Chemotherapeutic-Induced Cardiovascular Dysfunction: Physiological Effects, Early Detection—The Role of Telomerase to Counteract Mitochondrial Defects and Oxidative Stress

Abstract
:
1. Introduction
2. Emerging Roles of Telomerase
3. Role of Chemotherapeutic Agents in the Stimulation of Cardiovascular Diseases and Emerging Therapeutic Telomerase Countermeasures
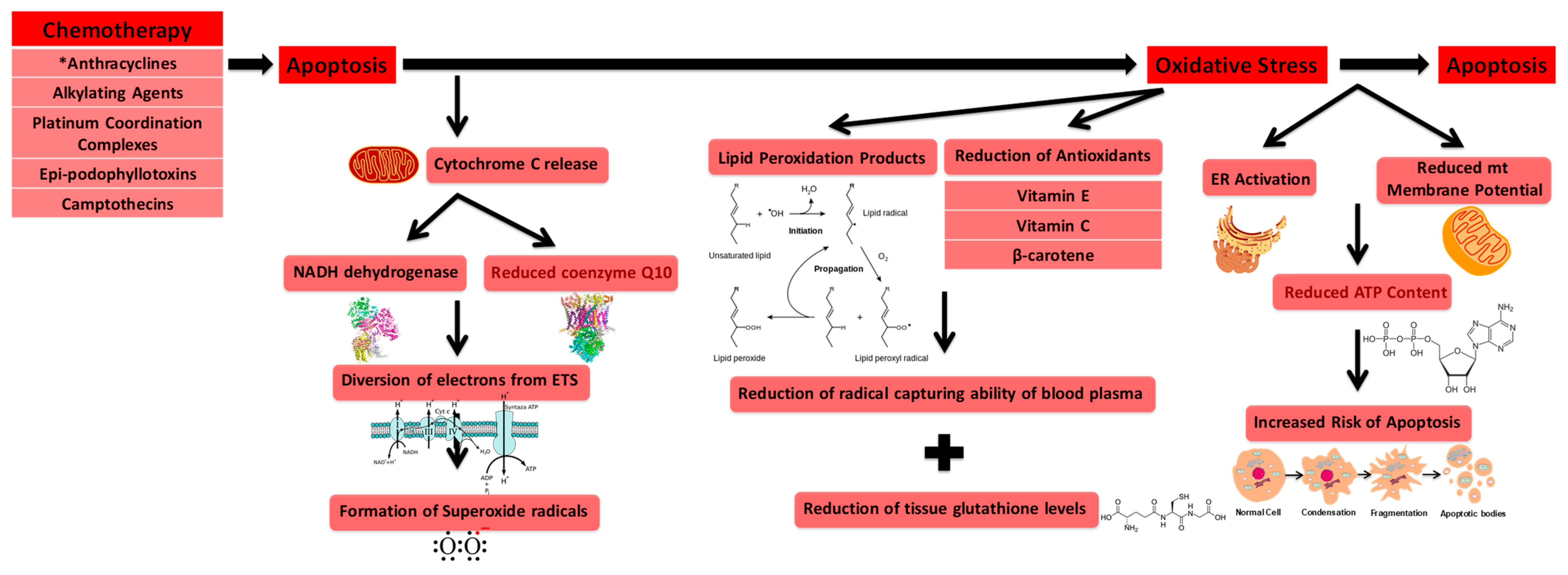
3.1. Oxidative Stress Involved in Cardiotoxicity
3.2. Free Radical Formation (Reactive Oxygen Species) during Cardiotoxicity
3.3. DNA Damage
3.4. TERT as a Regulator of mtROS
3.5. Role of TERT in Counteracting mtDNA Damage
4. Mechanisms of Chemotherapy-Induced Changes of Physiological and Cellular Dynamics—Emerging Therapeutic Role of Telomerase
Physiological Complications
5. Chemotherapeutic-Derived Physiological Effects
5.1. Cardiomyopathy
5.2. Heart Failure
5.3. Microvascular Defects
5.4. Molecular Changes
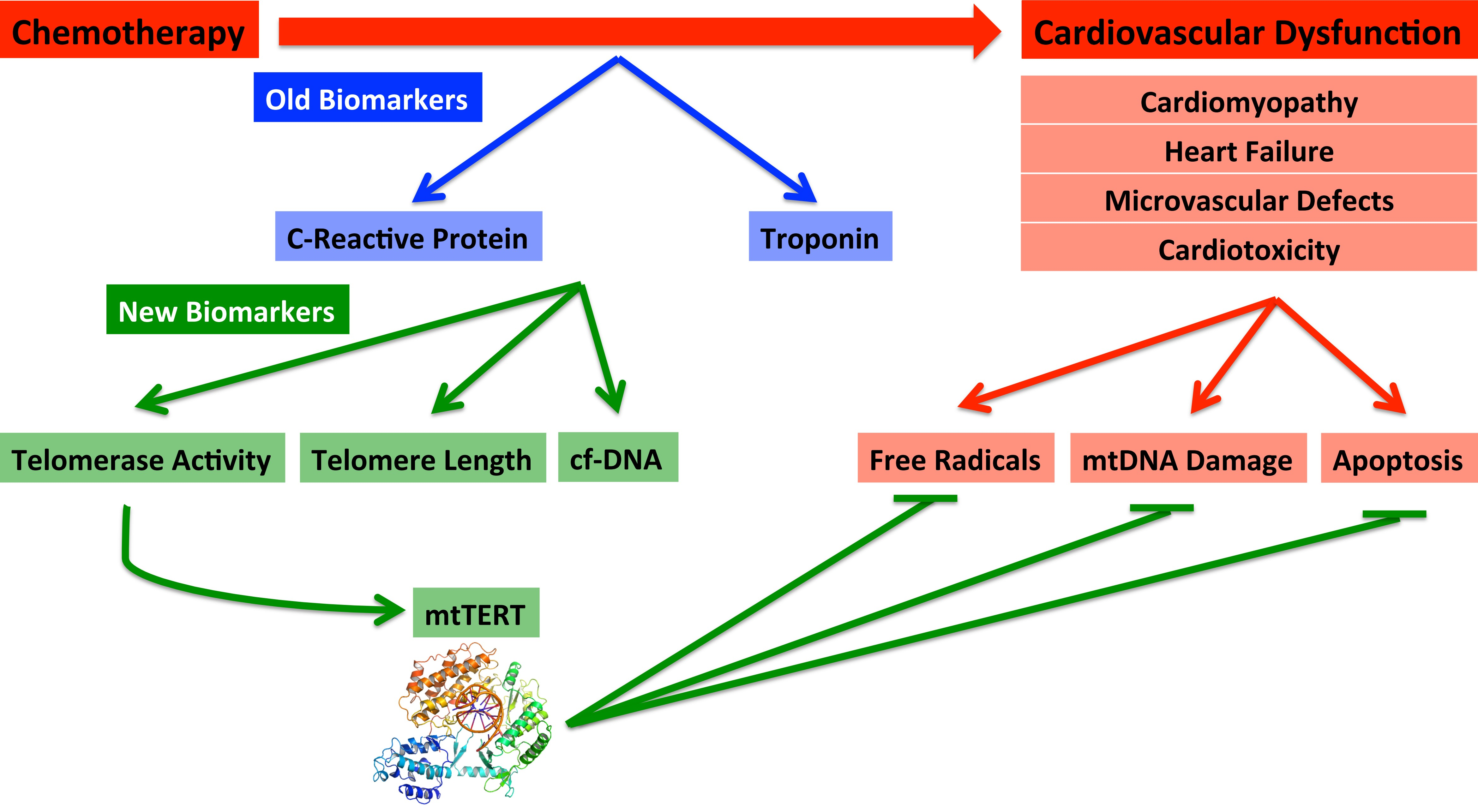
6. Biomarkers—Old and New
7. Established Biomarkers
7.1. Troponin
7.2. Inflammatory Markers/C-Reactive Protein (CRP)
7.3. New Biomarkers: Telomere Length and Telomerase Activity
7.4. New Biomarker: mtDNA Damage during Cardiovascular Dysfunction and Cardiotoxicity
8. Future Directions
Acknowledgments
Conflicts of Interest
References
- Barac, A. Improving prediction of cardiovascular complications of cancer therapy: What does the future hold? Future Cardiol. Future Sci. Group 2015, 11, 383–387. [Google Scholar] [CrossRef] [PubMed]
- Ai, D.; Banchs, J.; Owusu-Agyemang, P.; Cata, J.P. Chemotherapy-induced cardiovascular toxicity: Beyond anthracyclines. Minerva Anestesiol. 2014, 80, 586–594. [Google Scholar] [PubMed]
- Chawla, A.; Chawla, R.; Jaggi, S. Microvasular and macrovascular complications in diabetes mellitus: Distinct or continuum? Indian J. Endocrinol. Metab. 2016, 20, 546–551. [Google Scholar] [CrossRef] [PubMed]
- Serné, E.H.; de Jongh, R.T.; Eringa, E.C.; Ijzerman, R.G.; de Boer, M.P.; Stehouwer, C.D.A. Microvascular dysfunction: Causative role in the association between hypertension, insulin resistance and the metabolic syndrome? Essays Biochem. 2006, 42, 163–176. [Google Scholar] [CrossRef] [PubMed]
- Guarini, G.; Kiyooka, T.; Ohanyan, V.; Pung, Y.F.; Marzilli, M.; Chen, Y.R.; Chen, C.L.; Kang, P.T.; Hardwick, J.P.; Kolz, C.L.; et al. Impaired coronary metabolic dilation in the metabolic syndrome is linked to mitochondrial dysfunction and mitochondrial DNA damage. Basic Res. Cardiol. 2016, 111, 29. [Google Scholar] [CrossRef] [PubMed]
- UK Prospective Diabetes Study (UKPDS) Group. Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33). Lancet 1998, 352, 837–853. [Google Scholar]
- Diabetes Control and Complications Trial Research Group; Nathan, D.M.; Genuth, S.; Lachin, J.; Cleary, P.; Crofford, O.; Davis, M.; Rand, L.; Siebert, C. The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. N. Engl. J. Med. 1993, 329, 977–986. [Google Scholar] [CrossRef] [PubMed]
- Vithian, K.; Hurel, S. Microvascular complications: Pathophysiology and management. Clin. Med. 2010, 10, 505–509. [Google Scholar] [CrossRef]
- Chen, Z.I.; Ai, D.I. Cardiotoxicity associated with targeted cancer therapies. Mol. Clin. Oncol. 2016, 4, 675–681. [Google Scholar] [CrossRef] [PubMed]
- Cross, M.J.; Berridge, B.R.; Clements, P.J.M.; Cove-Smith, L.; Force, T.L.; Hoffmann, P.; Holbrook, M.; Lyon, A.R.; Mellor, H.R.; Norris, A.A.; et al. Physiological, pharmacological and toxicological considerations of drug-induced structural cardiac injury. Br. J. Pharmacol. 2015, 172, 957–974. [Google Scholar] [CrossRef] [PubMed]
- Volkova, M.; Russell, R., III. Anthracycline cardiotoxicity: Prevalence, pathogenesis and treatment. Curr. Cardiol. Rev. 2011, 7, 214–220. [Google Scholar] [CrossRef]
- Criscitiello, C.; Metzger-Filho, O.; Saini, K.S.; de Castro, G.; Diaz, M.; La Gerche, A.; de Azambuja, E.; Piccart-Gebhart, M.J. Targeted therapies in breast cancer: Are heart and vessels also being targeted? Breast Cancer Res. 2012, 14, 209. [Google Scholar] [CrossRef] [PubMed]
- Bostan, H.B.; Rezaee, R.; Valokala, M.G.; Tsarouhas, K.; Golokhvast, K.; Tsatsakis, A.M.; Karimi, G. Cardiotoxicity of nano-particles. Life Sci. 2016, 165, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Cheung-Ong, K.; Giaever, G.; Nislow, C. DNA-damaging agents in cancer chemotherapy: Serendipity and chemical biology. Chem. Biol. 2013, 20, 648–659. [Google Scholar] [CrossRef] [PubMed]
- Basu, A.; Bodycombe, N.E.; Cheah, J.H.; Price, E.V.; Liu, K.; Schaefer, G.I.; Ebright, R.Y.; Stewart, M.L.; Ito, D.; Wang, S.; et al. An interactive resource to identify cancer genetic and lineage dependencies targeted by small molecules. Cell 2013, 154, 1151–1161. [Google Scholar] [CrossRef] [PubMed]
- Cahill, L.E.; Bertoia, M.L.; Aroner, S.A.; Mukamal, K.J.; Jensen, M.K. New and emerging biomarkers in cardiovascular disease. Curr. Diabetes Rep. 2015, 15, 88. [Google Scholar] [CrossRef] [PubMed]
- Marian, A.; Nambi, V. Biomarkers of cardiac disease. Expert Rev. Mol. Diagn. 2004, 4, 805–820. [Google Scholar] [CrossRef] [PubMed]
- Shay, J.W.; Wright, W.E. Hayflick, his limit, and cellular ageing. Nat. Rev. Mol. Cell Biol. 2000, 1, 72–76. [Google Scholar] [CrossRef] [PubMed]
- Zvereva, M.I.; Shcherbakova, D.M.; Dontsova, O.A. Telomerase: Structure, functions, and activity regulation. Biochemistry 2010, 75, 1563–1583. [Google Scholar] [CrossRef] [PubMed]
- Zurek, M.; Altschmied, J.; Kohlgrüber, S.; Ale-Agha, N.; Haendeler, J. Role of telomerase in the cardiovascular system. Genes 2016, 7, 29. [Google Scholar] [CrossRef] [PubMed]
- Yester, J.W.; Kühn, B. Mechanisms of cardiomyocyte proliferation and differentiation in development and regeneration. Curr. Cardiol. Rep. 2017, 19, 13. [Google Scholar] [CrossRef] [PubMed]
- Seimiya, H.; Sawada, H.; Muramatsu, Y.; Shimizu, M.; Ohko, K.; Yamane, K.; Tsuruo, T. Involvement of 14-3-3 proteins in nuclear localization of telomerase. EMBO J. 2000, 19, 2652–2661. [Google Scholar] [CrossRef] [PubMed]
- Scuric, Z.; Carroll, J.E.; Bower, J.E.; Ramos-Perlberg, S.; Petersen, L.; Esquivel, S.; Hogan, M.; Chapman, A.M.; Irwin, M.R.; Breen, E.C.; et al. Biomarkers of aging associated with past treatments in breast cancer survivors. NPJ Breast Cancer 2017, 3, 50. [Google Scholar] [CrossRef] [PubMed]
- Santos, J.H.; Meyer, J.N.; Skorvaga, M.; Annab, L.A.; van Houten, B. Mitochondrial hTERT exacerbates free-radical-mediated mtDNA damage. Aging Cell 2004, 3, 399–411. [Google Scholar] [CrossRef] [PubMed]
- Armbruster, B.N.; Banik, S.S.; Guo, C.; Smith, A.C.; Counter, C.M. N-terminal domains of the human telomerase catalytic subunit required for enzyme activity in vivo. Mol. Cell. Biol. 2001, 21, 7775–7786. [Google Scholar] [CrossRef] [PubMed]
- Nugent, C.I.; Lundblad, V. The telomerase reverse transcriptase: Components and regulation. Genes Dev. 1998, 12, 1073–1085. [Google Scholar] [CrossRef] [PubMed]
- Martínez, P.; Blasco, M.A. Telomeric and extra-telomeric roles for telomerase and the telomere-binding proteins. Nat. Rev. Cancer 2011, 11. [Google Scholar] [CrossRef] [PubMed]
- Collins, K. The biogenesis and regulation of telomerase holoenzymes. Nat. Rev. Mol. Cell Biol. 2006, 7, 484–494. [Google Scholar] [CrossRef] [PubMed]
- Sarek, G.; Marzec, P.; Margalef, P.; Boulton, S.J. Molecular basis of telomere dysfunction in human genetic diseases. Nat. Struct. Mol. Biol. 2015, 22, 867–874. [Google Scholar] [CrossRef] [PubMed]
- Vassilopoulou, L. Biomolecular profile of colorectal cancer—The role of telomerase as a potent biomarker. Farmacia 2017, 65, 643–659. [Google Scholar]
- Bertorelle, R.; Rampazzo, E.; Pucciarelli, S.; Nitti, D.; de Rossi, A. Telomeres, telomerase and colorectal cancer. World J. 2014, 20, 1940–1950. [Google Scholar] [CrossRef] [PubMed]
- Niiyama, H.; Mizumoto, K.; Sato, N.; Nagai, E.; Mibu, R.; Fukui, T.; Kinoshita, M.; Tanaka, M. Quantitative analysis of hTERT mRNA expression in colorectal cancer. Am. J. Gastroenterol. 2001, 96, 1895–1900. [Google Scholar] [CrossRef] [PubMed]
- North, B.J.; Sinclair, D.A. The intersection between aging and cardiovascular disease. Circ. Res. 2012, 110, 1097–1108. [Google Scholar] [CrossRef] [PubMed]
- Hornsby, P.J. Telomerase and the aging process. Exp. Gerontol. 2007, 42, 575–581. [Google Scholar] [CrossRef] [PubMed]
- Chang, E.; Harley, C.B. Telomere length and replicative aging in human vascular tissues. Proc. Natl. Acad. Sci. USA 1995, 92, 11190–11194. [Google Scholar] [CrossRef] [PubMed]
- Yeh, J.-K.; Wang, C.-Y. Telomeres and telomerase in cardiovascular diseases. Genes 2016, 7, 58. [Google Scholar] [CrossRef] [PubMed]
- Beyer, A.M.; Freed, J.K.; Durand, M.J.; Riedel, M.; Ait-Aissa, K.; Green, P.; Hockenberry, J.C.; Morgan, R.G.; Donato, A.J.; Peleg, R.; et al. Critical role for telomerase in the mechanism of flow-mediated dilation in the human microcirculation. Circ. Res. 2016, 118, 856–866. [Google Scholar] [CrossRef] [PubMed]
- Saretzki, G. Telomerase beyond Telomeres: New Roles for an Old Enzyme; Nova Science Publishers: Hauppauge, NY, USA, 2010. [Google Scholar]
- Saretzki, G. Telomerase, mitochondria and oxidative stress. Exp. Gerontol. 2009, 44, 485–492. [Google Scholar] [CrossRef] [PubMed]
- Chiodi, I.; Mondello, C. Telomere-independent functions of telomerase in nuclei, cytoplasm, and mitochondria. Front. Oncol. 2012, 2, 133. [Google Scholar] [CrossRef] [PubMed]
- Kuo, L.; Chilian, W.M.; Davis, M.J. Interaction of pressure- and flow-induced responses in porcine coronary resistance vessels. Am. J. Physiol. 1991, 261, H1706–H1715. [Google Scholar] [CrossRef] [PubMed]
- Ait-Aissa, K.; Kadlec, A.O.; Hockenberry, J.; Gutterman, D.D.; Beyer, A.M. Telomerase reverse transcriptase protects against Angiotensin II induced microvascular endothelial dysfunction. Am. J. Physiol. Circ. Physiol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Sandin, S.; Rhodes, D. Telomerase structure. Curr. Opin. Struct. Biol. 2014, 25, 104–110. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Jin, R.; Zhang, B.; Chen, H.; Bai, Y.X.; Yang, P.X.; Han, S.W.; Xie, Y.H.; Huang, P.T.; Huang, C.; et al. Nucleolar localization of TERT is unrelated to telomerase function in human cells. J. Cell Sci. 2008, 121, 2169–2176. [Google Scholar] [CrossRef] [PubMed]
- Santos, J.H.; Meyer, J.N.; van Houten, B. Mitochondrial localization of telomerase as a determinant for hydrogen peroxide-induced mitochondrial DNA damage and apoptosis. Hum. Mol. Genet. 2006, 15, 1757–1768. [Google Scholar] [CrossRef] [PubMed]
- Ait-Aissa, K.; Ebben, J.D.; Kadlec, A.O.; Beyer, A.M. Friend or foe? Telomerase as a pharmacological target in cancer and cardiovascular disease. Pharmacol. Res. 2016, 111, 422–433. [Google Scholar] [CrossRef] [PubMed]
- Dhalla, N.S.; Temsah, R.M.; Netticadan, T. Role of oxidative stress in cardiovascular diseases. J. Hypertens. 2000, 18, 655–673. [Google Scholar] [CrossRef] [PubMed]
- Santos, J.; Gutterman, D.B.A. Mitochondrial telomerase regulates flow mediated dilation by suppressing mitochondrial derived free radical production. FASEB J. 2014, 28, 664.1. [Google Scholar]
- Florescu, M.; Cinteza, M.; Vinereanu, D. Chemotherapy-induced cardiotoxicity. Maedica 2013, 8, 59–67. [Google Scholar] [PubMed]
- Albini, A.; Pennesi, G.; Donatelli, F.; Cammarota, R.; de Flora, S.; Noonan, D.M. Cardiotoxicity of anticancer drugs: The need for cardio-oncology and cardio-oncological prevention. J. Natl. Cancer Inst. 2010, 102, 14–25. [Google Scholar] [CrossRef] [PubMed]
- Khakoo, A.Y.; Liu, P.P.; Force, T.; Lopez-Berestein, G.; Jones, L.W.; Schneider, J.; Hill, J. Cardiotoxicity due to cancer therapy. Texas Heart Inst. J. 2011, 38, 253–256. [Google Scholar]
- Huszno, J.; Leś, D.; Sarzyczny-Słota, D.; Nowara, E. Cardiac side effects of trastuzumab in breast cancer patients—Single centere experiences. Contemp. Oncol. 2013, 17, 190–195. [Google Scholar] [CrossRef] [PubMed]
- Cardinale, D.; Colombo, A.; Torrisi, R.; Sandri, M.T.; Civelli, M.; Salvatici, M.; Lamantia, G.; Colombo, N.; Cortinovis, S.; Dessanai, M.A.; et al. Trastuzumab-induced cardiotoxicity: Clinical and prognostic implications of troponin I evaluation. J. Clin. Oncol. 2010, 28, 3910–3916. [Google Scholar] [CrossRef] [PubMed]
- Ghobrial, I.M.; Rajkumar, S.V. Management of thalidomide toxicity. J. Support. Oncol. 2003, 1, 194–205. [Google Scholar] [PubMed]
- Di Lorenzo, G.; Autorino, R.; Bruni, G.; Carteni, G.; Ricevuto, E.; Tudini, M.; Ficorella, C.; Romano, C.; Aieta, M.; Giordano, A.; et al. Cardiovascular toxicity following sunitinib therapy in metastatic renal cell carcinoma: A multicenter analysis. Ann. Oncol. 2009, 20, 1535–1542. [Google Scholar] [CrossRef] [PubMed]
- Chintalgattu, V.; Rees, M.L.; Culver, J.C.; Goel, A.; Jiffar, T.; Zhang, J.; Dunner, K.; Pati, S.; Bankson, J.A.; Pasqualini, R.; et al. Coronary microvascular pericytes are the cellular target of sunitinib malate-induced cardiotoxicity. Sci. Transl. Med. 2013, 5, 187ra69. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Rahman, O.; Fouad, M. Risk of cardiovascular toxicities in patients with solid tumors treated with sorafenib: An updated systematic review and meta-analysis. Future Oncol. 2014, 10, 1981–1992. [Google Scholar] [CrossRef] [PubMed]
- Schmidinger, M.; Zielinski, C.C.; Vogl, U.M.; Bojic, A.; Bojic, M.; Schukro, C.; Ruhsam, M.; Hejna, M.; Schmidinger, H. Cardiac toxicity of sunitinib and sorafenib in patients with metastatic renal cell carcinoma. J. Clin. Oncol. 2008, 26, 5204–5212. [Google Scholar] [CrossRef] [PubMed]
- Pinkhas, D.; Ho, T.; Smith, S. Assessment of pazopanib-related hypertension, cardiac dysfunction and identification of clinical risk factors for their development. Cardio-Oncology 2017, 3, 5. [Google Scholar] [CrossRef] [PubMed]
- Paul, F.; Dörr, J.; Würfel, J.; Vogel, H.-P.; Zipp, F. Early mitoxantrone-induced cardiotoxicity in secondary progressive multiple sclerosis. J. Neurol. Neurosurg. Psychiatry 2007, 78, 198–200. [Google Scholar] [CrossRef] [PubMed]
- Brockstein, B.E.; Smiley, C.; Al-Sadir, J.; Williams, S.F. Cardiac and pulmonary toxicity in patients undergoing high-dose chemotherapy for lymphoma and breast cancer: Prognostic factors. Bone Marrow Transplant. 2000, 25, 885–894. [Google Scholar] [CrossRef] [PubMed]
- Dorr, R.T.; Shipp, N.G.; Liddil, J.D.; Iyengar, B.S.; Kunz, K.R.; Remers, W.A. Cardiotoxicity of mitomycin A, mitomycin C, and seven N7 analogs in vitro. Cancer Chemother. Pharmacol. 1992, 31, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Feliz, V.; Saiyad, S.; Ramarao, S.M.; Khan, H.; Leonelli, F.; Guglin, M. Melphalan-induced supraventricular tachycardia: Incidence and risk factors. Clin. Cardiol. 2011, 34, 356–359. [Google Scholar] [CrossRef] [PubMed]
- Carver, J.R.; Nasta, S.; Chong, E.A.; Stonecypher, M.; Wheeler, J.E.; Ahmadi, T.; Schuster, S.J. Myocarditis during lenalidomide therapy. Ann. Pharmacother. 2010, 44, 1840–1843. [Google Scholar] [CrossRef] [PubMed]
- Kloth, J.S.L.; Pagani, A.; Verboom, M.C.; Malovini, A.; Napolitano, C.; Kruit, W.H.J.; Sleijfer, S.; Steeghs, N.; Zambelli, A.; Mathijssen, R.H.J. Incidence and relevance of QTc-interval prolongation caused by tyrosine kinase inhibitors. Br. J. Cancer 2015, 112, 1011–1016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sobotka, P.A.; McMannis, J.; Fisher, R.I.; Stein, D.G.; Thomas, J.X., Jr. Effects of interleukin 2 on cardiac function in the isolated rat heart. J. Clin. Investig. 1990, 86, 845–850. [Google Scholar] [CrossRef] [PubMed]
- Turrisi, G.; Montagnani, F.; Grotti, S.; Marinozzi, C.; Bolognese, L.; Fiorentini, G. Congestive heart failure during imatinib mesylate treatment. Int. J. Cardiol. 2010, 145, 148–150. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, K.; Zhang, J.; Honbo, N.; Karliner, J.S. Doxorubicin cardiomyopathy. Cardiology 2010, 115, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Mitry, M.A.; Edwards, J.G. Doxorubicin induced heart failure: Phenotype and molecular mechanisms. Int. J. Cardiol. Hear. Vasc. 2016, 10, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Raja, W.; Mir, M.H.; Dar, I.; Banday, M.A.; Ahmad, I. Cisplatin induced paroxysmal supraventricular tachycardia. Indian J. Med. Paediatr. Oncol. 2013, 34, 330–332. [Google Scholar] [CrossRef] [PubMed]
- Unnikrishnan, D.; Dutcher, J.P.; Garl, S.; Varshneya, N.; Lucariello, R.; Wiernik, P.H. Cardiac monitoring of patients receiving arsenic trioxide therapy. Br. J. Haematol. 2004, 124, 610–617. [Google Scholar] [CrossRef] [PubMed]
- Economopoulou, P.; Kotsakis, A.; Kapiris, I.; Kentepozidis, N. Cancer therapy and cardiovascular risk: Focus on bevacizumab. Cancer Manag. Res. 2015, 7, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Orciuolo, E.; Buda, G.; Cecconi, N.; Galimberti, S.; Versari, D.; Cervetti, G.; Salvetti, A.; Petrini, M. Unexpected cardiotoxicity in haematological bortezomib treated patients. Br. J. Haematol. 2007, 138, 396–397. [Google Scholar] [CrossRef] [PubMed]
- Sendur, M.A.N.; Aksoy, S.; Altundag, K. Cardiotoxicity of novel HER2-targeted therapies. Curr. Med. Res. Opin. 2013, 29, 1015–1024. [Google Scholar] [CrossRef] [PubMed]
- Aleman, B.M.P.; Moser, E.C.; Nuver, J.; Suter, T.M.; Maraldo, M.V.; Specht, L.; Vrieling, C.; Darby, S.C. Cardiovascular disease after cancer therapy. EJC Suppl. 2014, 12, 18–28. [Google Scholar] [CrossRef] [PubMed]
- Fukumoto, M.; Nakaizumi, A.; Zhang, T.; Lentz, S.I.; Shibata, M.; Puro, D.G. Vulnerability of the retinal microvasculature to oxidative stress: Ion channel-dependent mechanisms. AJP Cell Physiol. 2012, 302, C1413–C1420. [Google Scholar] [CrossRef] [PubMed]
- Kanbay, M.; Sanchez-Lozada, L.-G.; Franco, M.; Madero, M.; Solak, Y.; Rodriguez-Iturbe, B.; Covic, A.; Johnson, R.J. Microvascular disease and its role in the brain and cardiovascular system: A potential role for uric acid as a cardiorenal toxin. Nephrol. Dial. Transplant. 2011, 26, 430–437. [Google Scholar] [CrossRef] [PubMed]
- Conklin, K.A. Chemotherapy-associated oxidative stress: Impact on chemotherapeutic effectiveness. Integr. Cancer Ther. 2004, 3, 294–300. [Google Scholar] [CrossRef] [PubMed]
- Trachootham, D.; Zhang, W.; Huang, P. Oxidative stress and drug resistance in cancer. In Drug Resistance in Cancer Cells; Springer: New York, NY, USA, 2009; pp. 137–175. [Google Scholar] [CrossRef]
- Markman, T.M.; Markman, M. Cardiotoxicity of antineoplastic agents: What is the present and future role for imaging? Curr. Oncol. Rep. 2014, 16, 396. [Google Scholar] [CrossRef] [PubMed]
- Alacacioglu, A.; Kebapcilar, L.; Onder Pamuk, B.; Sop, G.; Kucukiravul, C.; Bozkaya, G.; Yuksel, A.; Alacacioglu, I.; Sari, I.; Alacacioglu, A. Summary Oxidative and antioxidative status after anthracycline- based chemotherapy in breast cancer patients. J. BUON 2013, 18, 614–618. [Google Scholar] [PubMed]
- Amin, K.A.; Mohamed, B.M.; El-Wakil, M.A.M.; Ibrahem, S.O. Impact of breast cancer and combination chemotherapy on oxidative stress, hepatic and cardiac markers. J. Breast Cancer 2012, 15, 306–312. [Google Scholar] [CrossRef] [PubMed]
- Šimùnek, T.; Štìrba, M.; Popelová, O.; Adamcová, M.; Hrdina, R.; Geršl, V. Anthracycline-induced cardiotoxicity: Overview of studies examining the roles of oxidative stress and free cellular iron. Pharmacol. Rep. 2009, 61, 154–171. [Google Scholar] [CrossRef]
- Rtibi, K.; Grami, D.; Selmi, S.; Amri, M.; Sebai, H.; Marzouki, L. Vinblastine, an anticancer drug, causes constipation and oxidative stress as well as others disruptions in intestinal tract in rat. Toxicol. Rep. 2017, 4, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Vichaya, E.G.; Chiu, G.S.; Krukowski, K.; Lacourt, T.E.; Kavelaars, A.; Dantzer, R.; Heijnen, C.J.; Walker, A.K. Mechanisms of chemotherapy-induced behavioral toxicities. Front. Neurosci. 2015, 9, 131. [Google Scholar] [CrossRef] [PubMed]
- Canta, A.; Pozzi, E.; Carozzi, V.A. Mitochondrial dysfunction in chemotherapy-induced peripheral neuropathy (CIPN). Toxics 2015, 3, 198–223. [Google Scholar] [CrossRef] [PubMed]
- Csapo, M.; Lazar, L. Chemotherapy-induced cardiotoxicity: Pathophysiology and prevention. Clujul Med. 2014, 87, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Conklin, K.A. Coenzyme Q10 for prevention of anthracycline-induced cardiotoxicity. Integr. Cancer Ther. 2005, 4, 110–130. [Google Scholar] [CrossRef] [PubMed]
- Greenlee, H.; Shaw, J.; Lau, Y.-K.I.; Naini, A.; Maurer, M. Lack of effect of coenzyme q10 on doxorubicin cytotoxicity in breast cancer cell cultures. Integr. Cancer Ther. 2012, 11, 243–250. [Google Scholar] [CrossRef] [PubMed]
- Moreira, A.J.; Rodrigues, G.; Bona, S.; Cerski, C.T.; Marroni, C.A.; Mauriz, J.L.; González-Gallego, J.; Marroni, N.P. Oxidative stress and cell damage in a model of precancerous lesions and advanced hepatocellular carcinoma in rats. Toxicol. Rep. 2015, 2, 333–340. [Google Scholar] [CrossRef] [PubMed]
- Sugamura, K.; Keaney, J.F., Jr. Reactive oxygen species in cardiovascular disease. Free Radic. Biol. Med. 2011, 51, 978–992. [Google Scholar] [CrossRef] [PubMed]
- Hampton, M.B.; Fadeel, B.; Orrenius, S. Redox regulation of the caspases during apoptosis. Ann. N. Y. Acad. Sci. 1998, 854, 328–335. [Google Scholar] [CrossRef] [PubMed]
- Orient, A.; Donko, A.; Szabo, A.; Leto, T.L.; Geiszt, M. Novel sources of reactive oxygen species in the human body. Nephrol. Dial. Transplant. 2007, 22, 1281–1288. [Google Scholar] [CrossRef] [PubMed]
- Brown, G.C.; Borutaite, V. There is no evidence that mitochondria are the main source of reactive oxygen species in mammalian cells. Mitochondrion 2012, 12, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Ballinger, S.W. Mitochondrial dysfunction in cardiovascular disease. Free Radic. Biol. Med. 2005, 38, 1278–1295. [Google Scholar] [CrossRef] [PubMed]
- Weijl, N.I.; Cleton, F.J.; Osanto, S.; Kodama, T.; Naganuma, A.; Imura, N. Free radicals and antioxidants in chemotherapy-induced toxicity. Cancer Treat. Rev. 1997, 23, 209–240. [Google Scholar] [CrossRef]
- Asensio-López, M.C.; Soler, F.; Pascual-Figal, D.; Fernández-Belda, F.; Lax, A. Doxorubicin-induced oxidative stress: The protective effect of nicorandil on HL-1 cardiomyocytes. PLoS ONE 2017, 12, e0172803. [Google Scholar] [CrossRef] [PubMed]
- Stuehr, D.; Pou, S.; Rosen, G.M. Oxygen reduction by nitric-oxide synthases. J. Biol. Chem. 2001, 276, 14533–14536. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Evrovsky, Y.; Cole, D.; Trines, J.; Benson, L.N.; Lehotay, D.C. Doxorubicin-induced acute changes in cytotoxic aldehydes, antioxidant status and cardiac function in the rat. Biochim. Biophys. Acta 1997, 1360, 45–52. [Google Scholar] [PubMed]
- Casares, C.; Ramírez-Camacho, R.; Trinidad, A.; Roldán, A.; Jorge, E.; García-Berrocal, J.R. Reactive oxygen species in apoptosis induced by cisplatin: Review of physiopathological mechanisms in animal models. Eur. Arch. 2012, 269, 2455–2459. [Google Scholar] [CrossRef] [PubMed]
- Rybak, L.P. Mechanisms of cisplatin ototoxicity and progress in otoprotection. Curr. Opin. Otolaryngol. Head Neck Surg. 2007, 15, 364–369. [Google Scholar] [CrossRef] [PubMed]
- Anniko, M.; Sobin, A. Cisplatin: Evaluation of its ototoxic potential. Am. J. Otolaryngol. 1986, 7, 276–293. [Google Scholar] [CrossRef]
- Watanabe, K.; Hess, A.; Bloch, W.; Michel, O. Nitric oxide synthase inhibitor suppresses the ototoxic side effect of cisplatin in guinea pigs. Anticancer. Drugs 2000, 11, 401–406. [Google Scholar]
- Rybak, L.P.; Husain, K.; Whitworth, C.; Somani, S.M. Dose dependent protection by lipoic acid against cisplatin-induced ototoxicity in rats: Antioxidant defense system. Toxicol. Sci. 1999, 47, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Jutooru, I.; Guthrie, A.S.; Chadalapaka, G.; Pathi, S.; Kim, K.; Burghardt, R.; Jin, U.-H.; Safe, S. Mechanism of action of phenethylisothiocyanate and other reactive oxygen species-inducing anticancer agents. Mol. Cell. Biol. 2014, 34, 2382–2395. [Google Scholar] [CrossRef] [PubMed]
- Froelich-Ammon, S.J.; Osheroff, N. Topoisomerase poisons: Harnessing the dark side of enzyme mechanism. J. Biol. Chem. 1995, 270, 21429–21432. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.-Q.; Tian, J.; Qian, K.; Zhao, X.-B.; Morris-Natschke, S.L.; Yang, L.; Nan, X.; Tian, X.; Lee, K.-H. Recent progress on C-4-modified podophyllotoxin analogs as potent antitumor agents. Med. Res. Rev. 2015, 35, 1–62. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.L.; Yang, L.; Rowe, T.C.; Halligan, B.D.; Tewey, K.M.; Liu, L.F. Nonintercalative antitumor drugs interfere with the breakage-reunion reaction of mammalian DNA topoisomerase II. J. Biol. Chem. 1984, 259, 13560–13566. [Google Scholar] [PubMed]
- Burgess, D.J.; Doles, J.; Zender, L.; Xue, W.; Ma, B.; McCombie, W.R.; Hannon, G.J.; Lowe, S.W.; Hemann, M.T. Topoisomerase levels determine chemotherapy response in vitro and in vivo. Proc. Natl. Acad. Sci. USA 2008, 105, 9053–9058. [Google Scholar] [CrossRef] [PubMed]
- Hsiang, Y.H.; Hertzberg, R.; Hecht, S.; Liu, L.F. Camptothecin induces protein-linked DNA breaks via mammalian DNA topoisomerase I. J. Biol. Chem. 1985, 260, 14873–14878. [Google Scholar] [PubMed]
- Hertzberg, R.P.; Caranfa, M.J.; Hecht, S.M. On the mechanism of topoisomerase I inhibition by camptothecin: Evidence for binding to an enzyme-DNA complex. Biochemistry 1989, 28, 4629–4638. [Google Scholar] [CrossRef] [PubMed]
- Cutts, S.; Nudelman, A.; Rephaeli, A.; Phillips, D. The power and potential of doxorubicin-DNA adducts. IUBMB Life 2005, 57, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Teves, S.S.; Kemp, C.J.; Henikoff, S. Doxorubicin, DNA torsion, and chromatin dynamics. Biochim. Biophys. Acta 2014, 1845, 84–89. [Google Scholar] [CrossRef] [PubMed]
- Bancaud, A.; Conde e Silva, N.; Barbi, M.; Wagner, G.; Allemand, J.-F.; Mozziconacci, J.; Lavelle, C.; Croquette, V.; Victor, J.-M.; Prunell, A.; et al. Structural plasticity of single chromatin fibers revealed by torsional manipulation. Nat. Struct. Mol. Biol. 2006, 13, 444–450. [Google Scholar] [CrossRef] [PubMed]
- Pang, B.; Qiao, X.; Janssen, L.; Velds, A.; Groothuis, T.; Kerkhoven, R.; Nieuwland, M.; Ovaa, H.; Rottenberg, S.; van Tellingen, O.; et al. Drug-induced histone eviction from open chromatin contributes to the chemotherapeutic effects of doxorubicin. Nat. Commun. 2013, 4, 1908. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Kemp, C.J.; Henikoff, S. Doxorubicin enhances nucleosome turnover around promoters. Curr. Biol. 2013, 23, 782–787. [Google Scholar] [CrossRef] [PubMed]
- Minotti, G.; Menna, P.; Salvatorelli, E.; Cairo, G.; Gianni, L. Anthracyclines: Molecular advances and pharmacologic developments in antitumor activity and cardiotoxicity. Pharmacol. Rev. 2004, 56, 185–229. [Google Scholar] [CrossRef] [PubMed]
- Olson, R.D.; Mushlin, P.S. Doxorubicin cardiotoxicity: Analysis of prevailing hypotheses. FASEB J. 1990, 4, 3076–3086. [Google Scholar] [CrossRef] [PubMed]
- Cline, S.D. Mitochondrial DNA damage and its consequences for mitochondrial gene expression. Biochim. Biophys. Acta 2012, 1819, 979–991. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Lippard, S.J. Cellular processing of platinum anticancer drugs. Nat. Rev. Drug Discov. 2005, 4, 307–320. [Google Scholar] [CrossRef] [PubMed]
- Ayala-Torres, S.; Chen, Y.; Svoboda, T.; Rosenblatt, J.; van Houten, B. Analysis of gene-specific dna damage and repair using quantitative polymerase chain reaction. Methods 2000, 22, 135–147. [Google Scholar] [CrossRef] [PubMed]
- Todd, R.C.; Lippard, S.J. Inhibition of transcription by platinum antitumor compounds. Metallomics 2009, 1, 280–291. [Google Scholar] [CrossRef] [PubMed]
- Todd, R.C.; Lippard, S.J. Consequences of cisplatin binding on nucleosome structure and dynamics. Chem. Biol. 2010, 17, 1334–1343. [Google Scholar] [CrossRef] [PubMed]
- Zhu, G.; Myint, M.; Ang, W.H.; Song, L.; Lippard, S.J. Monofunctional platinum-DNA adducts are strong inhibitors of transcription and substrates for nucleotide excision repair in live mammalian cells. Cancer Res. 2012, 72, 790–800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giurgiovich, A.J.; Diwan, B.A.; Olivero, O.A.; Anderson, L.M.; Rice, J.M.; Poirier, M.C. Elevated mitochondrial cisplatin-DNA adduct levels in rat tissues after transplacental cisplatin exposure. Carcinogenesis 1997, 18, 93–96. [Google Scholar] [CrossRef] [PubMed]
- Giurgiovich, A.J.; Anderson, L.M.; Jones, A.B.; Dove, L.F.; Moskal, T.J.; Rice, J.M.; Olivero, O.A.; Poirier, M.C. Transplacental cisplatin exposure induces persistent fetal mitochondrial and genomic DNA damage in patas monkeys. Reprod. Toxicol. 1997, 11, 95–100. [Google Scholar] [CrossRef]
- Podratz, J.L.; Knight, A.M.; Ta, L.E.; Staff, N.P.; Gass, J.M.; Genelin, K.; Schlattau, A.; Lathroum, L.; Windebank, A.J. Cisplatin induced mitochondrial DNA damage in dorsal root ganglion neurons. Neurobiol. Dis. 2011, 41, 661–668. [Google Scholar] [CrossRef] [PubMed]
- Vaisman, A.; Lim, S.E.; Patrick, S.M.; Copeland, W.C.; Hinkle, D.C.; Turchi, J.J.; Chaney, S.G. Effect of DNA polymerases and high mobility group protein 1 on the carrier ligand specificity for translesion synthesis past platinum−dna adducts. Biochemistry 1999, 38, 11026–11039. [Google Scholar] [CrossRef] [PubMed]
- Ang, W.H.; Myint, M.; Lippard, S.J. Transcription inhibition by platinum-DNA cross-links in live mammalian cells. J. Am. Chem. Soc. 2010, 132, 7429–7435. [Google Scholar] [CrossRef] [PubMed]
- Le Doux, S.P.; Wilson, G.L.; Beecham, E.J.; Stevnsner, T.; Wassermann, K.; Bohr, V.A. Repair of mitochondrial DNA after various types of DNA damage in Chinese hamster ovary cells. Carcinogenesis 1992, 13, 1967–1973. [Google Scholar] [CrossRef]
- Vakonaki, E.; Tsarouhas, K.; Spandidos, D.A.; Tsatsakis, A.M. Complex interplay of DNA damage, DNA repair genes, and oxidative stress in coronary artery disease. Anatol. J. Cardiol. 2016, 16, 939. [Google Scholar] [CrossRef] [PubMed]
- Botto, N.; Rizza, A.; Colombo, M.G.; Mazzone, A.M.; Manfredi, S.; Masetti, S.; Clerico, A.; Biagini, A.; Andreassi, M.G. Evidence for DNA damage in patients with coronary artery disease. Mutat. Res. 2001, 493, 23–30. [Google Scholar] [CrossRef]
- Bhat, M.A.; Mahajan, N.; Gandhi, G. DNA and chromosomal damage in coronary artery disease patients. EXCLI J. 2013, 12, 872–884. [Google Scholar] [PubMed]
- Kadıoğlu, E.; Taçoy, G.; Özçağlı, E.; Okyay, K.; Akboğa, M.K.; Çengel, A.; Şardaş, S. The role of oxidative DNA damage and GSTM1, GSTT1, and hOGG1 gene polymorphisms in coronary artery disease risk. Anatol. J. Cardiol. 2016, 16, 931–938. [Google Scholar] [CrossRef] [PubMed]
- Balaban, R.S.; Nemoto, S.; Finkel, T. Mitochondria, Oxidants, and Aging. Cell 2005, 120, 483–495. [Google Scholar] [CrossRef] [PubMed]
- Passos, J.F.; Saretzki, G.; Ahmed, S.; Nelson, G.; Richter, T.; Peters, H.; Wappler, I.; Birket, M.J.; Harold, G.; Schaeuble, K.; et al. Mitochondrial dysfunction accounts for the stochastic heterogeneity in telomere-dependent senescence. PLoS Biol. 2007, 5, e110. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, S.; Passos, J.F.; Birket, M.J.; Beckmann, T.; Brings, S.; Peters, H.; Birch-Machin, M.A.; von Zglinicki, T.; Saretzki, G. Telomerase does not counteract telomere shortening but protects mitochondrial function under oxidative stress. J. Cell Sci. 2008, 121, 1046–1053. [Google Scholar] [CrossRef] [PubMed]
- Sharma, N.K.; Reyes, A.; Green, P.; Caron, M.J.; Bonini, M.G.; Gordon, D.M.; Holt, I.J.; Santos, J.H. Human telomerase acts as a hTR-independent reverse transcriptase in mitochondria. Nucleic Acids Res. 2012, 40, 712–725. [Google Scholar] [CrossRef] [PubMed]
- Haendeler, J.; Hoffmann, J.; Diehl, J.F.; Vasa, M.; Spyridopoulos, I.; Zeiher, A.M.; Dimmeler, S. Antioxidants inhibit nuclear export of telomerase reverse transcriptase and delay replicative senescence of endothelial cells. Circ. Res. 2004, 94, 768–775. [Google Scholar] [CrossRef] [PubMed]
- Haendeler, J.; Hoffmann, J.; Brandes, R.P.; Zeiher, A.M.; Dimmeler, S. Hydrogen peroxide triggers nuclear export of telomerase reverse transcriptase via SRC kinase family-dependent phosphorylation of tyrosine 707. Mol. Cell. Biol. 2003, 23, 4598–4610. [Google Scholar] [CrossRef] [PubMed]
- Taanman, J.-W. The mitochondrial genome: Structure, transcription, translation and replication. Biochim. Biophys. Acta Bioenerg. 1999, 1410, 103–123. [Google Scholar] [CrossRef]
- Haendeler, J.; Drose, S.; Buchner, N.; Jakob, S.; Altschmied, J.; Goy, C.; Spyridopoulos, I.; Zeiher, A.M.; Brandt, U.; Dimmeler, S. Mitochondrial telomerase reverse transcriptase binds to and protects mitochondrial dna and function from damage. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 929–935. [Google Scholar] [CrossRef] [PubMed]
- Swain, S.M.; Whaley, F.S.; Ewer, M.S. Congestive heart failure in patients treated with doxorubicin. Cancer 2003, 97, 2869–2879. [Google Scholar] [CrossRef] [PubMed]
- Berthiaume, J.M.; Wallace, K.B. Adriamycin-induced oxidative mitochondrial cardiotoxicity. Cell Biol. Toxicol. 2007, 23, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Goormaghtigh, E.; Huart, P.; Praet, M.; Brasseur, R.; Ruysschaert, J.M. Structure of the adriamycin-cardiolipin complex. Role in mitochondrial toxicity. Biophys. Chem. 1990, 35, 247–257. [Google Scholar] [CrossRef]
- Carvalho, C.; Santos, R.X.; Cardoso, S.; Correia, S.; Oliveira, P.J.; Santos, M.S.; Moreira, P.I. Doxorubicin: The good, the bad and the ugly effect. Curr. Med. Chem. 2009, 16, 3267–3285. [Google Scholar] [CrossRef] [PubMed]
- Tangpong, J.; Cole, M.P.; Sultana, R.; Joshi, G.; Estus, S.; Vore, M.; St. Clair, W.; Ratanachaiyavong, S.; St. Clair, D.K.; Butterfield, D.A. Adriamycin-induced, TNF-mediated central nervous system toxicity. Neurobiol. Dis. 2006, 23, 127–139. [Google Scholar] [CrossRef] [PubMed]
- Kalender, Y.; Yel, M.; Kalender, S. Doxorubicin hepatotoxicity and hepatic free radical metabolism in rats. Toxicology 2005, 209, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Perez, E.A. Paclitaxel in Breast Cancer. Oncologist 1998, 3, 373–389. [Google Scholar] [PubMed]
- Brana, I.; Tabernero, J. Cardiotoxicity. Ann. Oncol. 2010, 21, vii173–vii179. [Google Scholar] [CrossRef] [PubMed]
- Lebedinsky, C.; Gómez, J.; Park, Y.C.; Nieto, A.; Soto-Matos, A.; Parekh, T.; Alfaro, V.; Roy, E.; Lardelli, P.; Kahatt, C. Trabectedin has a low cardiac risk profile: A comprehensive cardiac safety analysis. Cancer Chemother. Pharmacol. 2011, 68, 1223–1231. [Google Scholar] [CrossRef] [PubMed]
- Sentürk, T.; Kanat, O.; Evrensel, T.; Aydinlar, A. Capecitabine-induced cardiotoxicity mimicking myocardial infarction. Neth. Heart J. 2009, 17, 277–280. [Google Scholar] [CrossRef] [PubMed]
- Tascilar, M.; Loos, W.J.; Seynaeve, C.; Verweij, J.; Sleijfer, S. The Pharmacologic basis of ifosfamide use in adult patients with advanced soft tissue sarcomas. Oncologist 2007, 12, 1351–1360. [Google Scholar] [CrossRef] [PubMed]
- Curigliano, G.; Cardinale, D.; Dent, S.; Criscitiello, C.; Aseyev, O.; Lenihan, D.; Cipolla, C.M. Cardiotoxicity of anticancer treatments: Epidemiology, detection, and management. CA Cancer J. Clin. 2016, 66, 309–325. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, M.A.; Antin, J.H.; Guinan, E.C.; Rappeport, J.M. Cyclophosphamide cardiotoxicity: An analysis of dosing as a risk factor. Blood 1986, 68, 1114–1118. [Google Scholar] [PubMed]
- Anderlini, P.; Benjamin, R.S.; Wong, F.C.; Kantarjian, H.M.; Andreeff, M.; Kornblau, S.M.; O’Brien, S.; Mackay, B.; Ewer, M.S.; Pierce, S.A. Idarubicin cardiotoxicity: A retrospective study in acute myeloid leukemia and myelodysplasia. J. Clin. Oncol. 1995, 13, 2827–2834. [Google Scholar] [CrossRef] [PubMed]
- Tjuljandin, S.A.; Doig, R.G.; Sobol, M.M.; Watson, D.M.; Sheridan, W.P.; Morstyn, G.; Mihaly, G.; Green, M.D. Pharmacokinetics and toxicity of two schedules of high dose epirubicin. Cancer Res. 1990, 50, 5095–5101. [Google Scholar] [PubMed]
- Chlebowski, R.T. Adriamycin (doxorubicin) cardiotoxicity: A review. West. J. Med. 1979, 131, 364–368. [Google Scholar] [PubMed]
- Saidi, A.; Alharethi, R. Management of chemotherapy induced cardiomyopathy. Curr. Cardiol. Rev. 2011, 7, 245–249. [Google Scholar] [CrossRef] [PubMed]
- Yoon, H.J.; Kim, K.H.; Kim, J.Y.; Park, H.J.; Cho, J.Y.; Hong, Y.J.; Park, H.W.; Kim, J.H.; Ahn, Y.; Jeong, M.H.; et al. Chemotherapy-induced left ventricular dysfunction in patients with breast cancer. J. Breast Cancer 2016, 19, 402–409. [Google Scholar] [CrossRef] [PubMed]
- Kwok, J.C.; Richardson, D.R. Anthracyclines induce accumulation of iron in ferritin in myocardial and neoplastic cells: Inhibition of the ferritin iron mobilization pathway. Mol. Pharmacol. 2003, 63, 849–861. [Google Scholar] [CrossRef] [PubMed]
- Yeh, E.T.H.; Tong, A.T.; Lenihan, D.J.; Yusuf, S.W.; Swafford, J.; Champion, C.; Durand, J.-B.; Gibbs, H.; Zafarmand, A.A.; Ewer, M.S. Cardiovascular complications of cancer therapy: Diagnosis, pathogenesis, and management. Circulation 2004, 109, 3122–3131. [Google Scholar] [CrossRef] [PubMed]
- Ewer, M.S.; Lippman, S.M. Type II chemotherapy-related cardiac dysfunction: Time to recognize a new entity. J. Clin. Oncol. 2005, 23, 2900–2902. [Google Scholar] [CrossRef] [PubMed]
- Perik, P.J.; de Vries, E.G.E.; Gietema, J.A.; van der Graaf, W.T.A.; Smilde, T.D.J.; Sleijfer, D.T.; van Veldhuisen, D.J. Serum HER2 levels are increased in patients with chronic heart failure. Eur. J. Heart Fail. 2007, 9, 173–177. [Google Scholar] [CrossRef] [PubMed]
- Von Hoff, D.D.; Layard, M.W.; Basa, P.; Davis, H.L.; von Hoff, A.L.; Rozencweig, M.; Muggia, F.M. Risk factors for doxorubicin-induced congestive heart failure. Ann. c Med. 1979, 91, 710–717. [Google Scholar] [CrossRef]
- Bosch, X.; Rovira, M.; Sitges, M.; Domènech, A.; Ortiz-Pérez, J.T.; de Caralt, T.M.; Morales-Ruiz, M.; Perea, R.J.; Monzó, M.; Esteve, J. Enalapril and carvedilol for preventing chemotherapy-induced left ventricular systolic dysfunction in patients with malignant hemopathies: The OVERCOME Trial (preventiOn of left Ventricular dysfunction with Enalapril and caRvedilol in patients submitted to intensive ChemOtherapy for the treatment of Malignant hEmopathies). J. Am. Coll. Cardiol. 2013, 61, 2355–2362. [Google Scholar] [CrossRef] [PubMed]
- Kaya, M.G.; Ozkan, M.; Gunebakmaz, O.; Akkaya, H.; Kaya, E.G.; Akpek, M.; Kalay, N.; Dikilitas, M.; Yarlioglues, M.; Karaca, H.; et al. Protective effects of nebivolol against anthracycline-induced cardiomyopathy: A randomized control study. Int. J. Cardiol. 2013, 167, 2306–2310. [Google Scholar] [CrossRef] [PubMed]
- Kalay, N.; Basar, E.; Ozdogru, I.; Er, O.; Cetinkaya, Y.; Dogan, A.; Oguzhan, A.; Eryol, N.K.; Topsakal, R.; Ergin, A.; et al. Protective effects of carvedilol against anthracycline-induced cardiomyopathy. J. Am. Coll. Cardiol. 2006, 48, 2258–2262. [Google Scholar] [CrossRef] [PubMed]
- Vejpongsa, P.; Yeh, E.T.H. Prevention of anthracycline-induced cardiotoxicity: challenges and opportunities. J. Am. Coll. Cardiol. 2014, 64, 938–945. [Google Scholar] [CrossRef] [PubMed]
- Georgakopoulos, P.; Roussou, P.; Matsakas, E.; Karavidas, A.; Anagnostopoulos, N.; Marinakis, T.; Galanopoulos, A.; Georgiakodis, F.; Zimeras, S.; Kyriakidis, M.; et al. Cardioprotective effect of metoprolol and enalapril in doxorubicin-treated lymphoma patients: A prospective, parallel-group, randomized, controlled study with 36-month follow-up. Am. J. Hematol. 2010, 85, 894–896. [Google Scholar] [CrossRef] [PubMed]
- Räsänen, M.; Degerman, J.; Nissinen, T.A.; Miinalainen, I.; Kerkelä, R.; Siltanen, A.; Backman, J.T.; Mervaala, E.; Hulmi, J.J.; Kivelä, R.; et al. VEGF-B gene therapy inhibits doxorubicin-induced cardiotoxicity by endothelial protection. Proc. Natl. Acad. Sci. USA 2016, 113, 13144–13149. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, E.L.; Sidaway, J.E.; Cross, M.J. Cardiotoxic drugs Herceptin and doxorubicin inhibit cardiac microvascular endothelial cell barrier formation resulting in increased drug permeability. Biol. Open 2016, 5, 1362–1370. [Google Scholar] [CrossRef] [PubMed]
- Renehan, A.G.; Booth, C.; Potten, C.S. What is apoptosis, and why is it important? BMJ 2001, 322, 1536–1538. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.-H.; Kang, P.M. Apoptosis in cardiovascular diseases: Mechanism and clinical implications. Korean Circ. J. 2010, 40, 299–305. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, S.H.; Earnshaw, W.C. Induction of apoptosis by cancer chemotherapy. Exp. Cell Res. 2000, 256, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Debatin, K. Activation of apoptosis pathways by anticancer treatment. Toxicol. Lett. 2000, 112–113, 41–48. [Google Scholar] [CrossRef]
- Solary, E.; Droin, N.; Bettaieb, A.; Corcos, L.; Dimanche-Boitrel, M.T.; Garrido, C. Positive and negative regulation of apoptotic pathways by cytotoxic agents in hematological malignancies. Leukemia 2000, 14, 1833–1849. [Google Scholar] [CrossRef] [PubMed]
- Thornberry, N.A.; Lazebnik, Y. Caspases: Enemies within. Science 1998, 281, 1312–1316. [Google Scholar] [CrossRef] [PubMed]
- Johnson, T.M.; Yu, Z.X.; Ferrans, V.J.; Lowenstein, R.A.; Finkel, T. Reactive oxygen species are downstream mediators of p53-dependent apoptosis. Proc. Natl. Acad. Sci. USA 1996, 93, 11848–11852. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, M.D. Reactive oxygen species and programmed cell death. Trends Biochem. Sci. 1996, 21, 83–86. [Google Scholar] [CrossRef]
- Clutton, S. The importance of oxidative stress in apoptosis. Br. Med. Bull. 1997, 53, 662–668. [Google Scholar] [CrossRef] [PubMed]
- Chandra, J.; Samali, A.; Orrenius, S. Triggering and modulation of apoptosis by oxidative stress. Free Radic. Biol. Med. 2000, 29, 323–333. [Google Scholar] [CrossRef]
- Hampton, M.B.; Orrenius, S. Dual regulation of caspase activity by hydrogen peroxide: Implications for apoptosis. FEBS Lett. 1997, 414, 552–556. [Google Scholar] [CrossRef]
- Shacter, E.; Williams, J.A.; Hinson, R.M.; Sentürker, S.; Lee, Y.J. Oxidative stress interferes with cancer chemotherapy: Inhibition of lymphoma cell apoptosis and phagocytosis. Blood 2000, 96, 307–313. [Google Scholar] [PubMed]
- Lee, Y.J.; Shacter, E. Oxidative stress inhibits apoptosis in human lymphoma cells. J. Biol. Chem. 1999, 274, 19792–19798. [Google Scholar] [CrossRef] [PubMed]
- Antoku, K.; Liu, Z.; Johnson, D.E. Inhibition of caspase proteases by CrmA enhances the resistance of human leukemic cells to multiple chemotherapeutic agents. Leukemia 1997, 11, 1665–1672. [Google Scholar] [CrossRef] [PubMed]
- Vincent, D.T.; Ibrahim, Y.F.; Espey, M.G.; Suzuki, Y.J. The role of antioxidants in the era of cardio-oncology. Cancer Chemother. Pharmacol. 2013, 72, 1157–1168. [Google Scholar] [CrossRef] [PubMed]
- Molenaar, C.; Wiesmeijer, K.; Verwoerd, N.P.; Khazen, S.; Eils, R.; Tanke, H.J.; Dirks, R.W. Visualizing telomere dynamics in living mammalian cells using PNA probes. EMBO J. 2003, 22, 6631–6641. [Google Scholar] [CrossRef] [PubMed]
- Henson, J.D.; Neumann, A.A.; Yeager, T.R.; Reddel, R.R. Alternative lengthening of telomeres in mammalian cells. Oncogene 2002, 21, 598–610. [Google Scholar] [CrossRef] [PubMed]
- Flynn, R.L.; Cox, K.E.; Jeitany, M.; Wakimoto, H.; Bryll, A.R.; Ganem, N.J.; Bersani, F.; Pineda, J.R.; Suvà, M.L.; Benes, C.H.; et al. Alternative lengthening of telomeres renders cancer cells hypersensitive to ATR inhibitors. Science 2015, 347, 273–277. [Google Scholar] [CrossRef] [PubMed]
- Listerman, I.; Sun, J.; Gazzaniga, F.S.; Lukas, J.L.; Blackburn, E.H. The major reverse transcriptase-incompetent splice variant of the human telomerase protein inhibits telomerase activity but protects from apoptosis. Cancer Res. 2013, 73, 2817–2828. [Google Scholar] [CrossRef] [PubMed]
- Ling, X.; Wen, L.; Zhou, Y. Role of mitochondrial translocation of telomerase in hepatocellular carcinoma cells with multidrug resistance. Int. J. Med. Sci. 2012, 9, 545–554. [Google Scholar] [CrossRef] [PubMed]
- Christenson, E.S.; James, T.; Agrawal, V.; Park, B.H. Use of biomarkers for the assessment of chemotherapy-induced cardiac toxicity. Clin. Biochem. 2015, 48, 223–235. [Google Scholar] [CrossRef] [PubMed]
- Morrow, D.A.; Cannon, C.P.; Jesse, R.L.; Newby, L.K.; Ravkilde, J.; Storrow, A.B.; Wu, A.H.B.; Christenson, R.H.; National Academy of Clinical Biochemistry. National academy of clinical biochemistry laboratory medicine practice guidelines: Clinical characteristics and utilization of biochemical markers in acute coronary syndromes. Circulation 2007, 115, e356–e375. [Google Scholar] [CrossRef] [PubMed]
- Yancy, C.W.; Jessup, M.; Bozkurt, B.; Butler, J.; Casey, D.E.; Drazner, M.H.; Fonarow, G.C.; Geraci, S.A.; Horwich, T.; Januzzi, J.L.; et al. 2013 ACCF/AHA guideline for the management of heart failure. J. Am. Coll. Cardiol. 2013, 62, e147–e239. [Google Scholar] [CrossRef] [PubMed]
- Herman, E.H.; Lipshultz, S.E.; Rifai, N.; Zhang, J.; Papoian, T.; Yu, Z.X.; Takeda, K.; Ferrans, V.J. Use of cardiac troponin T levels as an indicator of doxorubicin-induced cardiotoxicity. Cancer Res. 1998, 58, d195–d197. [Google Scholar]
- Herman, E.H.; Zhang, J.; Lipshultz, S.E.; Rifai, N.; Chadwick, D.; Takeda, K.; Yu, Z.-X.; Ferrans, V.J. Correlation between serum levels of cardiac troponin-T and the severity of the chronic cardiomyopathy induced by doxorubicin. J. Clin. Oncol. 1999, 17, 2237. [Google Scholar] [CrossRef] [PubMed]
- Cardinale, D.; Sandri, M.T.; Colombo, A.; Colombo, N.; Boeri, M.; Lamantia, G.; Civelli, M.; Peccatori, F.; Martinelli, G.; Fiorentini, C.; et al. Prognostic value of troponin I in cardiac risk stratification of cancer patients undergoing high-dose chemotherapy. Circulation 2004, 109, 2749–2754. [Google Scholar] [CrossRef] [PubMed]
- Kilickap, S.; Barista, I.; Akgul, E.; Aytemir, K.; Aksoyek, S.; Aksoy, S.; Celik, I.; Kes, S.; Tekuzman, G. cTnT can be a useful marker for early detection of anthracycline cardiotoxicity. Ann. Oncol. 2005, 16, 798–804. [Google Scholar] [CrossRef] [PubMed]
- Ruggiero, A.; de Rosa, G.; Rizzo, D.; Leo, A.; Maurizi, P.; de Nisco, A.; Vendittelli, F.; Zuppi, C.; Mordente, A.; Riccardi, R. Myocardial performance index and biochemical markers for early detection of doxorubicin-induced cardiotoxicity in children with acute lymphoblastic leukaemia. Int. J. Clin. Oncol. 2013, 18, 927–933. [Google Scholar] [CrossRef] [PubMed]
- Dolci, A.; Dominici, R.; Cardinale, D.; Sandri, M.T.; Panteghini, M. Biochemical markers for prediction of chemotherapy-induced cardiotoxicity. Am. J. Clin. Pathol. 2008, 130, 688–695. [Google Scholar] [CrossRef] [PubMed]
- Ky, B.; Putt, M.; Sawaya, H.; French, B.; Januzzi, J.L.; Sebag, I.A.; Plana, J.C.; Cohen, V.; Banchs, J.; Carver, J.R.; et al. Early increases in multiple biomarkers predict subsequent cardiotoxicity in patients with breast cancer treated with doxorubicin, taxanes, and trastuzumab. J. Am. Coll. Cardiol. 2014, 63, 809–816. [Google Scholar] [CrossRef] [PubMed]
- Cardinale, D.; Sandri, M.T.; Martinoni, A.; Borghini, E.; Civelli, M.; Lamantia, G.; Cinieri, S.; Martinelli, G.; Fiorentini, C.; Cipolla, C.M. Myocardial injury revealed by plasma troponin I in breast cancer treated with high-dose chemotherapy. Ann. Oncol. 2002, 13, 710–715. [Google Scholar] [CrossRef] [PubMed]
- Garrone, O.; Crosetto, N.; lo Nigro, C.; Catzeddu, T.; Vivenza, D.; Monteverde, M.; Merlano, M.; Feola, M. Prediction of anthracycline cardiotoxicity after chemotherapy by biomarkers kinetic analysis. Cardiovasc. Toxicol. 2012, 12, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Morno, C.; Petrescu, L. Early detection of anthracycline-mediated cardiotoxicity: The value of considering both global longitudinal left ventricular strain and twist 1. Can. J. Physiol. Pharmacol. 2013, 91, 601–607. [Google Scholar] [CrossRef] [PubMed]
- Sawaya, H.; Sebag, I.A.; Plana, J.C.; Januzzi, J.L.; Ky, B.; Cohen, V.; Gosavi, S.; Carver, J.R.; Wiegers, S.E.; Martin, R.P.; et al. Early detection and prediction of cardiotoxicity in chemotherapy-treated patients. Am. J. Cardiol. 2011, 107, 1375–1380. [Google Scholar] [CrossRef] [PubMed]
- Freda, B.J.; Tang, W.H.W.; van Lente, F.; Peacock, W.F.; Francis, G.S. Cardiac troponins in renal insufficiency: Review and clinical implications. J. Am. Coll. Cardiol. 2002, 40, 2065–2071. [Google Scholar] [CrossRef]
- Tanindi, A.; Cemri, M. Troponin elevation in conditions other than acute coronary syndromes. Vasc. Health Risk Manag. 2011, 7, 597–603. [Google Scholar] [CrossRef] [PubMed]
- Tian, S.; Hirshfield, K.M.; Jabbour, S.K.; Toppmeyer, D.; Haffty, B.G.; Khan, A.J.; Goyal, S. Serum biomarkers for the detection of cardiac toxicity after chemotherapy and radiation therapy in breast cancer patients. Front. Oncol. 2014, 4, 277. [Google Scholar] [CrossRef] [PubMed]
- Shrivastava, A.K.; Singh, H.V.; Raizada, A.; Singh, S.K. C-reactive protein, inflammation and coronary heart disease. Egypt. Heart J. 2015, 67, 89–97. [Google Scholar] [CrossRef]
- Morris, P.G.; Chen, C.; Steingart, R.; Fleisher, M.; Lin, N.; Moy, B.; Come, S.; Sugarman, S.; Abbruzzi, A.; Lehman, R.; et al. Troponin I and C-reactive protein are commonly detected in patients with breast cancer treated with dose-dense chemotherapy incorporating trastuzumab and lapatinib. Clin. Cancer Res. 2011, 17, 3490–3499. [Google Scholar] [CrossRef] [PubMed]
- Lipshultz, S.E.; Miller, T.L.; Scully, R.E.; Lipsitz, S.R.; Rifai, N.; Silverman, L.B.; Colan, S.D.; Neuberg, D.S.; Dahlberg, S.E.; Henkel, J.M.; et al. Changes in cardiac biomarkers during doxorubicin treatment of pediatric patients with high-risk acute lymphoblastic leukemia: Associations with long-term echocardiographic outcomes. J. Clin. Oncol. 2012, 30, 1042–1049. [Google Scholar] [CrossRef] [PubMed]
- Balduzzi, S.; Mantarro, S.; Guarneri, V.; Tagliabue, L.; Pistotti, V.; Moja, L.; D’Amico, R. Trastuzumab-containing regimens for metastatic breast cancer. In Cochrane Database of Systematic Reviews; Moja, L., Ed.; John Wiley & Sons, Ltd.: Chichester, UK, 2014. [Google Scholar] [CrossRef]
- Von Zglinicki, T. Oxidative stress shortens telomeres. Trends Biochem. Sci. 2002, 27, 339–344. [Google Scholar] [CrossRef]
- Bekaert, S.; de Meyer, T.; Rietzschel, E.R.; de Buyzere, M.L.; de Bacquer, D.; Langlois, M.; Segers, P.; Cooman, L.; van Damme, P.; Cassiman, P.; et al. Telomere length and cardiovascular risk factors in a middle-aged population free of overt cardiovascular disease. Aging Cell 2007, 6, 639–647. [Google Scholar] [CrossRef] [PubMed]
- Valdes, A.; Andrew, T.; Gardner, J.; Kimura, M.; Oelsner, E.; Cherkas, L.; Aviv, A.; Spector, T. Obesity, cigarette smoking, and telomere length in women. Lancet 2005, 366, 662–664. [Google Scholar] [CrossRef]
- Strandberg, T.E.; Saijonmaa, O.; Tilvis, R.S.; Pitkl, K.H.; Strandberg, A.Y.; Salomaa, V.; Miettinen, T.A.; Fyhrquist, F. Telomere length in old age and cholesterol across the life course. J. Am. Geriatr. Soc. 2011, 59, 1979–1981. [Google Scholar] [CrossRef] [PubMed]
- Benetos, A.; Okuda, K.; Lajemi, M.; Kimura, M.; Thomas, F.; Skurnick, J.; Labat, C.; Bean, K.; Aviv, A. Telomere length as an indicator of biological aging: The gender effect and relation with pulse pressure and pulse wave velocity. Hypertension 2001, 37, 381–385. [Google Scholar] [CrossRef] [PubMed]
- Muezzinler, A.; Zaineddin, A.K.; Brenner, H. Body mass index and leukocyte telomere length in adults: A systematic review and meta-analysis. Obes. Rev. 2014, 15, 192–201. [Google Scholar] [CrossRef] [PubMed]
- Sampson, M.J.; Winterbone, M.S.; Hughes, J.C.; Dozio, N.; Hughes, D.A. Monocyte telomere shortening and oxidative DNA damage in type 2 diabetes. Diabetes Care 2006, 29, 283–289. [Google Scholar] [CrossRef] [PubMed]
- Cherkas, L.F.; Hunkin, J.L.; Kato, B.S.; Richards, J.B.; Gardner, J.P.; Surdulescu, G.L.; Kimura, M.; Lu, X.; Spector, T.D.; Aviv, A. The association between physical activity in leisure time and leukocyte telomere length. Arch. Intern. Med. 2008, 168, 154. [Google Scholar] [CrossRef] [PubMed]
- Cherkas, L.F.; Aviv, A.; Valdes, A.M.; Hunkin, J.L.; Gardner, J.P.; Surdulescu, G.L.; Kimura, M.; Spector, T.D. The effects of social status on biological aging as measured by white-blood-cell telomere length. Aging Cell 2006, 5, 361–365. [Google Scholar] [CrossRef] [PubMed]
- Strandberg, T.E.; Strandberg, A.Y.; Saijonmaa, O.; Tilvis, R.S.; Pitkälä, K.H.; Fyhrquist, F. Association between alcohol consumption in healthy midlife and telomere length in older men. The Helsinki Businessmen Study. Eur. J. Epidemiol. 2012, 27, 815–822. [Google Scholar] [CrossRef] [PubMed]
- Wolkowitz, O.M.; Mellon, S.H.; Epel, E.S.; Lin, J.; Dhabhar, F.S.; Su, Y.; Reus, V.I.; Rosser, R.; Burke, H.M.; Kupferman, E.; et al. Leukocyte telomere length in major depression: Correlations with chronicity, inflammation and oxidative stress—Preliminary findings. PLoS ONE 2011, 6, e17837. [Google Scholar] [CrossRef] [PubMed]
- Comporti, M.; Signorini, C.; Leoncini, S.; Gardi, C.; Ciccoli, L.; Giardini, A.; Vecchio, D.; Arezzini, B. Ethanol-induced oxidative stress: Basic knowledge. Genes Nutr. 2010, 5, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Strandberg, T.E.; Saijonmaa, O.; Tilvis, R.S.; Pitkala, K.H.; Strandberg, A.Y.; Miettinen, T.A.; Fyhrquist, F. Association of telomere length in older men with mortality and midlife body mass index and smoking. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2011, 66A, 815–820. [Google Scholar] [CrossRef] [PubMed]
- Sano, H.; Nagai, R.; Matsumoto, K.; Horiuchi, S. Receptors for proteins modified by advanced glycation endproducts (AGE)—Their functional role in atherosclerosis. Mech. Ageing Dev. 1999, 107, 333–346. [Google Scholar] [CrossRef]
- Xu, Q.; Parks, C.G.; DeRoo, L.A.; Cawthon, R.M.; Sandler, D.P.; Chen, H. Multivitamin use and telomere length in women. Am. J. Clin. Nutr. 2009, 89, 1857–1863. [Google Scholar] [CrossRef] [PubMed]
- Ornish, D.; Lin, J.; Daubenmier, J.; Weidner, G.; Epel, E.; Kemp, C.; Magbanua, M.J.M.; Marlin, R.; Yglecias, L.; Carroll, P.R.; et al. Increased telomerase activity and comprehensive lifestyle changes: A pilot study. Lancet Oncol. 2008, 9, 1048–1057. [Google Scholar] [CrossRef]
- Berezin, A.E. Circulating cell-free mitochondrial DNA as biomarker of cardiovascular risk: new challenges of old findings. Angiology 2015, 3, 161–163. [Google Scholar] [CrossRef]
- Chen, S.; Xie, X.; Wang, Y.; Gao, Y.; Xie, X.; Yang, J.; Ye, J. Association between leukocyte mitochondrial DNA content and risk of coronary heart disease: A case-control study. Atherosclerosis 2014, 237, 220–226. [Google Scholar] [CrossRef] [PubMed]
- Alvarado-Vasquez, N. Circulating cell-free mitochondrial DNA as the probable inducer of early endothelial dysfunction in the prediabetic patient. Exp. Gerontol. 2015, 69, 70–78. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Cai, X.; Xie, L.; Tang, Y.; Cheng, J.; Wang, J.; Wang, L.; Gong, J. Circulating cell free mitochondrial DNA is a biomarker in the development of coronary heart disease in the patients with type 2 diabetes. Clin. Lab. 2015, 61, 661–667. [Google Scholar] [CrossRef] [PubMed]
- Borghini, A.; Mercuri, A.; Turchi, S.; Chiesa, M.R.; Piccaluga, E.; Andreassi, M.G. Increased circulating cell-free DNA levels and mtDNA fragments in interventional cardiologists occupationally exposed to low levels of ionizing radiation. Environ. Mol. Mutagen. 2015, 56, 293–300. [Google Scholar] [CrossRef] [PubMed]
- Nakahira, K.; Kyung, S.-Y.; Rogers, A.J.; Gazourian, L.; Youn, S.; Massaro, A.F.; Quintana, C.; Osorio, J.C.; Wang, Z.; Zhao, Y.; et al. Circulating mitochondrial DNA in patients in the ICU as a marker of mortality: Derivation and validation. PLoS Med. 2013, 10, e1001577. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug | Cardiovacular Side Effect | References |
|---|---|---|
| Trastuzumab | Heart Failure, Cardiotoxicity, LVEF reduction, Troponin 1 elevation | Huszno et al. [52], Cardinale et al. [53] |
| Thalidomide | Sinus Bradycardia, Peripheral Edema, Orthostatic Hypotension | Ghobrial et al. [54] |
| Sunitinib | Hypertension, LVEF dysfunction, CHF, depletion of coronary microvascular pericytes | Lorenzo et al. [55], Chintalgattu et al. [56] |
| Sorafenib | Hypertension, left ventricle dysfunction, cardiac ischemia, hypercholesterolemia, hypertriglyceridemia | Abdel-Rahman et al. [57], Schmidinger et al. [58] |
| Pazopanib | Hypertension, cardiomyopathy, cardiac dysrhythmias | Pinkhas et al. [59] |
| Mitoxantrone | Cardiotoxicity, LVEF reduction, CHF, diastolic dysfunction | Paul et al. [60] |
| Mitomycin | Cardiotoxicity, heart-cell toxicity, low reduction potentials | Brockstein et al. [61], Dorr et al. [62] |
| Melphalan | Atrial Fibrillation | Feliz et al. [63] |
| Lenalidomide | Myocarditis | Carver et al. [64] |
| Lapatinib | Cardiotoxicity, QTc Elongation | Kloth et al. [65] |
| Interleukin-2 | Edema, hypotension, increased heart rate, increased cardiac index | Sobotka et al. [66] |
| Imatinib Mesylate | Cardiotoxicity, heart failure, cardiomyocyte dysfunction | Turrisi et al. [67], Schmidinger et al. [58] |
| Doxorubicin | Cardiomyopathy [68], heart failure [69] | Chatterjee et al. [68], Mitry et al. [69] |
| Cisplatin | Hypertension, heart failure, myocarditis, cardiomyopathy, cardiac arrhythmias: supraventricular tachycardia, bradycardia, block | Raja et al. [70] |
| Arsenic trioxide | Prolonged QTc | Unnikrishnan et al. [71] |
| Bevacizumab | Hypertension, heart failure, thromboembolic events | Economopoulou et al. [72] |
| Bortezomib | Heart block, heart failure | Orciuolo et al. [73] |
| Pertuzumab | Cardiotoxicity (during co-treatment with trastuzumab), myocardial dysfunction | Sendur et al. [74] |
| Drug | Dosage Range (Toxic) | Cardiovascular Damage | Frequency of Cardiovascular Damage | Reference |
|---|---|---|---|---|
| Paclitaxel | Standard dose | QTc elongation | Uncommon | Perez [149] |
| Arsenic trioxide | Standard dose | QTc elongation | Common | Brana et al. [150] |
| Trabectedin | Standard dose | Cardiac ischemia | Intermediate | Lebedinsky et al. [151] |
| Paclitaxel | Standard dose | Cardiac ischemia | Uncommon | Perez [149] |
| Capecitabine | Standard dose | Cardiac ischemia | Intermediate | Sentürk et al. [152] |
| Ifosfamide | >10 mg/m2 | Uncommon | Tascilar et al. [153], Curigliano et al. [154] | |
| Cyclophosphamide | >100–120 mg/kg | Left ventricular dysfunction | Intermediate | Goldberg et al. [155] |
| Paclitaxel | Standard dose | Left ventricular dysfunction | Intermediate | Perez [149] |
| Idarubicin | 150–290 mg/m2 | Intermediate | Anderlini et al. [156] | |
| Epirubicin | >900 mg/m2 | Common | Tjuljandin et al. [157] | |
| Doxorubicin | >450 mg/m2 | Left ventricular dysfunction | Common | Chlebowski [158] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Quryshi, N.; Norwood Toro, L.E.; Ait-Aissa, K.; Kong, A.; Beyer, A.M. Chemotherapeutic-Induced Cardiovascular Dysfunction: Physiological Effects, Early Detection—The Role of Telomerase to Counteract Mitochondrial Defects and Oxidative Stress. Int. J. Mol. Sci. 2018, 19, 797. https://doi.org/10.3390/ijms19030797
Quryshi N, Norwood Toro LE, Ait-Aissa K, Kong A, Beyer AM. Chemotherapeutic-Induced Cardiovascular Dysfunction: Physiological Effects, Early Detection—The Role of Telomerase to Counteract Mitochondrial Defects and Oxidative Stress. International Journal of Molecular Sciences. 2018; 19(3):797. https://doi.org/10.3390/ijms19030797
Chicago/Turabian StyleQuryshi, Nabeel, Laura E. Norwood Toro, Karima Ait-Aissa, Amanda Kong, and Andreas M. Beyer. 2018. "Chemotherapeutic-Induced Cardiovascular Dysfunction: Physiological Effects, Early Detection—The Role of Telomerase to Counteract Mitochondrial Defects and Oxidative Stress" International Journal of Molecular Sciences 19, no. 3: 797. https://doi.org/10.3390/ijms19030797