [CrossRef] [PubMed]
- Zanin, J.P.; Abercrombie, E.; Friedman, W.J. Proneurotrophin-3 promotes cell cycle withdrawal of developing cerebellar granule cell progenitors via the p75 neurotrophin receptor. Elife 2016, 5, e16654. [Google Scholar] [CrossRef] [PubMed]
- Kisiswa, L.; Fernàndez-Suàrez, D.; Sergaki, M.C.; Ibáñez, C. RIP2 Gates TRAF6 Interaction with death receptor p75NTR to regulate cerebellar granule neuron survival. Cell Rep. 2018, 24, 1013–1024. [Google Scholar] [CrossRef] [PubMed]
- Miale, I.L.; Sidman, R.L. An autoradiographic analysis of histogenesis in the mouse cerebellum. Exp. Neurol. 1961, 4, 277–296. [Google Scholar] [CrossRef]
- D’Arcangelo, G.; Miao, G.G.; Chen, S.C.; Soares, H.D.; Morgan, J.I.; Curran, T. A protein related to extracellular matrix proteins deleted in the mouse mutant reeler. Nature 1995, 374, 719–723. [Google Scholar] [CrossRef] [PubMed]
- D’Arcangelo, G. Reelin mouse mutants as models of cortical development disorders. Epilepsy Behav. 2006, 8, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Jankowski, J.; Miething, A.; Schilling, K.; Oberdick, J.; Baader, S. Cell death as a regulator of cerebellar histogenesis and compartmentation. Cerebellum 2011, 10, 373–392. [Google Scholar] [CrossRef]
- Sarna, J.R.; Hawkes, R. Patterned Purkinje cell death in the cerebellum. Prog. Neurobiol. 2003, 70, 473–507. [Google Scholar] [CrossRef]
- Madalosso, S.H.; Pèrez-Villegas, E.M.; Armengol, J.A. Naturally occurring neuronal death during the postnatal development of Purkinje cells and their precerebellar afferent projections. Brain Res. Rev. 2005, 49, 267–279. [Google Scholar] [CrossRef] [PubMed]
- Jankowski, J.; Miething, A.; Schilling, K.; Baader, S.L. Physiological Purkinje cell death is spatiotemporally organized in the developing mouse cerebellum. Cerebellum 2009, 8, 277–290. [Google Scholar] [CrossRef] [PubMed]
- Marti-Clua, J. Natural apoptosis in developing mice dopamine midbrain neurons and vermal Purkinje cells. Folia Neuropathol. 2016, 54, 180–189. [Google Scholar] [CrossRef] [PubMed]
- Krajewska, M.; Mai, J.K.; Zapata, J.M.; Ashwell, K.W.; Schendel, S.L.; Reed, J.C.; Krajewski, S. Dynamics of expression of apoptosis-regulatory proteins Bid, Bcl-2, Bcl-X, Bax and Bak during development of murine nervous system. Cell Death Differ. 2002, 9, 145–157. [Google Scholar] [CrossRef] [PubMed]
- Zecevic, N.; Rakic, P. Differentiation of Purkinje cells and their relationship to other components of the developing cerebellar cortex in man. J. Comp. Neurol. 1976, 167, 27–48. [Google Scholar] [CrossRef] [PubMed]
- Dusart, I.; Guenet, J.; Sotelo, C. Purkinje cell death: Differences between developmental cell death and neurodegenerative death in mutant mice. Cerebellum 2006, 5, 163–173. [Google Scholar] [CrossRef] [PubMed]
- Caddy, K.W.; Biscoe, T.J. Structural and quantitative studies on the normal C3H and Lurcher mutant mouse. Philos. Trans. R. Soc. Lond. B Biol. Sci. 1979, 287, 167–201. [Google Scholar] [CrossRef]
- Zuo, J.; De Jager, P.L.; Takahashi, K.A.; Jiang, W.; Linden, D.J.; Heintz, N. Neurodegeneration in Lurcher mice caused by mutation in [delta]2 glutamate receptor gene. Nature 1997, 388, 769–773. [Google Scholar] [CrossRef]
- Chakrabarti, L.; Eng, J.; Ivanov, N.; Garden, G.A.; La Spada, A.R. Autophagy activation and enhanced mitophagy characterize the Purkinje cells of pcd mice prior to neuronal death. Mol. Brain 2009, 2, 24. [Google Scholar] [CrossRef]
- Duchala, C.S.; Shick, H.E.; Garcia, J.; Deweese, D.M.; Sun, X.; Stewart, V.J.; Macklin, W.B. The toppler mouse: A novel mutant exhibiting loss of Purkinje cells. J. Comp. Neurol. 2004, 476, 113–129. [Google Scholar] [CrossRef] [PubMed]
- Zanjani, H.S.; Vogel, M.W.; Mariani, J. Deletion of the GluRd2 receptor in the Hotfoot mouse mutant causes granule cell loss, delayed Purkinje cell death, and reductions in Purkinje cell dendritic tree area. Cerebellum 2016, 15, 755–766. [Google Scholar] [CrossRef] [PubMed]
- White, F.A.; Keller-Peck, C.R.; Knudson, C.M.; Korsmeyer, S.J.; Snider, W.D. Widespread elimination of naturally occurring neuronal death in Bax-deficient mice. J. Neurosci. 1998, 18, 1428–1439. [Google Scholar] [CrossRef] [PubMed]
- Goswami, J.; Martin, L.A.; Goldowitz, D.; Beitz, A.J.; Feddersen, R.M. Enhanced Purkinje cell survival but compromised cerebellar function in targeted anti-apoptotic protein transgenic mice. Mol. Cell. Neurosci. 2005, 29, 202–221. [Google Scholar] [CrossRef] [PubMed]
- Rakic, P. Extrinsic cytological determinants of basket and stellate cell dendrite pattern in the cerebellar molecular layer. J. Comp. Neurol. 1972, 146, 335–354. [Google Scholar] [CrossRef] [PubMed]
- Yamanaka, H.; Yanagawa, Y.; Obata, K. Development of stellate and basket cells and their apoptosis in mouse cerebellar cortex. Neurosci. Res. 2004, 50, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Oomman, S.; Strahlendorf, H.; Finckbone, V.; Strahlendorf, J. Non-lethal active caspase-3 expression in Bergmann glia of postnatal rat cerebellum. Dev. Brain Res. 2005, 160, 130–145. [Google Scholar] [CrossRef] [PubMed]
- Hyman, B.T.; Yuan, J. Apoptotic and non-apoptotic roles of caspases in neuronal physiology and pathophysiology. Nat. Rev. Neurosci. 2012, 13, 395–406. [Google Scholar] [CrossRef]
- Dekkers, M.P.; Nikoletopoulou, V.; Barde, Y.A. Cell biology in neuroscience: Death of developing neurons: New insights and implications for connectivity. J. Cell Biol. 2013, 203, 385–393. [Google Scholar] [CrossRef] [PubMed]
- D’Amelio, M.; Cavallucci, V.; Cecconi, F. Neuronal caspase-3 signaling: Not only cell death. Cell Death Differ. 2010, 17, 1104–1114. [Google Scholar] [CrossRef] [PubMed]
- D’Amelio, M.; Sheng, M.; Cecconi, F. Caspase-3 in the central nervous system: Beyond apoptosis. Trends Neurosci. 2012, 35, 700–709. [Google Scholar] [CrossRef] [PubMed]


© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lossi, L.; Castagna, C.; Merighi, A. Caspase-3 Mediated Cell Death in the Normal Development of the Mammalian Cerebellum. Int. J. Mol. Sci. 2018, 19, 3999. https://doi.org/10.3390/ijms19123999
Lossi L, Castagna C, Merighi A. Caspase-3 Mediated Cell Death in the Normal Development of the Mammalian Cerebellum. International Journal of Molecular Sciences. 2018; 19(12):3999. https://doi.org/10.3390/ijms19123999
Chicago/Turabian StyleLossi, Laura, Claudia Castagna, and Adalberto Merighi. 2018. "Caspase-3 Mediated Cell Death in the Normal Development of the Mammalian Cerebellum" International Journal of Molecular Sciences 19, no. 12: 3999. https://doi.org/10.3390/ijms19123999
APA StyleLossi, L., Castagna, C., & Merighi, A. (2018). Caspase-3 Mediated Cell Death in the Normal Development of the Mammalian Cerebellum. International Journal of Molecular Sciences, 19(12), 3999. https://doi.org/10.3390/ijms19123999