Current Insights into Oral Cancer Epigenetics
by
 , , ,
, , ,
Alexandra Iulia Irimie
1, 
Cristina Ciocan
2,
Diana Gulei
2,
Nikolay Mehterov
3,4,
Atanas G. Atanasov
5,6,
Diana Dudea
1 and
Ioana Berindan-Neagoe
2,7,8,* 1
Department of Prosthetic Dentistry and Dental Materials, Division Dental Propaedeutics, Aesthetics, Faculty of Dentistry, “Iuliu Hatieganu” University of Medicine and Pharmacy, 400001 Cluj-Napoca, Romania
2
MEDFUTURE—Research Center for Advanced Medicine, “Iuliu Hatieganu” University of Medicine and Pharmacy, Marinescu 23 Street, 400001 Cluj-Napoca, Romania
3
Department of Medical Biology, Faculty of Medicine, Medical University-Plovdiv, 15-А Vassil Aprilov Blvd., 4000 Plovdiv, Bulgaria
4
Technological Center for Emergency Medicine, 15-АVassil Aprilov Blvd., 4000 Plovdiv, Bulgaria
5
Institute of Genetics and Animal Breeding of the Polish Academy of Sciences, 05-552 Jastrzebiec, Poland
6
Department of Pharmacognosy, University of Vienna, Althanstrasse 14, A-1090 Vienna, Austria
7
Research Center for Functional Genomics, Biomedicine and Translational Medicine, “Iuliu Hatieganu” University of Medicine and Pharmacy, Marinescu 23 Street, 400337 Cluj-Napoca, Romania
8
Department of Functional Genomics and Experimental Pathology, The Oncology Institute “Prof. Dr. Ion Chiricuţă”, 400015 Cluj-Napoca, Romania
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2018, 19(3), 670; https://doi.org/10.3390/ijms19030670
Submission received: 30 January 2018
/
Revised: 20 February 2018
/
Accepted: 22 February 2018
/
Published: 27 February 2018
(This article belongs to the Section Molecular Pathology, Diagnostics, and Therapeutics)
Abstract
:Epigenetic modifications have emerged into one of the cancer hallmarks, replacing the concept of malignant pathologies as being solely genetic-based conditions. The epigenetic landscape is responsible for normal development but also for the heterogeneity among tissues in terms of gene expression patterns. Dysregulation in these mechanisms has been associated with disease stage, and increased attention is now granted to cancer in order to take advantage of these modifications in terms of novel therapeutic strategies or diagnosis/prognosis tools. Oral cancer has also been subjected to epigenetic analysis with numerous studies revealing that the development and progression of this malignancy are partially induced by an altered epigenetic substrate together with genetic alterations and prolonged exposure to environmental risk factors. The present review summarizes the most important epigenetic modifications associated with oral cancer and also their potential to be used as new therapeutic targets.
1. Introduction
Cancer stands among the most complex human diseases with multiple levels of regulation. Its complexity and heterogeneity sustain the development and advancement of carcinogenesis. Despite the significant progress that has been made in fundamental and clinical research, the incidence and mortality rates associated with malignant pathologies are still at high levels with an expected increased rate in the next 15 years [1]. Head and neck cancers rank as the sixth most common malignancy, where oral cancer represents the most frequent subtype within this spectrum [2,3]. Moreover, oral squamous cell carcinoma (OSCC) accounts for approximately 90% of the oral subtype, being by far the most common type of malignancy within the oral cavity [4,5].
The induction and development of oral cancer are due to a sum of genetic changes combined with environmental risk factors (especially tobacco and alcohol consumption but also viral infections and chronic inflammations) that in the end lead to alterations in the activity of oncogenes and tumor suppressor genes [6,7]. The level of complexity behind the abnormal expression and function of these genes is extremely high, where epigenetic mechanisms are standing as another segment of regulation besides the actual changes in the DNA sequence, which include mutations, deletions, and amplifications [8]. While epigenetic mechanisms are essential for sustaining the development and tissue-specific homeostasis of the organism, a deregulation in these processes can lead to the installation of pathological states, and increased attention is being granted nowadays toward cancer [9]. Therefore, the classical concept where cancer is controlled mainly by genetic modifications has now shifted toward a more comprehensive picture where DNA methylation, modifications of histones, and nucleosome positioning are now considered to play a crucial role. Also, the expression of non-coding RNAs (ncRNAs), especially microRNAs (miRNAs), may be influenced and at the same time is also influencing the epigenetic mechanisms [10].
Along with other types of cancers, oral malignancies have also been subjected to intense research in the last years regarding the specific modifications within the epigenome. The goal has been to obtain a more detailed profile of this disease and also to possibly develop novel diagnostic, prognostic, and therapeutic tools. The present review aims to present the most important aspects regarding the epigenetic changes that have been found until now in oral cancers and how these modifications can be used in the patient’s favor.
2. Epigenetics—An Emerging Concept
The basic characterization of the epigenetic concept states that these mechanisms are reversible changes that are not related to modifications in the structure of DNA and can be inherited and preserved for multiple generations. Increased research has extended the initial characterization made in 1942 by C. H. Waddington: “the causal interactions between genes and their products, which bring the phenotype into being.” The definition was primarily referring to embryonic development—“the study of heritable changes in gene expression that occur independent of changes in the primary DNA sequence,”—a characterization that is further applicable to the whole organism and stages of development, including disease states [9]. Actually, epigenetic modifications enable the possibility of heterogeneity between cells despite the same genetic material and similar cellular signaling patterns.
Research advances in the study of cancer complexity have revealed that the epigenetic landscape is different between malignant and normal cells, a fact that influences the progression of the disease and is also involved in all stages of malignant development [11,12]. Nevertheless, the reality that these non-genetic modifications are reversible is opening the possibility of new therapeutic strategies, where targeted agents can be used to restore what is considered the normal conduit in cell signaling and impair cancer development [13].
The structure of chromatin consists of nucleosome units that in turn are composed of DNA wrapped around eight histone proteins [14]. This structure can be modified through epigenetic mechanisms that are mainly divided into four sectors: DNA methylation, covalent modifications of histones, non-covalent modifications—such as the introduction of histone variants and remodeling of nucleosome structure—and non-coding RNA-related modifications, especially miRNAs (Figure 1). These mechanisms act together to create the specific epigenetic landscape associated with a certain population of cells or a process within the organism. Modifications in the epigenetic signature can contribute to the induction of abnormal cellular signaling where cells lose their initial identity and become pathogenic. This is the case of malignant diseases where, besides the complex genetic background, epigenetic processes are adding an additional level of complexity in terms of cancer regulatory mechanisms.
3. DNA Methylation
Within the main mechanisms responsible for epigenetic regulation, DNA methylation is by far the most studied, being responsible for gene silencing and chromatin architecture. These processes take place in concordance with other epigenetic modifications in order to ensure the specific regulatory landscape and to control the phenotype of the target cell. DNA methylation primarily materializes within the so-called “CpG islands” that are composed of a series of CpG dinucleotide structures located usually at the 5′ end of genes. The CpG dinucleotides can also be found in genome regions that contain repetitive sequences, such as in the case of retrotransposon elements, rDNA, and centromeric repeats [15,16,17]. While most of the CpG sites within repetitive sequences are methylated, in the case of the grouped ones (CpG islands) that usually occupy more than half of the promoter of a certain gene, the pattern is consistently unmethylated for both undifferentiated and differentiated tissues [18]. There are also some isolated cases where CpG islands are methylated, as in the case of the X-chromosome that is inactivated during development and also for some genes associated with incipient development stages that are silenced through methylation in adult tissues [15,17,19]. For the case of dispersed CpG islands, the heavily methylated status is used to impair chromosomal instability by inhibiting the expression of transposable elements and also non-coding sequences [18].
Global analysis of CpG status in terms of methylation marks has proven to be useful for profiling the cancer epigenetic landscape. Therefore, it has been shown that approximately 5–10% of the CpG islands are aberrantly methylated within malignant pathologies, a fact that leads to the silencing of specific coding and non-coding genes (e.g., tumor suppressor genes) and implicit propagation of altered signals within specific pathways [20]. However, there is data involving gene bodies and CpG “shores” (regions situated near CpG islands—upstream or downstream) that despite the abnormal methylated status within cancer, the downstream effect does not consist of silencing the transcriptional and translational processes, a fact that suggests that the spatial context (localization of the CpG sites within the genome) is actually a crucial element in the establishment of gene silencing [20].
Although the hypermethylation of CpG islands is the most common subject in cancer epigenetics, the actual malignant epigenetic profile is associated with a global hypomethylation trend. Also, restrictive data has been presented about the status of the genes responsible for the methylation process—DNA methyltransferases (DNMTs)—and also of those responsible for the expression of methyl-binding proteins accountable for recruiting histone-modifying enzymes [21]. Mutations in these genes can heavily impact the epigenetic signature [22] and implicitly lead to the induction of different diseases. As said, extensive analysis regarding the integrity of the reminded genes in cancer samples is still at the beginning but with the potential of revealing new layers of regulation in terms of cancer epigenetics. One such study conducted by Ley et al. found that DNMT3A is mutated in approximately 25% of patients diagnosed with acute myeloid leukemia (AML); those authors also associated the mutations with an impact on the prognosis of such individuals [23].
4. Histone Modifications
Histone proteins are predisposed to different modifications comprising ubiquitylation, sumoylation, methylation, acetylation, and also phosphorylation. These modifications take place preferentially at the N-terminal tails, affecting gene transcription and vital signaling pathways [24]. As opposed to DNA methylation, histone covalent modifications can also promote transcription and not only silence the expression of specific genes. As a practical example, lysine acetylation is majorly associated with the accessibility of transcription machinery to the chromatin and implicit transcription promotion, where lysine methylation does not follow necessarily the same pattern. Depending on the specific localization of methylation, the epigenetic modifications can be correlated with transcription activation or repression. More specifically, methylation of H3 lysine 9 (H3K9), H3 lysine 27 (H3K27), and H4 lysine 20 (H4K20) is associated with inhibitory mechanisms, where methylation of histone H3 lysine 4 (H3K4) and H3 lysine 36 are conducted towards activation of chromatin transcription [9,25]. The landscape of histone modification is different depending on the cellular context and these differences contribute to specific cell behaviors, including cancer cells that lose the homeostatic epigenetic pattern. This pattern is coordinated through the activity of enzymes that are able to add or remove methyl and acetyl groups and also functionally interact in order to establish the specific epigenetic profile [9,26,27]. Modifications in the structure and activity of these enzymes were previously associated with cancer susceptibility and also development. For example, p300 HATs (histone acetyltransferases) were found as being mutated in gastrointestinal cancers where the counterpart molecules, HDACs (histone deacetylases), were less frequently encountered as being deregulated. For the case of methyltransferases, it was assessed that mice lacking the Suv39 family of enzymes responsible for the methylation of H3K9 are predisposed to malignant pathologies, especially B cell lymphomas. Aurora kinases, enzymes with histone phosphorylation capacity, have also been correlated with cancer mechanisms [28]. Oral cancer is characterized by increased histone acetylation where experimental impairment of p300 acetyltransferase significantly decreased the tumor parameters in mice models [29].
5. Nucleosome Positioning and Histone Variants
Nucleosome positioning and also histone variants are framed in the non-covalent modifications category, but also with important roles regarding the establishment of the cell-specific epigenome. The position of nucleosomes is especially important when looking at the accessibility of transcription factors to specific regions within the chromatin structure, a process that has direct effects on the inhibition or activation of gene expression [30]. Genomic mapping of these positions showed that the pattern is strictly controlled around gene promoters where the actual gene sequence is characterized by a more random distribution of nucleosomes. There are also nucleosome-free regions (NFR) that are considered to provide the “space” for the transcription machinery to self-assemble and also to disconnect from the gene. Modifications in nucleosome architecture, together with other regulatory epigenetic mechanisms, can lead to transcription impairment or stimulation where the addition of an NFR or the loss of another can drastically modify the cell phenotype [31,32,33,34,35].
6. Non-Coding RNAs—miRNAs
miRNAs belong to the non-coding group of sequences and are able to modulate the expression of specific genes based on complementarity rules. These small molecules of approximately 22 nucleotides in length bind to the 3′UTR of the mRNA and block the translation of the target gene or, even more, can induce complete degradation of the transcript [38,39,40]. Therefore, miRNAs have emerged as key molecules within the regulation of signaling pathways, being able to modulate the expression of their target gene and implicitly maintain the normal state of the organism [41,42,43]. In terms of malignant pathologies, there are a great number of studies that have demonstrated that miRNAs are aberrantly expressed in cancer cells, a fact that contributes to the maintenance and also the development of the pathology. More specifically, the pattern consists of downregulated tumor suppressor miRNAs that in normal states inhibit the activity of oncogenic genes, and upregulated tumor promoting miRNAs that are able to impair the translation of the tumor suppressor ones. Subsequent to these discoveries, researchers have implemented experimental protocols where the pathological miRNA pattern is modulated for therapeutic purposes through administration of exogenous sequences (miRNA mimics or inhibitors) or for implementation of novel diagnosis and prognosis tools (considering the fact that miRNAs are differentially expressed in tumor or fluid samples vs. control ones) [44,45].
However, how is the global expression of the miRNA panel (miRNome) maintained during the normal scenarios and how is it lost in pathological ones? Studies have shown that miRNAs are also subjected to epigenetic regulation, such as protein-coding genes, and can also target specific effectors within the epigenetic machinery, such as enzymes for DNA methylation (DNMT3A and DNMT3B) and also those for histone modification (EZH2) [46,47,48,49,50,51]. Therefore, these regulatory pathways once again demonstrate the complexity of the epigenetic landscape and also the elaborated pathways involved in cancer induction, maintenance, and development.
7. Epigenetic Landscape in Oral Cancer
As previously discussed, the epigenetic marks represent an important layer of regulation within normal cells that maintain the homeostatic state and also the specific phenotype. An emerging number of studies have demonstrated that this strict regulatory mechanism is lost in cancer cells with consequences that favor the malignant niche and the advancement toward metastatic spread. Studies of these mechanisms have provided new etiologic perspectives for oral cancer genesis with an increased focus on DNA methylation, histone modifications, and miRNA regulation (Figure 2) [52,53,54].
8. DNA Methylation in Oral Cancer
A comparison of normal versus tumor samples of oral cancer has demonstrated that there are significant differences between the epigenetic signatures, where the tumor samples exhibit increased genome-wide hypomethylation and also hypermethylation of promoter regions [52,55]. Hypomethylation can result in increased chromosome instability due to the release of repetitive elements within the genome and also possible activation of silenced proto-oncogenes by the removal of promoter hypermethylation. The opposite mechanism, hypermethylation, is also present in oral cancer where CpG sites within the promoter sequence of tumor suppressor genes are methylated, a fact that impedes the access of transcription factors to these segments and, implicitly, the expression of specific genes.
For oral cancer, certain risk factors such as tobacco and alcohol consumption and also chronic inflammation of the oral mucosa have been linked to dysregulations in the epigenetic pattern regarding the methylation status. For example, smokers have been associated with an increased global hypomethylation [56,57] where, on the other hand, alcohol consumers are characterized by CpG hypermethylation according to clinical studies effectuated on patient samples and also in murine models for oral malignancies. Furthermore, analysis of specific gene hypermethylation in 47 OSCC patients (p16, DAPK, RASSF1A, APC, WIF1, RUNX3, E-cad, MGMT, and hMLH1) revealed that the epigenetic status can be used as a prognosis tool, where DAPK promoter hypermethylation by its own can function as an independent parameter for overall survival (HR = 4.105) [58].
Much attention has been granted to epigenetic modifications of specific genes in order to unravel the molecular mechanism behind their behavior in oral cancer and also to find therapeutic alternatives to restore the normal epigenetic mechanisms and inhibit cancer development.
APC (adenomatous polyposis coli) is a tumor suppressor gene that normally is translated into a multi-domain protein able to bind different molecules including β-catenin, which functions in adherence junctions and Wnt signaling. Mutations (loss) in (of) APC have been linked to the inability to bind β-catenin, which further accumulates in the nucleus and triggers the canonical Wnt signaling [59,60,61] pathway that is essential for tumorigenesis (cell proliferation and differentiation). The mutational profile of APC was also investigated in oral cancer using basic research protocols. It was found that this gene is mutated in the SAS oral cancer cell line but with no amino acid changes. Moreover, no changes were observed in the tumor samples from patients. However, in cell lines (75%) as well as clinical specimens (90%) an increased accumulation of β-catenin was found [62]. Even if this scenario seems like a disruption from the previous association between WT APC (wild-type APC) and β-catenin in oral cancer, the further evidence has demonstrated that in fact the promoter of the APC gene is increasingly methylated in OSCC patient tissue samples and also in cell lines compared to controls [63,64,65,66]. This could explain the accumulation of β-catenin in oral cancer even in the absence of a functional mutation in the APC gene (considering the fact that promoter methylation concludes with silencing of gene expression).
Survivin plays a major role in oral cancer and also in other types of malignancies by inhibiting the apoptotic mechanisms and also regulating cell cycle, favoring the overcoming of checkpoints. The expression of the gene has been found to be increased in different tumor samples, including the ones collected from oral cancer patients, and was also correlated with clinicopathological features; moreover, this gene is usually not expressed in normal tissue [67,68]. Overexpression of survivin in basic cancer research has been previously linked with the presence of several single nucleotide polymorphisms (SNPs) [69]. However, there is evidence that in oral malignancies this gene is actually hypomethylated [52,70,71], contrary to normal samples where hypermethylation prevents the increased expression.
E-cadherin is synthesized from the CDH1 gene and is responsible for keeping the adhesion between cells intact. The loss of CDH1 expression is frequent in numerous types of cancers, an event that facilitates the epithelial to mesenchymal transition (EMT) and implicitly the colonization of secondary sites (metastasis) [72]. CDH1 silencing has been observed in OSCC and was also correlated with a highly aggressive pattern and poor prognosis in the clinical environment (OSCC patients). One of the proposed mechanisms for this downregulated pattern of expression consists of aberrant epigenetic regulation through increased hypermethylation [73,74,75,76,77]. However, the results are quite inconsistent, with values ranging from 7% to 46%; it was previously proposed in the literature that there is the need for standardization of the IHC-based methods and further assessment of the epigenetic-related modifications of CDH1 [78]. Considering the strong impact of environmental risk factors on the epigenetic pattern and also the heterogeneity of these outside modulators and the phenotypic differences among different individuals, it is possible that these results will keep rolling in an unstable manner. A possible approach could be the development of a large multi-centric study for the evaluation of the epigenetic pattern in oral cancer patients where the outside values could be significantly attenuated.
PTEN (phosphatase and tensin homolog deleted on chromosome 10) has been called the “new guardian of the genome,” playing a major role in the suppression of cell survival and proliferation but also differentiation, apoptosis, and invasion, ranking in second place regarding the frequency of mutations in cancer (after TP53) [79]. Hypermethylation of promoter regions has been linked to different types of malignancies (observations made in both clinical samples and cancer-specific cell lines), including cervical [80], gastric [81], endometrial [82], and also non-small-cell lung cancer [83]. Downregulation of PTEN was also observed in oral cancer with a possible connection to epigenetic modification: hypermethylation. Kurasawa and colleagues are one of the groups that sustain this mechanism for PTEN regulation in OSCC where an increased number of patients exhibited hypermethylation patterns, a mechanism persistent also in 4 out of 6 cell lines, where the transcript levels were found as significantly downregulated but with no associated mutations [84]. The exact mechanisms behind PTEN expression in oral cancer are still incompletely deciphered, although the cancer inhibitory role of this sequence is well established in other cancers. Patients lacking this functional gene are characterized by more aggressive forms of malignancies and also a poor prognosis. In this sense there are some contradictory opinions regarding the PTEN gene in both clinical and basic research [85,86,87,88,89]; some groups support the downregulated pattern where others do not follow the same data.
There are also several other genes proposed as being epigenetically regulated and direct participants in oral cancer progression through their aberrant level of expression in clinical specimens and different oral cancer cell lines. Table 1 presents a comprehensive list of these genes together with their role in carcinogenesis.
9. Histone Modifications in Oral Cancer
The process of aberrant histone modification in oral cancer is not as extensively studied as DNA hypermethylation/hypomethylation, although these two mechanisms work in close connection. Mancuso et al. found that OSCC tissue differs from its normal counterparts through H3K4 histone methylation patterns, where H3K4me2 levels were increased and H3K4me3 were at lower levels in clinical samples. They also investigated the status of H3K4me1 but no important changes were observed. This differential epigenetic signature between tumor and normal samples sustains the idea of aberrant histone modifications in cancer that further contribute to the preservation of the malignant phenotype [109]. A more recent study evaluated the global histone modifications in 186 patients with OSCC and found several epigenetic alterations that were also associated with tumor status and stage and also invasion. More specifically, H3K4ac was decreased whereas H3K27me3 was at higher levels in OSCC clinical samples. These two markers are correlated with advanced forms of oral cancer and also cancer-specific survival (CSS) and disease-free survival (DFS). The authors also proposed this pathological pattern to be a valuable prognosis tool in OSCC [110]. Besides the actual histone modifications, there are also enzymes that are accountable for the post-transcriptional modifications, such as histone deacetylase (HDAC) that is responsible for the acetylation degree of the histone tails and is implicitly involved in numerous processes associated with cancer: apoptosis, differentiation, and growth arrest [111]. As a result, HDAC 6 was associated with increased expression levels in the majority of the malignant samples when compared to TAMs, an increase that was also proportional to the cancer stage (in OSCC-derived cell lines and also clinical specimens) [112]. CAF-1 (chromatin assembly factor-1) is responsible for the organization of the nuclear chromatin status within DNA replication, being also found as deregulated in different oral cancer subtypes [113,114]. Moreover, the status of CAF-1 is considered as a possible prognosis marker in OSCC and is associated with specific clinical parameters: advanced stages and metastasis [113,114].
10. Epigenetic Alterations of miRNAs in Oral Cancer
Modification of the miRNAs profile represents a crucial event for cancer induction, development, and also invasion and metastasis, where the altered expression of these sequences is associated with malignant signaling pathways. Sethi et al. have comprehensively presented the most important miRNAs involved in head and neck cancers [115] together with their possible translated clinical role; however, in terms of epigenetic modulation of miRNAs, the data are much more restrictive for oral cancers. The miR-34 family is one of the best-studied groups with tumor suppressor functions in numerous types of malignancies, including oral cancer. Kozaki and colleagues found that miR-34a together with three other miRNAs, miR-137, miR-193a, and miR-203, are characterized by a constant downregulated expression in OSCC cell lines (RT7 as a control). Considering the fact that all of the four miRNAs are located near CpG islands, one of the hypotheses for the impaired expression involves the hypermethylation of the promoter regions. Treatment of the cells with 5-aza-2′-deoxycytidine, a potent demethylation agent, restored the expression of the four miRNAs, sustaining the epigenetic regulation of the reminded sequences. Moreover, ectopic expression of miR-137 or miR-193a in oral cancer basic research significantly improved cell growth, enforcing their tumor suppressor role [115]. Epigenetic modifications are also responsible for the cell-specific expression within different cellular entities. miR-200 s/miR-205 that are expressed in oral cancer compared with normal samples are actually inhibited in CD44high oral CSCs (cancer stem cells) due to the lack of DNA hypermethylation [116]. These data from multiple sample types in OSCC reinforce the complexity of epigenetic and genetic regulations within cancer and cell microenvironments, regulations that are aberrantly driven towards the maintenance of the malignant invasion. The most important dysregulated miRNAs in oral cancer are presented in Table 2.
Another possible field of study could be represented by the miRNAs that possibly target the enzymes involved in establishing the epigenetic landscape. These sequences could pathologically modify the expression of the epigenetic-related enzymes and lead to the establishment of differential epigenetic profiles. Even so, this idea of study is still limited within the scientific literature, especially in the context of oral cancer.
11. Epigenetic Therapies in Oral Cancer
The reversible character of epigenetic modifications is currently explored in preclinical research for therapeutic purposes in different types of malignancies, including oral cancer. Zebularine is an inhibitor of DNA methyltransferase, where treatment of HSC-3 cell line (OSCC model) resulted in impaired cell growth and also a decreased population of cells situated in the G2/M cell cycle phase [128]. Even so, the untargeted character of zebularine remains a constant problem for the future implementation of this type of agent in the clinic. Zebularine combined with cisplatin promoted cell death via an apoptotic mechanism while the combination of the same compound with 5-fluorouracil minimized the action of the chemotherapeutic agent [129]. This type of strategy has been clinically tested in patients with head and neck cancer where cisplatin was combined with azacitidine (hypomethylation activity). Both of the two studies involving these types of therapeutic formulations have been terminated with no clear results (accrual problems) (ClinicalTrials.gov Identifier: NCT00901537 and NCT00443261). Natural agents are also being explored for potential epigenetic modifying abilities; green tea administrated in in vitro models of oral cancer revealed inhibitory effects that were associated with a reversal of RECK gene hypermethylation and increased expression [130].
Histone deacetylase inhibitors are gaining momentum for oral cancer treatment with the purpose of promoting the activity of tumor suppressor genes by suppressing the acetylation process and preserving the loose structure of chromatin. The most promising results were shown in the case of combined strategies where histone deacetylase inhibitors were administrated with chemotherapeutic agents in preclinical models in order to synergistically decrease the malignant development (e.g., cisplatin combined with MS-275) [131]. Other similar agents that were tested for histone epigenetic landscape modulation consist of trichostatin A, butyric acid derivatives, and romidepsin [131].
miRNAs also have an increased potential in terms of oral cancer therapeutics for the modification of the pathological epigenetic profile. As in the case of other types of malignancies, the non-coding profile of oral cancer is also altered, an event that contributes to the maintenance of the cancer hallmarks [51,132,133]. Even if the “classical” approach consists of introducing exogenous tumor suppressor sequences for their reinforced expression (and also inhibitor sequences for oncogenic miRNAs), some of the downregulated sequences could also be restored at their basal level with epigenetic modifiers. Saito et al. applied this type of strategy by administrating epigenetic drugs (4-phenylbutyric acid and 5-aza-2′-deoxycytidine) in human cancer cell lines for the promotion of miR-127 levels and downstream inhibition of BCL6 (B-cell lymphoma 6) [134]. Even if miR-127-specific therapy is not valid for oral cancer (found as overexpressed OSCC tissue samples compared to control [116]), similar specific tumor suppressor miRNA could be restored to their homeostatic level through this strategy. For example, miR-200s and miR-205 were associated with decreased expression in oral malignant tissue versus healthy controls where their activation corresponded to low CpG methylation [116]. Another perspective could be represented by the restoration of miRNAs that normally inhibit the aberrant activity of enzymes such as DNA methyltransferase, as in the case of zebularine administration. However, the constant issue remains the nonspecificity of these epigenetic modifier drugs where healthy cells could also be affected or, moreover, oncogenic genes previously suppressed even in cancer cells could be activated (an event that could decrease the efficiency of the experimentally activated tumor suppressor genes).
12. Conclusions
Epigenetic mechanisms have gained their role in the cancer hallmarks; it is now clear that these types of alterations are also part of the heterogeneous cancer signaling. One steady advantage is the reversible feature of epigenetics, where the aberrant signature can be modified through the administration of exogenous inhibitors of DNA methyltransferase or histone deacetylase. The idea of combining the standard therapy with new epigenetic modulatory agents could improve significantly the clinical outcome considering that, for example, genes related to chemoresistance have been found as hypermethylated [135]. In this way, the efficiency of the classical treatment could be significantly improved. These preliminary results in oral and also other types of cancer are promising where most probably it is just a matter of time before these types of therapeutic agents (epigenetic modifiers) will gather more attention in clinical practice.
Acknowledgments
This study was supported by an internal grant (4944/6/08.03.2016) from Iuliu Hatieganu University of Medicine and Pharmacy, entitled “Identification of the methylation profile for genes related to oral squamous cancer”.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Irimie, A.I.; Braicu, C.; Cojocneanu-Petric, R.; Berindan-Neagoe, I.; Campian, R.S. Novel technologies for oral squamous carcinoma biomarkers in diagnostics and prognostics. Acta Odontol. Scand. 2015, 73, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Hema, K.N.; Smitha, T.; Sheethal, H.S.; Mirnalini, S.A. Epigenetics in oral squamous cell carcinoma. J. Oral Maxillofac. Pathol. 2017, 21, 252–259. [Google Scholar] [CrossRef] [PubMed]
- Parkin, D.M.; Bray, F.; Ferlay, J.; Pisani, P. Global cancer statistics, 2002. CA Cancer J. Clin. 2005, 55, 74–108. [Google Scholar] [CrossRef] [PubMed]
- Stewart, B.W.; Greim, H.; Shuker, D.; Kauppinen, T. Defence of iarc monographs. Lancet 2003, 361, 1300. [Google Scholar] [CrossRef]
- Irimie, A.I.; Braicu, C.; Zanoaga, O.; Pileczki, V.; Gherman, C.; Berindan-Neagoe, I.; Campian, R.S. Epigallocatechin-3-gallate suppresses cell proliferation and promotes apoptosis and autophagy in oral cancer ssc-4 cells. Onco Targets Ther. 2015, 8, 461–470. [Google Scholar] [PubMed]
- Perez-Sayans, M.; Somoza-Martin, J.M.; Barros-Angueira, F.; Reboiras-Lopez, M.D.; Gandara Rey, J.M.; Garcia-Garcia, A. Genetic and molecular alterations associated with oral squamous cell cancer (review). Oncol. Rep. 2009, 22, 1277–1282. [Google Scholar] [CrossRef] [PubMed]
- Irimie, A.I.; Braicu, C.; Pileczki, V.; Petrushev, B.; Soritau, O.; Campian, R.S.; Berindan-Neagoe, I. Knocking down of p53 triggers apoptosis and autophagy, concomitantly with inhibition of migration on ssc-4 oral squamous carcinoma cells. Mol. Cell. Biochem. 2016, 419, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Lingen, M.W.; Pinto, A.; Mendes, R.A.; Franchini, R.; Czerninski, R.; Tilakaratne, W.M.; Partridge, M.; Peterson, D.E.; Woo, S.B. Genetics/epigenetics of oral premalignancy: Current status and future research. Oral Dis. 2011, 17 (Suppl. 1), 7–22. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Kelly, T.K.; Jones, P.A. Epigenetics in cancer. Carcinogenesis 2010, 31, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Heyn, H.; Esteller, M. DNA methylation profiling in the clinic: Applications and challenges. Nat. Rev. Genet. 2012, 13, 679–692. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.A.; Baylin, S.B. The fundamental role of epigenetic events in cancer. Nat. Rev. Genet. 2002, 3, 415–428. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.A.; Baylin, S.B. The epigenomics of cancer. Cell 2007, 128, 683–692. [Google Scholar] [CrossRef] [PubMed]
- Yoo, C.B.; Jones, P.A. Epigenetic therapy of cancer: Past, present and future. Nat. Rev. Drug Discov. 2006, 5, 37–50. [Google Scholar] [CrossRef] [PubMed]
- Kornberg, R.D. Chromatin structure: A repeating unit of histones and DNA. Science 1974, 184, 868–871. [Google Scholar] [CrossRef] [PubMed]
- Bird, A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002, 16, 6–21. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Leung, F.C. An evaluation of new criteria for cpg islands in the human genome as gene markers. Bioinformatics 2004, 20, 1170–1177. [Google Scholar] [CrossRef] [PubMed]
- Illingworth, R.; Kerr, A.; Desousa, D.; Jorgensen, H.; Ellis, P.; Stalker, J.; Jackson, D.; Clee, C.; Plumb, R.; Rogers, J.; et al. A novel cpg island set identifies tissue-specific methylation at developmental gene loci. PLoS Biol. 2008, 6, e22. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.M.; Bird, A. DNA methylation landscapes: Provocative insights from epigenomics. Nat. Rev. Genet. 2008, 9, 465–476. [Google Scholar] [CrossRef] [PubMed]
- Eckhardt, F.; Lewin, J.; Cortese, R.; Rakyan, V.K.; Attwood, J.; Burger, M.; Burton, J.; Cox, T.V.; Davies, R.; Down, T.A.; et al. DNA methylation profiling of human chromosomes 6, 20 and 22. Nat. Genet. 2006, 38, 1378–1385. [Google Scholar] [CrossRef] [PubMed]
- Baylin, S.B.; Jones, P.A. A decade of exploring the cancer epigenome-biological and translational implications. Nat. Rev. Cancer 2011, 11, 726–734. [Google Scholar] [CrossRef] [PubMed]
- Dawson, M.A.; Kouzarides, T. Cancer epigenetics: From mechanism to therapy. Cell 2012, 150, 12–27. [Google Scholar] [CrossRef] [PubMed]
- Robertson, K.D. DNA methylation and human disease. Nat. Rev. Genet. 2005, 6, 597–610. [Google Scholar] [CrossRef] [PubMed]
- Ley, T.J.; Ding, L.; Walter, M.J.; McLellan, M.D.; Lamprecht, T.; Larson, D.E.; Kandoth, C.; Payton, J.E.; Baty, J.; Welch, J.; et al. Dnmt3a mutations in acute myeloid leukemia. N. Engl. J. Med. 2010, 363, 2424–2433. [Google Scholar] [CrossRef] [PubMed]
- Kouzarides, T. Chromatin modifications and their function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, B.E.; Meissner, A.; Lander, E.S. The mammalian epigenome. Cell 2007, 128, 669–681. [Google Scholar] [CrossRef] [PubMed]
- Haberland, M.; Montgomery, R.L.; Olson, E.N. The many roles of histone deacetylases in development and physiology: Implications for disease and therapy. Nat. Rev. Genet. 2009, 10, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y. Histone lysine demethylases: Emerging roles in development, physiology and disease. Nat. Rev. Genet. 2007, 8, 829–833. [Google Scholar] [CrossRef] [PubMed]
- Bannister, A. The Role of Epigenetics in Cancer. Available online: http://www.abcam.com/index.html?pageconfig=resource&rid=10755 (accessed on 27 February 2018).
- Arif, M.; Vedamurthy, B.M.; Choudhari, R.; Ostwal, Y.B.; Mantelingu, K.; Kodaganur, G.S.; Kundu, T.K. Nitric oxide-mediated histone hyperacetylation in oral cancer: Target for a water-soluble hat inhibitor, ctk7a. Chem. Biol. 2010, 17, 903–913. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.; Pugh, B.F. Nucleosome positioning and gene regulation: Advances through genomics. Nat. Rev. Genet. 2009, 10, 161–172. [Google Scholar] [CrossRef] [PubMed]
- Yuan, G.C.; Liu, Y.J.; Dion, M.F.; Slack, M.D.; Wu, L.F.; Altschuler, S.J.; Rando, O.J. Genome-scale identification of nucleosome positions in s. Cerevisiae. Science 2005, 309, 626–630. [Google Scholar] [CrossRef] [PubMed]
- Shivaswamy, S.; Bhinge, A.; Zhao, Y.; Jones, S.; Hirst, M.; Iyer, V.R. Dynamic remodeling of individual nucleosomes across a eukaryotic genome in response to transcriptional perturbation. PLoS Biol. 2008, 6, e65. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.C.; Jeong, S.; Liang, G.; Takai, D.; Fatemi, M.; Tsai, Y.C.; Egger, G.; Gal-Yam, E.N.; Jones, P.A. Role of nucleosomal occupancy in the epigenetic silencing of the mlh1 cpg island. Cancer Cell 2007, 12, 432–444. [Google Scholar] [CrossRef] [PubMed]
- Gal-Yam, E.N.; Jeong, S.; Tanay, A.; Egger, G.; Lee, A.S.; Jones, P.A. Constitutive nucleosome depletion and ordered factor assembly at the grp78 promoter revealed by single molecule footprinting. PLoS Genet. 2006, 2, e160. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.L.; Peterson, C.L. Atp-dependent chromatin remodeling. Curr. Top. Dev. Biol. 2005, 65, 115–148. [Google Scholar] [PubMed]
- Santenard, A.; Torres-Padilla, M.E. Epigenetic reprogramming in mammalian reproduction: Contribution from histone variants. Epigenetics 2009, 4, 80–84. [Google Scholar] [CrossRef] [PubMed]
- Sarma, K.; Reinberg, D. Histone variants meet their match. Nat. Rev. Mol. Cell Biol. 2005, 6, 139–149. [Google Scholar] [CrossRef] [PubMed]
- Budisan, L.; Gulei, D.; Zanoaga, O.M.; Irimie, A.I.; Sergiu, C.; Braicu, C.; Gherman, C.D.; Berindan-Neagoe, I. Dietary intervention by phytochemicals and their role in modulating coding and non-coding genes in cancer. Int. J. Mol. Sci. 2017, 18. [Google Scholar] [CrossRef] [PubMed]
- Gulei, D.; Magdo, L.; Jurj, A.; Raduly, L.; Cojocneanu-Petric, R.; Moldovan, A.; Moldovan, C.; Florea, A.; Pasca, S.; Pop, L.A.; et al. The silent healer: Mir-205-5p up-regulation inhibits epithelial to mesenchymal transition in colon cancer cells by indirectly up-regulating e-cadherin expression. Cell Death Dis. 2018, 9, 66. [Google Scholar] [CrossRef] [PubMed]
- Gulei, D.; Mehterov, N.; Nabavi, S.M.; Atanasov, A.G.; Berindan-Neagoe, I. Targeting ncrnas by plant secondary metabolites: The ncrnas game in the balance towards malignancy inhibition. Biotechnol. Adv. 2017. [Google Scholar] [CrossRef] [PubMed]
- Pop-Bica, C.; Gulei, D.; Cojocneanu-Petric, R.; Braicu, C.; Petrut, B.; Berindan-Neagoe, I. Understanding the role of non-coding rnas in bladder cancer: From dark matter to valuable therapeutic targets. Int. J. Mol. Sci. 2017, 18. [Google Scholar] [CrossRef] [PubMed]
- Berindan-Neagoe, I.; Calin, G.A. Molecular pathways: Micrornas, cancer cells, and microenvironment. Clin. Cancer Res. 2014, 20, 6247–6253. [Google Scholar] [CrossRef] [PubMed]
- Braicu, C.; Calin, G.A.; Berindan-Neagoe, I. Micrornas and cancer therapy—From bystanders to major players. Curr. Med. Chem. 2013, 20, 3561–3573. [Google Scholar] [CrossRef] [PubMed]
- Berindan-Neagoe, I.; Monroig Pdel, C.; Pasculli, B.; Calin, G.A. Micrornaome genome: A treasure for cancer diagnosis and therapy. CA Cancer J. Clin. 2014, 64, 311–336. [Google Scholar] [CrossRef] [PubMed]
- Zaharie, F.; Muresan, M.S.; Petrushev, B.; Berce, C.; Gafencu, G.A.; Selicean, S.; Jurj, A.; Cojocneanu-Petric, R.; Lisencu, C.I.; Pop, L.A.; et al. Exosome-carried microrna-375 inhibits cell progression and dissemination via bcl-2 blocking in colon cancer. J. Gastrointest. Liver Dis. 2015, 24, 435–443. [Google Scholar]
- Fabbri, M.; Garzon, R.; Cimmino, A.; Liu, Z.; Zanesi, N.; Callegari, E.; Liu, S.; Alder, H.; Costinean, S.; Fernandez-Cymering, C.; et al. Microrna-29 family reverts aberrant methylation in lung cancer by targeting DNA methyltransferases 3a and 3b. Proc. Natl. Acad. Sci. USA 2007, 104, 15805–15810. [Google Scholar] [CrossRef] [PubMed]
- Friedman, J.M.; Liang, G.; Liu, C.C.; Wolff, E.M.; Tsai, Y.C.; Ye, W.; Zhou, X.; Jones, P.A. The putative tumor suppressor microrna-101 modulates the cancer epigenome by repressing the polycomb group protein ezh2. Cancer Res. 2009, 69, 2623–2629. [Google Scholar] [CrossRef] [PubMed]
- Fabbri, M.; Calin, G.A. Epigenetics and mirnas in human cancer. Adv. Genet. 2010, 70, 87–99. [Google Scholar] [PubMed]
- Kala, R.; Peek, G.W.; Hardy, T.M.; Tollefsbol, T.O. Micrornas: An emerging science in cancer epigenetics. J. Clin. Bioinform. 2013, 3, 6. [Google Scholar] [CrossRef] [PubMed]
- Kozaki, K.; Imoto, I.; Mogi, S.; Omura, K.; Inazawa, J. Exploration of tumor-suppressive micrornas silenced by DNA hypermethylation in oral cancer. Cancer Res. 2008, 68, 2094–2105. [Google Scholar] [CrossRef] [PubMed]
- Gomes, C.C.; Gomez, R.S. Microrna and oral cancer: Future perspectives. Oral Oncol. 2008, 44, 910–914. [Google Scholar] [CrossRef] [PubMed]
- Gasche, J.A.; Goel, A. Epigenetic mechanisms in oral carcinogenesis. Future Oncol. 2012, 8, 1407–1425. [Google Scholar] [CrossRef] [PubMed]
- Mascolo, M.; Siano, M.; Ilardi, G.; Russo, D.; Merolla, F.; De Rosa, G.; Staibano, S. Epigenetic disregulation in oral cancer. Int. J. Mol. Sci. 2012, 13, 2331–2353. [Google Scholar] [CrossRef] [PubMed]
- Jithesh, P.V.; Risk, J.M.; Schache, A.G.; Dhanda, J.; Lane, B.; Liloglou, T.; Shaw, R.J. The epigenetic landscape of oral squamous cell carcinoma. Br. J. Cancer 2013, 108, 370–379. [Google Scholar] [CrossRef] [PubMed]
- Piyathilake, C.J.; Bell, W.C.; Jones, J.; Henao, O.L.; Heimburger, D.C.; Niveleau, A.; Grizzle, W.E. Pattern of nonspecific (or global) DNA methylation in oral carcinogenesis. Head Neck 2005, 27, 1061–1067. [Google Scholar] [CrossRef] [PubMed]
- Guerrero-Preston, R.; Baez, A.; Blanco, A.; Berdasco, M.; Fraga, M.; Esteller, M. Global DNA methylation: A common early event in oral cancer cases with exposure to environmental carcinogens or viral agents. P. R. Health Sci. J. 2009, 28, 24–29. [Google Scholar] [PubMed]
- Baba, S.; Yamada, Y.; Hatano, Y.; Miyazaki, Y.; Mori, H.; Shibata, T.; Hara, A. Global DNA hypomethylation suppresses squamous carcinogenesis in the tongue and esophagus. Cancer Sci. 2009, 100, 1186–1191. [Google Scholar] [CrossRef] [PubMed]
- Supic, G.; Kozomara, R.; Jovic, N.; Zeljic, K.; Magic, Z. Prognostic significance of tumor-related genes hypermethylation detected in cancer-free surgical margins of oral squamous cell carcinomas. Oral Oncol. 2011, 47, 702–708. [Google Scholar] [CrossRef] [PubMed]
- Aoki, K.; Taketo, M.M. Adenomatous polyposis coli (apc): A multi-functional tumor suppressor gene. J. Cell. Sci. 2007, 120, 3327–3335. [Google Scholar] [CrossRef] [PubMed]
- Reya, T.; Clevers, H. Wnt signalling in stem cells and cancer. Nature 2005, 434, 843–850. [Google Scholar] [CrossRef] [PubMed]
- Barker, N.; Clevers, H. Mining the wnt pathway for cancer therapeutics. Nat. Rev. Drug Discov. 2006, 5, 997–1014. [Google Scholar] [CrossRef] [PubMed]
- Iwai, S.; Katagiri, W.; Kong, C.; Amekawa, S.; Nakazawa, M.; Yura, Y. Mutations of the apc, beta-catenin, and axin 1 genes and cytoplasmic accumulation of beta-catenin in oral squamous cell carcinoma. J. Cancer Res. Clin. Oncol. 2005, 131, 773–782. [Google Scholar] [CrossRef] [PubMed]
- Rigi-Ladiz, M.A.; Kordi-Tamandani, D.M.; Torkamanzehi, A. Analysis of hypermethylation and expression profiles of apc and atm genes in patients with oral squamous cell carcinoma. Clin. Epigenet. 2011, 3, 6. [Google Scholar] [CrossRef] [PubMed]
- Uesugi, H.; Uzawa, K.; Kawasaki, K.; Shimada, K.; Moriya, T.; Tada, A.; Shiiba, M.; Tanzawa, H. Status of reduced expression and hypermethylation of the apc tumor suppressor gene in human oral squamous cell carcinoma. Int. J. Mol. Med. 2005, 15, 597–602. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.; Eiberg, H.; Krogdahl, A.; Liu, C.J.; Sorensen, J.A. Cytoplasmic expression of e-cadherin and beta-catenin correlated with loh and hypermethylation of the apc gene in oral squamous cell carcinomas. J. Oral Pathol. Med. 2005, 34, 116–119. [Google Scholar] [CrossRef] [PubMed]
- Ali, J.; Sabiha, B.; Jan, H.U.; Haider, S.A.; Khan, A.A.; Ali, S.S. Genetic etiology of oral cancer. Oral Oncol. 2017, 70, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Lo Muzio, L.; Pannone, G.; Staibano, S.; Mignogna, M.D.; Rubini, C.; Mariggio, M.A.; Procaccini, M.; Ferrari, F.; De Rosa, G.; Altieri, D.C. Survivin expression in oral squamous cell carcinoma. Br. J. Cancer 2003, 89, 2244–2248. [Google Scholar] [CrossRef] [PubMed]
- Khan, Z.; Tiwari, R.P.; Mulherkar, R.; Sah, N.K.; Prasad, G.B.; Shrivastava, B.R.; Bisen, P.S. Detection of survivin and p53 in human oral cancer: Correlation with clinicopathologic findings. Head Neck 2009, 31, 1039–1048. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, P.K.; Goel, A.; Mittal, R.D. Survivin: A molecular biomarker in cancer. Indian J. Med. Res. 2015, 141, 389–397. [Google Scholar] [PubMed]
- Chen, Y.K.; Hsue, S.S.; Lin, L.M. Survivin expression is regulated by an epigenetic mechanism for dmba-induced hamster buccal-pouch squamous-cell carcinomas. Arch. Oral Biol. 2005, 50, 593–598. [Google Scholar] [CrossRef] [PubMed]
- Hsue, S.S.; Wang, W.C.; Chen, Y.K.; Lin, L.M. Expression of inhibitors of apoptosis family protein in 7,12-dimethylbenz[a]anthracene-induced hamster buccal-pouch squamous-cell carcinogenesis is associated with mutant p53 accumulation and epigenetic changes. Int. J. Exp. Pathol. 2008, 89, 309–320. [Google Scholar] [CrossRef] [PubMed]
- Gulei, D.; Mehterov, N.; Ling, H.; Stanta, G.; Braicu, C.; Berindan-Neagoe, I. The “good-cop bad-cop” tgf-beta role in breast cancer modulated by non-coding rnas. Biochim. Biophys. Acta 2017, 1861, 1661–1675. [Google Scholar] [CrossRef] [PubMed]
- Viswanathan, M.; Tsuchida, N.; Shanmugam, G. Promoter hypermethylation profile of tumor-associated genes p16, p15, hmlh1, mgmt and e-cadherin in oral squamous cell carcinoma. Int. J. Cancer 2003, 105, 41–46. [Google Scholar] [CrossRef] [PubMed]
- Maruya, S.; Issa, J.P.; Weber, R.S.; Rosenthal, D.I.; Haviland, J.C.; Lotan, R.; El-Naggar, A.K. Differential methylation status of tumor-associated genes in head and neck squamous carcinoma: Incidence and potential implications. Clin. Cancer Res. 2004, 10, 3825–3830. [Google Scholar] [CrossRef] [PubMed]
- Saito, Y.; Takazawa, H.; Uzawa, K.; Tanzawa, H.; Sato, K. Reduced expression of e-cadherin in oral squamous cell carcinoma: Relationship with DNA methylation of 5′ cpg island. Int. J. Oncol. 1998, 12, 293–298. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, S.; Sasaki, A.; Mese, H.; Alcalde, R.E.; Tsuji, T.; Matsumura, T. The e-cadherin gene is silenced by cpg methylation in human oral squamous cell carcinomas. Int. J. Cancer 2001, 93, 667–673. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.W.; Chow, V.; Lam, K.Y.; Wei, W.I.; Yuen, A. Loss of e-cadherin expression resulting from promoter hypermethylation in oral tongue carcinoma and its prognostic significance. Cancer 2002, 94, 386–392. [Google Scholar] [CrossRef] [PubMed]
- Vered, M.; Allon, I.; Buchner, A.; Dayan, D. E-cadherin in oral scc: An analysis of the confusing literature and new insights related to its immunohistochemical expression. Histol. Histopathol. 2012, 27, 141–150. [Google Scholar] [PubMed]
- Yin, Y.; Shen, W.H. Pten: A new guardian of the genome. Oncogene 2008, 27, 5443–5453. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.J.; Liu, V.W.; Wang, Y.; Tsang, P.C.; Ngan, H.Y. Differential DNA methylation profiles in gynecological cancers and correlation with clinico-pathological data. BMC Cancer 2006, 6, 212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, Y.H.; Lee, H.S.; Kim, W.H. Promoter methylation and silencing of pten in gastric carcinoma. Lab. Investig. 2002, 82, 285–291. [Google Scholar] [CrossRef] [PubMed]
- Salvesen, H.B.; MacDonald, N.; Ryan, A.; Jacobs, I.J.; Lynch, E.D.; Akslen, L.A.; Das, S. Pten methylation is associated with advanced stage and microsatellite instability in endometrial carcinoma. Int. J. Cancer 2001, 91, 22–26. [Google Scholar] [CrossRef]
- Soria, J.C.; Lee, H.Y.; Lee, J.I.; Wang, L.; Issa, J.P.; Kemp, B.L.; Liu, D.D.; Kurie, J.M.; Mao, L.; Khuri, F.R. Lack of pten expression in non-small cell lung cancer could be related to promoter methylation. Clin. Cancer Res. 2002, 8, 1178–1184. [Google Scholar] [PubMed]
- Kurasawa, Y.; Shiiba, M.; Nakamura, M.; Fushimi, K.; Ishigami, T.; Bukawa, H.; Yokoe, H.; Uzawa, K.; Tanzawa, H. Pten expression and methylation status in oral squamous cell carcinoma. Oncol. Rep. 2008, 19, 1429–1434. [Google Scholar] [PubMed]
- Lee, J.I.; Soria, J.C.; Hassan, K.A.; El-Naggar, A.K.; Tang, X.; Liu, D.D.; Hong, W.K.; Mao, L. Loss of pten expression as a prognostic marker for tongue cancer. Arch. Otolaryngol. Head Neck Surg. 2001, 127, 1441–1445. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Samaranayake, L.P.; Zhou, H.; Xiao, L. Homozygous deletion of the pten tumor-suppressor gene is not a feature in oral squamous cell carcinoma. Oral Oncol. 2000, 36, 95–99. [Google Scholar] [CrossRef]
- Mavros, A.; Hahn, M.; Wieland, I.; Koy, S.; Koufaki, O.N.; Strelocke, K.; Koch, R.; Haroske, G.; Schackert, H.K.; Eckelt, U. Infrequent genetic alterations of the tumor suppressor gene pten/mmac1 in squamous cell carcinoma of the oral cavity. J. Oral Pathol. Med. 2002, 31, 270–276. [Google Scholar] [CrossRef] [PubMed]
- Squarize, C.H.; Castilho, R.M.; Santos Pinto, D., Jr. Immunohistochemical evidence of pten in oral squamous cell carcinoma and its correlation with the histological malignancy grading system. J. Oral Pathol. Med. 2002, 31, 379–384. [Google Scholar] [CrossRef] [PubMed]
- Shin, K.H.; Kim, J.M.; Rho, K.S.; Park, K.H.; Oh, J.E.; Min, B.M. Inactivation of the pten gene by mutation, exonic deletion, and loss of transcript in human oral squamous cell carcinomas. Int. J. Oncol. 2002, 21, 997–1001. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, V.; Saranath, D. Concurrent hypermethylation of multiple regulatory genes in chewing tobacco associated oral squamous cell carcinomas and adjacent normal tissues. Oral Oncol. 2004, 40, 145–153. [Google Scholar] [CrossRef]
- Huang, M.J.; Yeh, K.T.; Shih, H.C.; Wang, Y.F.; Lin, T.H.; Chang, J.Y.; Shih, M.C.; Chang, J.G. The correlation between cpg methylation and protein expression of p16 in oral squamous cell carcinomas. Int. J. Mol. Med. 2002, 10, 551–554. [Google Scholar] [PubMed]
- Kato, K.; Hara, A.; Kuno, T.; Mori, H.; Yamashita, T.; Toida, M.; Shibata, T. Aberrant promoter hypermethylation of p16 and mgmt genes in oral squamous cell carcinomas and the surrounding normal mucosa. J. Cancer Res. Clin. Oncol. 2006, 132, 735–743. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, M.; Nelson, H.H.; Peters, E.; Ringstrom, E.; Posner, M.; Kelsey, K.T. Patterns of gene promoter methylation in squamous cell cancer of the head and neck. Oncogene 2002, 21, 4231–4236. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Cespedes, M.; Esteller, M.; Wu, L.; Nawroz-Danish, H.; Yoo, G.H.; Koch, W.M.; Jen, J.; Herman, J.G.; Sidransky, D. Gene promoter hypermethylation in tumors and serum of head and neck cancer patients. Cancer Res. 2000, 60, 892–895. [Google Scholar] [PubMed]
- Rosas, S.L.; Koch, W.; da Costa Carvalho, M.G.; Wu, L.; Califano, J.; Westra, W.; Jen, J.; Sidransky, D. Promoter hypermethylation patterns of p16, o6-methylguanine-DNA-methyltransferase, and death-associated protein kinase in tumors and saliva of head and neck cancer patients. Cancer Res. 2001, 61, 939–942. [Google Scholar] [PubMed]
- Viet, C.T.; Jordan, R.C.; Schmidt, B.L. DNA promoter hypermethylation in saliva for the early diagnosis of oral cancer. J. Calif. Dent. Assoc. 2007, 35, 844–849. [Google Scholar] [PubMed]
- Youssef, E.M.; Lotan, D.; Issa, J.P.; Wakasa, K.; Fan, Y.H.; Mao, L.; Hassan, K.; Feng, L.; Lee, J.J.; Lippman, S.M.; et al. Hypermethylation of the retinoic acid receptor-beta(2) gene in head and neck carcinogenesis. Clin. Cancer Res. 2004, 10, 1733–1742. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.S.; Issa, J.P.; Roberts, D.B.; Williams, M.D.; Weber, R.S.; Kies, M.S.; El-Naggar, A.K. Quantitative promoter hypermethylation analysis of cancer-related genes in salivary gland carcinomas: Comparison with methylation-specific pcr technique and clinical significance. Clin. Cancer Res. 2008, 14, 2664–2672. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.H.; Huang, S.F.; Chen, I.H.; Liao, C.T.; Wang, H.M.; Hsieh, L.L. Methylation of rassf1a, rassf2a, and hin-1 is associated with poor outcome after radiotherapy, but not surgery, in oral squamous cell carcinoma. Clin. Cancer Res. 2009, 15, 4174–4180. [Google Scholar] [CrossRef] [PubMed]
- Imai, T.; Toyota, M.; Suzuki, H.; Akino, K.; Ogi, K.; Sogabe, Y.; Kashima, L.; Maruyama, R.; Nojima, M.; Mita, H.; et al. Epigenetic inactivation of rassf2 in oral squamous cell carcinoma. Cancer Sci. 2008, 99, 958–966. [Google Scholar] [CrossRef] [PubMed]
- Lo Muzio, L.; Staibano, S.; Pannone, G.; Mignogna, M.D.; Mariggio, A.; Salvatore, G.; Chieffi, P.; Tramontano, D.; De Rosa, G.; Altieri, D.C. Expression of the apoptosis inhibitor survivin in aggressive squamous cell carcinoma. Exp. Mol. Pathol. 2001, 70, 249–254. [Google Scholar] [CrossRef] [PubMed]
- Czerninski, R.; Krichevsky, S.; Ashhab, Y.; Gazit, D.; Patel, V.; Ben-Yehuda, D. Promoter hypermethylation of mismatch repair genes, hmlh1 and hmsh2 in oral squamous cell carcinoma. Oral Dis. 2009, 15, 206–213. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Ramirez, I.; Ramirez-Amador, V.; Irigoyen-Camacho, M.E.; Sanchez-Perez, Y.; Anaya-Saavedra, G.; Granados-Garcia, M.; Garcia-Vazquez, F.; Garcia-Cuellar, C.M. Hmlh1 promoter methylation is an early event in oral cancer. Oral Oncol. 2011, 47, 22–26. [Google Scholar] [CrossRef] [PubMed]
- Takeshima, M.; Saitoh, M.; Kusano, K.; Nagayasu, H.; Kurashige, Y.; Malsantha, M.; Arakawa, T.; Takuma, T.; Chiba, I.; Kaku, T.; et al. High frequency of hypermethylation of p14, p15 and p16 in oral pre-cancerous lesions associated with betel-quid chewing in sri lanka. J. Oral Pathol. Med. 2008, 37, 475–479. [Google Scholar] [CrossRef] [PubMed]
- Ishida, E.; Nakamura, M.; Ikuta, M.; Shimada, K.; Matsuyoshi, S.; Kirita, T.; Konishi, N. Promotor hypermethylation of p14arf is a key alteration for progression of oral squamous cell carcinoma. Oral Oncol. 2005, 41, 614–622. [Google Scholar] [CrossRef] [PubMed]
- Sailasree, R.; Abhilash, A.; Sathyan, K.M.; Nalinakumari, K.R.; Thomas, S.; Kannan, S. Differential roles of p16ink4a and p14arf genes in prognosis of oral carcinoma. Cancer Epidemiol. Biomark. Prev. 2008, 17, 414–420. [Google Scholar] [CrossRef] [PubMed]
- Yeh, K.T.; Chang, J.G.; Lin, T.H.; Wang, Y.F.; Tien, N.; Chang, J.Y.; Chen, J.C.; Shih, M.C. Epigenetic changes of tumor suppressor genes, p15, p16, vhl and p53 in oral cancer. Oncol. Rep. 2003, 10, 659–663. [Google Scholar] [PubMed]
- Shaw, R.J.; Liloglou, T.; Rogers, S.N.; Brown, J.S.; Vaughan, E.D.; Lowe, D.; Field, J.K.; Risk, J.M. Promoter methylation of p16, rarbeta, e-cadherin, cyclin a1 and cytoglobin in oral cancer: Quantitative evaluation using pyrosequencing. Br. J. Cancer 2006, 94, 561–568. [Google Scholar] [CrossRef] [PubMed]
- Mancuso, M.; Matassa, D.S.; Conte, M.; Colella, G.; Rana, G.; Fucci, L.; Piscopo, M. H3k4 histone methylation in oral squamous cell carcinoma. Acta Biochim. Pol. 2009, 56, 405–410. [Google Scholar] [PubMed]
- Chen, Y.W.; Kao, S.Y.; Wang, H.J.; Yang, M.H. Histone modification patterns correlate with patient outcome in oral squamous cell carcinoma. Cancer 2013, 119, 4259–4267. [Google Scholar] [CrossRef] [PubMed]
- De Ruijter, A.J.; van Gennip, A.H.; Caron, H.N.; Kemp, S.; van Kuilenburg, A.B. Histone deacetylases (hdacs): Characterization of the classical hdac family. Biochem. J. 2003, 370, 737–749. [Google Scholar] [CrossRef] [PubMed]
- Sakuma, T.; Uzawa, K.; Onda, T.; Shiiba, M.; Yokoe, H.; Shibahara, T.; Tanzawa, H. Aberrant expression of histone deacetylase 6 in oral squamous cell carcinoma. Int. J. Oncol. 2006, 29, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Staibano, S.; Mascolo, M.; Rocco, A.; Lo Muzio, L.; Ilardi, G.; Siano, M.; Pannone, G.; Vecchione, M.L.; Nugnes, L.; Califano, L.; et al. The proliferation marker chromatin assembly factor-1 is of clinical value in predicting the biological behaviour of salivary gland tumours. Oncol. Rep. 2011, 25, 13–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Staibano, S.; Mignogna, C.; Lo Muzio, L.; Mascolo, M.; Salvatore, G.; Di Benedetto, M.; Califano, L.; Rubini, C.; De Rosa, G. Chromatin assembly factor-1 (caf-1)-mediated regulation of cell proliferation and DNA repair: A link with the biological behaviour of squamous cell carcinoma of the tongue? Histopathology 2007, 50, 911–919. [Google Scholar] [CrossRef] [PubMed]
- Sethi, N.; Wright, A.; Wood, H.; Rabbitts, P. Micrornas and head and neck cancer: Reviewing the first decade of research. Eur. J. Cancer 2014, 50, 2619–2635. [Google Scholar] [CrossRef] [PubMed]
- Wiklund, E.D.; Gao, S.; Hulf, T.; Sibbritt, T.; Nair, S.; Costea, D.E.; Villadsen, S.B.; Bakholdt, V.; Bramsen, J.B.; Sorensen, J.A.; et al. Microrna alterations and associated aberrant DNA methylation patterns across multiple sample types in oral squamous cell carcinoma. PLoS ONE 2011, 6, e27840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, C.M.; Lin, P.M.; Wang, Y.M.; Chen, Z.J.; Lin, S.F.; Yang, M.Y. Circulating mirna is a novel marker for head and neck squamous cell carcinoma. Tumour Biol. 2012, 33, 1933–1942. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.M.; Phillips, B.L.; Patel, R.S.; Cohen, D.M.; Jakymiw, A.; Kong, W.W.; Cheng, J.Q.; Chan, E.K. Keratinization-associated mir-7 and mir-21 regulate tumor suppressor reversion-inducing cysteine-rich protein with kazal motifs (reck) in oral cancer. J. Biol. Chem. 2012, 287, 29261–29272. [Google Scholar] [CrossRef] [PubMed]
- Siow, M.Y.; Ng, L.P.; Vincent-Chong, V.K.; Jamaludin, M.; Abraham, M.T.; Abdul Rahman, Z.A.; Kallarakkal, T.G.; Yang, Y.H.; Cheong, S.C.; Zain, R.B. Dysregulation of mir-31 and mir-375 expression is associated with clinical outcomes in oral carcinoma. Oral Dis. 2014, 20, 345–351. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.J.; Lin, S.C.; Yang, C.C.; Cheng, H.W.; Chang, K.W. Exploiting salivary mir-31 as a clinical biomarker of oral squamous cell carcinoma. Head Neck 2012, 34, 219–224. [Google Scholar] [CrossRef] [PubMed]
- Momen-Heravi, F.; Trachtenberg, A.J.; Kuo, W.P.; Cheng, Y.S. Genomewide study of salivary micrornas for detection of oral cancer. J. Dent. Res. 2014, 93, 86S–93S. [Google Scholar] [CrossRef] [PubMed]
- Shiiba, M.; Shinozuka, K.; Saito, K.; Fushimi, K.; Kasamatsu, A.; Ogawara, K.; Uzawa, K.; Ito, H.; Takiguchi, Y.; Tanzawa, H. Microrna-125b regulates proliferation and radioresistance of oral squamous cell carcinoma. Br. J. Cancer 2013, 108, 1817–1821. [Google Scholar] [CrossRef] [PubMed]
- Ni, Y.H.; Huang, X.F.; Wang, Z.Y.; Han, W.; Deng, R.Z.; Mou, Y.B.; Ding, L.; Hou, Y.Y.; Hu, Q.G. Upregulation of a potential prognostic biomarker, mir-155, enhances cell proliferation in patients with oral squamous cell carcinoma. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. 2014, 117, 227–233. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.J.; Zhang, C.Y.; Zhou, Z.T.; Ma, J.Y.; Liu, Y.; Bao, Z.X.; Jiang, W.W. Microrna-155 in oral squamous cell carcinoma: Overexpression, localization, and prognostic potential. Head Neck 2015, 37, 970–976. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.C.; Hung, P.S.; Wang, P.W.; Liu, C.J.; Chu, T.H.; Cheng, H.W.; Lin, S.C. Mir-181 as a putative biomarker for lymph-node metastasis of oral squamous cell carcinoma. J. Oral Pathol. Med. 2011, 40, 397–404. [Google Scholar] [CrossRef] [PubMed]
- Chang, K.W.; Liu, C.J.; Chu, T.H.; Cheng, H.W.; Hung, P.S.; Hu, W.Y.; Lin, S.C. Association between high mir-211 microrna expression and the poor prognosis of oral carcinoma. J. Dent. Res. 2008, 87, 1063–1068. [Google Scholar] [CrossRef] [PubMed]
- Min, A.; Zhu, C.; Peng, S.; Rajthala, S.; Costea, D.E.; Sapkota, D. Micrornas as important players and biomarkers in oral carcinogenesis. BioMed Res. Int. 2015, 2015, 186904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, M.; Shinohara, F.; Nishimura, K.; Echigo, S.; Rikiishi, H. Epigenetic regulation of chemosensitivity to 5-fluorouracil and cisplatin by zebularine in oral squamous cell carcinoma. Int. J. Oncol. 2007, 31, 1449–1456. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.; Shinohara, F.; Endo, M.; Sugazaki, M.; Echigo, S.; Rikiishi, H. Zebularine suppresses the apoptotic potential of 5-fluorouracil via camp/pka/creb pathway against human oral squamous cell carcinoma cells. Cancer Chemother. Pharmacol. 2009, 64, 223–232. [Google Scholar] [CrossRef] [PubMed]
- Kato, K.; Long, N.K.; Makita, H.; Toida, M.; Yamashita, T.; Hatakeyama, D.; Hara, A.; Mori, H.; Shibata, T. Effects of green tea polyphenol on methylation status of reck gene and cancer cell invasion in oral squamous cell carcinoma cells. Br. J. Cancer 2008, 99, 647–654. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Suzuki, M.; Sato, Y.; Echigo, S.; Rikiishi, H. Sequence-dependent interaction between cisplatin and histone deacetylase inhibitors in human oral squamous cell carcinoma cells. Int. J. Oncol. 2006, 28, 1233–1241. [Google Scholar] [CrossRef] [PubMed]
- Manikandan, M.; Deva Magendhra Rao, A.K.; Arunkumar, G.; Manickavasagam, M.; Rajkumar, K.S.; Rajaraman, R.; Munirajan, A.K. Oral squamous cell carcinoma: Microrna expression profiling and integrative analyses for elucidation of tumourigenesis mechanism. Mol. Cancer 2016, 15, 28. [Google Scholar] [CrossRef] [PubMed]
- Manasa, V.G.; Kannan, S. Impact of microrna dynamics on cancer hallmarks: An oral cancer scenario. Tumour Biol. 2017, 39. [Google Scholar] [CrossRef] [PubMed]
- Saito, Y.; Liang, G.; Egger, G.; Friedman, J.M.; Chuang, J.C.; Coetzee, G.A.; Jones, P.A. Specific activation of microrna-127 with downregulation of the proto-oncogene bcl6 by chromatin-modifying drugs in human cancer cells. Cancer Cell 2006, 9, 435–443. [Google Scholar] [CrossRef] [PubMed]
- Balch, C.; Yan, P.; Craft, T.; Young, S.; Skalnik, D.G.; Huang, T.H.; Nephew, K.P. Antimitogenic and chemosensitizing effects of the methylation inhibitor zebularine in ovarian cancer. Mol. Cancer Ther. 2005, 4, 1505–1514. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
The landscape of epigenetic mechanisms. Epigenetic changes consist of reversible modifications affecting the structure of the DNA/chromosome that are further translated at the protein level through expressional changes. One of the most studied epigenetic mechanisms consists of DNA methylation that occurs mainly within the CpG islands that are located in different repetitive genome regions or, more often, within promoter regions. The methylation pattern is different depending on the region, where most of the islands located in the promoter area are hypomethylated, the other ones, from repetitive segments, are methylated. Histone modifications are also part of the epigenetic machinery and mainly consist of ubiquitylation, sumoylation, methylation, acetylation, and also phosphorylation of the histone tails. Depending on the type of modification, these mechanisms can result in increased activity of the specific DNA segment or inversely blockage of function. Not in the least, miRNAs also play a crucial part in the establishment of the epigenetic landscape where these sequences are differentially expressed between different cellular entities and also between normal and pathological cells. Their ability to target and inhibit the translation of specific genes makes them crucial players within homeostatic signaling pathways and also therapeutic targets in disease states.
Figure 1.
The landscape of epigenetic mechanisms. Epigenetic changes consist of reversible modifications affecting the structure of the DNA/chromosome that are further translated at the protein level through expressional changes. One of the most studied epigenetic mechanisms consists of DNA methylation that occurs mainly within the CpG islands that are located in different repetitive genome regions or, more often, within promoter regions. The methylation pattern is different depending on the region, where most of the islands located in the promoter area are hypomethylated, the other ones, from repetitive segments, are methylated. Histone modifications are also part of the epigenetic machinery and mainly consist of ubiquitylation, sumoylation, methylation, acetylation, and also phosphorylation of the histone tails. Depending on the type of modification, these mechanisms can result in increased activity of the specific DNA segment or inversely blockage of function. Not in the least, miRNAs also play a crucial part in the establishment of the epigenetic landscape where these sequences are differentially expressed between different cellular entities and also between normal and pathological cells. Their ability to target and inhibit the translation of specific genes makes them crucial players within homeostatic signaling pathways and also therapeutic targets in disease states.

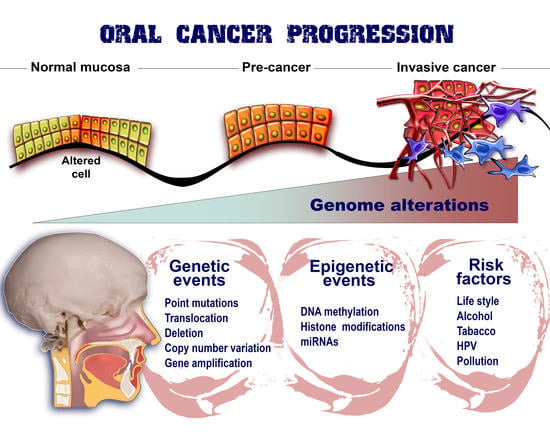
Figure 2.
Epigenetic marks upon oral cancer. The evolution of oral cancer from a few altered cells to invasive phenotypes able to metastasize and populate secondary sites is the consequence of numerous factors that are interplaying their roles. Therefore, genetic events together with risk factors are combined with epigenetic mechanisms in order to ensure the proper environment for malignant development. All these factors gradually contribute to the organization of an unstable genome and also implicitly to the promotion of cancer advancement.
Figure 2.
Epigenetic marks upon oral cancer. The evolution of oral cancer from a few altered cells to invasive phenotypes able to metastasize and populate secondary sites is the consequence of numerous factors that are interplaying their roles. Therefore, genetic events together with risk factors are combined with epigenetic mechanisms in order to ensure the proper environment for malignant development. All these factors gradually contribute to the organization of an unstable genome and also implicitly to the promotion of cancer advancement.


{kind=link}
{kind=link}
{kind=link}
Table 1.
Epigenetically regulated genes with implications in oral cancer progression.
| Gene | Mechanism | Locus | Epigenetic Modification | Ref |
|---|---|---|---|---|
| CDKN2A/p16 | Cell cycle, senescence | 9p21 | Hypermethylation | [73,74,90,91,92] |
| CDH1/E-cadherin | EMT, adhesion | 16q22.1 | Hypermethylation * | [73,74,75,77] |
| PTEN | Differentiation, survival, proliferation, invasion, apoptosis | 10q23.3 | Hypermethylation * | [84,89] |
| DAPK1 | Apoptosis | 9q34.1 | Hypermethylation | [93,94,95] |
| MGMT | DNA repair | 10q26 | Hypermethylation | [90,92,94,96] |
| RARB2 | Cell proliferation | 3p24 | Hypermethylation | [97,98] |
| RASSF1/2 | Cell cycle, apoptosis, and microtubule formation | 3p21.3/20p13 | Hypermethylation | [99,100] |
| APC | Cell proliferation | 5q22.2 | Hypermethylation | [64,65] |
| Survivin | Cell proliferation and apoptosis | 17q25 | Hypomethylation | [70,71,101] |
| MLH1 | DNA repair | 3p22.2 | Hypermethylation | [73,102,103] |
| p14(ARF) | Cell proliferation, division, angiogenesis | 9p21 | Hypermethylation | [104,105,106] |
| p15INK4B | Cell cycle | 9p21.3 | Hypermethylation | [104,107] |
| p16INK4A | Cell cycle, senescence | 9p21 | Hypermethylation | [73,74,90,91,92] |
| RARβ | Cell growth and differentiation | 3p24 | Hypermethylation * | [97,108] |
* Controversial results or unclear role in oral cancer.
Table 2.
Most significant dysregulated miRNAs in oral cancer.
| miRNA | Type of Malignancy | Possible Clinical Utility | Expression | Reference |
|---|---|---|---|---|
| miR-21 | Head and neck squamous cell carcinoma, Oral Squamous cell carcinoma | Diagnosis/prognosis utility (detected also in plasma) | Upregulated | [117,118] |
| miR-375 | Oral squamous cell carcinoma | Associated with clinical parameters | Downregulated | [119] |
| miR-31 | Oral carcinoma | Non-invasive and early diagnosis tool (saliva) and also prognosis marker | Upregulated | [120] |
| miR-7 | Oral squamous cell carcinoma | Upregulated | [118] | |
| miR-27b | Oral squamous cell carcinoma | Saliva biomarker for OSCC | Upregulated | [121] |
| miR-125b | Oral squamous cell carcinoma | Therapeutic target | Downregulated | [122] |
| miR-155 | Oral squamous cell carcinoma | Prognosis value | Upregulated | [123,124] |
| miR-181 | Oral squamous cell carcinoma | Lymph node metastasis marker | Upregulated | [125] |
| miR-211 | Oral carcinoma | Poor prognosis marker | Upregulated | [126] |
| Additional upregulated miRNAs | miR-9*, miR-424, miR-7–1*, miR-15b, miR-9, miR-155, and miR-196a, miR-24, miR-18a, miR-221, miR-16, let-7b, | [118,121,127] | ||
| Additional downregulated miRNAs | miR-486-5p, miR-136, miR-147, miR-1250, miR-148a, miR-632, miR-646, miR-668, miR-877, miR-503, miR-220a, miR-323-5p, miR-223, miR-29a | [118,121,127] | ||
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Irimie, A.I.; Ciocan, C.; Gulei, D.; Mehterov, N.; Atanasov, A.G.; Dudea, D.; Berindan-Neagoe, I. Current Insights into Oral Cancer Epigenetics. Int. J. Mol. Sci. 2018, 19, 670. https://doi.org/10.3390/ijms19030670
AMA Style
Irimie AI, Ciocan C, Gulei D, Mehterov N, Atanasov AG, Dudea D, Berindan-Neagoe I. Current Insights into Oral Cancer Epigenetics. International Journal of Molecular Sciences. 2018; 19(3):670. https://doi.org/10.3390/ijms19030670
Chicago/Turabian StyleIrimie, Alexandra Iulia, Cristina Ciocan, Diana Gulei, Nikolay Mehterov, Atanas G. Atanasov, Diana Dudea, and Ioana Berindan-Neagoe. 2018. "Current Insights into Oral Cancer Epigenetics" International Journal of Molecular Sciences 19, no. 3: 670. https://doi.org/10.3390/ijms19030670
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.