PON-tstab: Protein Variant Stability Predictor. Importance of Training Data Quality

 ,
,
Abstract
:1. Introduction
2. Results
2.1. Cleaning and Pruning Stability Data
2.2. Dataset Properties
2.3. Novel Stability Predictor
2.4. Predictor Training
2.5. Testing
3. Discussion
4. Materials and Methods
4.1. Variation Data
4.2. Features
4.3. Predictor Training
4.4. Method Quality Assessment
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| ΔΔG | Gibb’s free energy |
| AAS | Amino acid substitution |
| BLAST | Basic Local Alignment Search Tool |
| CPR | Correct prediction ratio |
| DSC | Differential scanning calorimetry |
| FN | False negative |
| FP | False positive |
| GC2 | Generalized squared correlation |
| MCC | Matthews correlation coefficient |
| ML | Machine learning |
| MPRA | Massively parallel reporter assay |
| MSA | Multiple sequence alignment |
| NPV | Negative predictive value |
| PPV | Positive predictive value |
| QM | Quantum mechanics |
| TN | True negative |
| TNR | True negative rate |
| TP | True positive |
| TPR | True positive rate |
References
- O’Fagain, C. Engineering protein stability. Methods Mol. Biol. 2011, 681, 103–136. [Google Scholar] [CrossRef] [PubMed]
- Socha, R.D.; Tokuriki, N. Modulating protein stability—Directed evolution strategies for improved protein function. FEBS J. 2013, 280, 5582–5595. [Google Scholar] [CrossRef] [PubMed]
- Poultney, C.S.; Butterfoss, G.L.; Gutwein, M.R.; Drew, K.; Gresham, D.; Gunsalus, K.C.; Shasha, D.E.; Bonneau, R. Rational design of temperature-sensitive alleles using computational structure prediction. PLoS ONE 2011, 6, e23947. [Google Scholar] [CrossRef] [PubMed]
- Tan, K.P.; Khare, S.; Varadarajan, R.; Madhusudhan, M.S. TSpred: A web server for the rational design of temperature-sensitive mutants. Nucleic Acids Res. 2014, 42, W277–W284. [Google Scholar] [CrossRef] [PubMed]
- Chakshusmathi, G.; Mondal, K.; Lakshmi, G.S.; Singh, G.; Roy, A.; Ch, R.B.; Madhusudhanan, S.; Varadarajan, R. Design of temperature-sensitive mutants solely from amino acid sequence. Proc. Natl. Acad. Sci. USA 2004, 101, 7925–7930. [Google Scholar] [CrossRef] [PubMed]
- Ferrer-Costa, C.; Orozco, M.; de la Cruz, X. Characterization of disease-associated single amino acid polymorphisms in terms of sequence and structure properties. J. Mol. Biol. 2002, 315, 771–786. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Chen, B.; Tan, G.; Vihinen, M.; Shen, B. Structure-based prediction of the effects of a missense variant on protein stability. Amino Acids 2013, 44, 847–855. [Google Scholar] [CrossRef] [PubMed]
- Folkman, L.; Stantic, B.; Sattar, A.; Zhou, Y. EASE-MM: Sequence-Based Prediction of Mutation-Induced Stability Changes with Feature-Based Multiple Models. J. Mol. Biol. 2016, 428, 1394–1405. [Google Scholar] [CrossRef] [PubMed]
- Capriotti, E.; Fariselli, P.; Casadio, R. I-Mutant2.0: Predicting stability changes upon mutation from the protein sequence or structure. Nucleic Acids Res. 2005, 33, W306–W310. [Google Scholar] [CrossRef] [PubMed]
- Fariselli, P.; Martelli, P.L.; Savojardo, C.; Casadio, R. INPS: Predicting the impact of non-synonymous variations on protein stability from sequence. Bioinformatics 2015, 31, 2816–2821. [Google Scholar] [CrossRef] [PubMed]
- Dehouck, Y.; Kwasigroch, J.M.; Gilis, D.; Rooman, M. PoPMuSiC 2.1: A web server for the estimation of protein stability changes upon mutation and sequence optimality. BMC Bioinform. 2011, 12, 151. [Google Scholar] [CrossRef] [PubMed]
- Giollo, M.; Martin, A.J.; Walsh, I.; Ferrari, C.; Tosatto, S.C. NeEMO: A method using residue interaction networks to improve prediction of protein stability upon mutation. BMC Genom. 2014, 15 (Suppl. 4), S7. [Google Scholar] [CrossRef] [PubMed]
- Quan, L.; Lv, Q.; Zhang, Y. STRUM: Structure-based prediction of protein stability changes upon single-point mutation. Bioinformatics 2016, 32, 2936–2946. [Google Scholar] [CrossRef] [PubMed]
- Masso, M.; Vaisman, I.I. AUTO-MUTE 2.0: A Portable Framework with Enhanced Capabilities for Predicting Protein Functional Consequences upon Mutation. Adv. Bioinform. 2014, 2014, 278385. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Fang, J. PROTS-RF: A robust model for predicting mutation-induced protein stability changes. PLoS ONE 2012, 7, e47247. [Google Scholar] [CrossRef] [PubMed]
- Pires, D.E.; Ascher, D.B.; Blundell, T.L. DUET: A server for predicting effects of mutations on protein stability using an integrated computational approach. Nucleic Acids Res. 2014, 42, W314–W319. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.D.; Bava, K.A.; Gromiha, M.M.; Prabakaran, P.; Kitajima, K.; Uedaira, H.; Sarai, A. ProTherm and ProNIT: Thermodynamic databases for proteins and protein-nucleic acid interactions. Nucleic Acids Res. 2006, 34, D204–D206. [Google Scholar] [CrossRef] [PubMed]
- Niroula, A.; Vihinen, M. Variation interpretation predictors: Principles, types, performance, and choice. Hum. Mutat. 2016, 37, 579–597. [Google Scholar] [CrossRef] [PubMed]
- Walsh, I.; Pollastri, G.; Tosatto, S.C. Correct machine learning on protein sequences: A peer-reviewing perspective. Brief. Bioinform. 2016, 17, 831–840. [Google Scholar] [CrossRef] [PubMed]
- Nair, P.S.; Vihinen, M. VariBench: A benchmark database for variations. Hum. Mutat. 2013, 34, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Vihinen, M. How to evaluate performance of prediction methods? Measures and their interpretation in variation effect analysis. BMC Genom. 2012, 13 (Suppl. 4), S2. [Google Scholar] [CrossRef] [PubMed]
- Vihinen, M. Guidelines for reporting and using prediction tools for genetic variation analysis. Hum. Mutat. 2013, 34, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Vihinen, M. Performance of protein stability predictors. Hum. Mutat. 2010, 31, 675–684. [Google Scholar] [CrossRef] [PubMed]
- Potapov, V.; Cohen, M.; Schreiber, G. Assessing computational methods for predicting protein stability upon mutation: Good on average but not in the details. Protein Eng. Des. Sel. 2009, 22, 553–560. [Google Scholar] [CrossRef] [PubMed]
- Tsuji, T.; Chrunyk, B.A.; Chen, X.; Matthews, C.R. Mutagenic analysis of the interior packing of an alpha/beta barrel protein. Effects on the stabilities and rates of interconversion of the native and partially folded forms of the alpha subunit of tryptophan synthase. Biochemistry 1993, 32, 5566–5575. [Google Scholar] [CrossRef] [PubMed]
- Tweedy, N.B.; Hurle, M.R.; Chrunyk, B.A.; Matthews, C.R. Multiple replacements at position 211 in the alpha subunit of tryptophan synthase as a probe of the folding unit association reaction. Biochemistry 1990, 29, 1539–1545. [Google Scholar] [CrossRef] [PubMed]
- Campos, L.A.; Bueno, M.; Lopez-Llano, J.; Jimenez, M.A.; Sancho, J. Structure of stable protein folding intermediates by equilibrium phi-analysis: The apoflavodoxin thermal intermediate. J. Mol. Biol. 2004, 344, 239–255. [Google Scholar] [CrossRef] [PubMed]
- Matthews, J.M.; Ward, L.D.; Hammacher, A.; Norton, R.S.; Simpson, R.J. Roles of histidine 31 and tryptophan 34 in the structure, self-association, and folding of murine interleukin-6. Biochemistry 1997, 36, 6187–6196. [Google Scholar] [CrossRef] [PubMed]
- Isom, D.G.; Vardy, E.; Oas, T.G.; Hellinga, H.W. Picomole-scale characterization of protein stability and function by quantitative cysteine reactivity. Proc. Natl. Acad. Sci. USA 2010, 107, 4908–4913. [Google Scholar] [CrossRef] [PubMed]
- Schultz, D.A.; Baldwin, R.L. Cis proline mutants of ribonuclease A. I. Thermal stability. Protein Sci. 1992, 1, 910–916. [Google Scholar] [CrossRef] [PubMed]
- Matsumura, M.; Matthews, B.W. Control of enzyme activity by an engineered disulfide bond. Science 1989, 243, 792–794. [Google Scholar] [CrossRef] [PubMed]
- Ruvinov, S.; Wang, L.; Ruan, B.; Almog, O.; Gilliland, G.L.; Eisenstein, E.; Bryan, P.N. Engineering the independent folding of the subtilisin BPN’ prodomain: Analysis of two-state folding versus protein stability. Biochemistry 1997, 36, 10414–10421. [Google Scholar] [CrossRef] [PubMed]
- Khatun, J.; Khare, S.D.; Dokholyan, N.V. Can contact potentials reliably predict stability of proteins? J. Mol. Biol. 2004, 336, 1223–1238. [Google Scholar] [CrossRef] [PubMed]
- Niroula, A.; Urolagin, S.; Vihinen, M. PON-P2: Prediction method for fast and reliable identification of harmful variants. PLoS ONE 2015, 10, e0117380. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Niroula, A.; Shen, B.; Vihinen, M. PON-Sol: Prediction of effects of amino acid substitutions on protein solubility. Bioinformatics 2016, 32, 2032–2034. [Google Scholar] [CrossRef] [PubMed]
- Niroula, A.; Vihinen, M. Predicting severity of disease-causing variants. Hum. Mutat. 2017, 38, 357–364. [Google Scholar] [CrossRef] [PubMed]
- Niroula, A.; Vihinen, M. Classification of Amino Acid Substitutions in Mismatch Repair Proteins Using PON-MMR2. Hum. Mutat. 2015, 36, 1128–1134. [Google Scholar] [CrossRef] [PubMed]
- Wei, Q.; Dunbrack, R.L., Jr. The role of balanced training and testing data sets for binary classifiers in bioinformatics. PLoS ONE 2013, 8, e67863. [Google Scholar] [CrossRef] [PubMed]
- Capriotti, E.; Fariselli, P.; Rossi, I.; Casadio, R. A three-state prediction of single point mutations on protein stability changes. BMC Bioinform. 2008, 9 (Suppl. 2), S6. [Google Scholar] [CrossRef] [PubMed]
- Pakula, A.A.; Sauer, R.T. Genetic analysis of protein stability and function. Annu. Rev. Genet. 1989, 23, 289–310. [Google Scholar] [CrossRef] [PubMed]
- Olatubosun, A.; Väliaho, J.; Härkönen, J.; Thusberg, J.; Vihinen, M. PON-P: Integrated predictor for pathogenicity of missense variants. Hum. Mutat. 2012, 33, 1166–1174. [Google Scholar] [CrossRef] [PubMed]
- Consortium, T.U. UniProt: The universal protein knowledgebase. Nucleic Acids Res. 2017, 45, D158–D169. [Google Scholar] [CrossRef]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [PubMed]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal W and Clustal X version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef] [PubMed]
- Fares, M.A.; McNally, D. CAPS: Coevolution analysis using protein sequences. Bioinformatics 2006, 22, 2821–2822. [Google Scholar] [CrossRef] [PubMed]
- Kawashima, S.; Kanehisa, M. AAindex: Amino acid index database. Nucleic Acids Res. 2000, 28, 374. [Google Scholar] [CrossRef] [PubMed]
- Shen, B.; Vihinen, M. Conservation and covariance in PH domain sequences: Physicochemical profile and information theoretical analysis of XLA-causing mutations in the Btk PH domain. Protein Eng. Des. Sel. 2004, 17, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Lockwood, S.; Krishnamoorthy, B.; Ye, P. Neighborhood properties are important determinants of temperature sensitive mutations. PLoS ONE 2011, 6, e28507. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Blanco, Y.B.; Paz, W.; Green, J.; Marrero-Ponce, Y. ProtDCal: A program to compute general-purpose-numerical descriptors for sequences and 3D-structures of proteins. BMC Bioinform. 2015, 16, 162. [Google Scholar] [CrossRef] [PubMed]
- Breiman, L. Random forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef]
- Vihinen, M.; den Dunnen, J.T.; Dalgleish, R.; Cotton, R.G. Guidelines for establishing locus specific databases. Hum. Mutat. 2012, 33, 298–305. [Google Scholar] [CrossRef] [PubMed]
- Baldi, P.; Brunak, S.; Chauvin, Y.; Andersen, C.A.; Nielsen, H. Assessing the accuracy of prediction algorithms for classification: An overview. Bioinformatics 2000, 16, 412–424. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
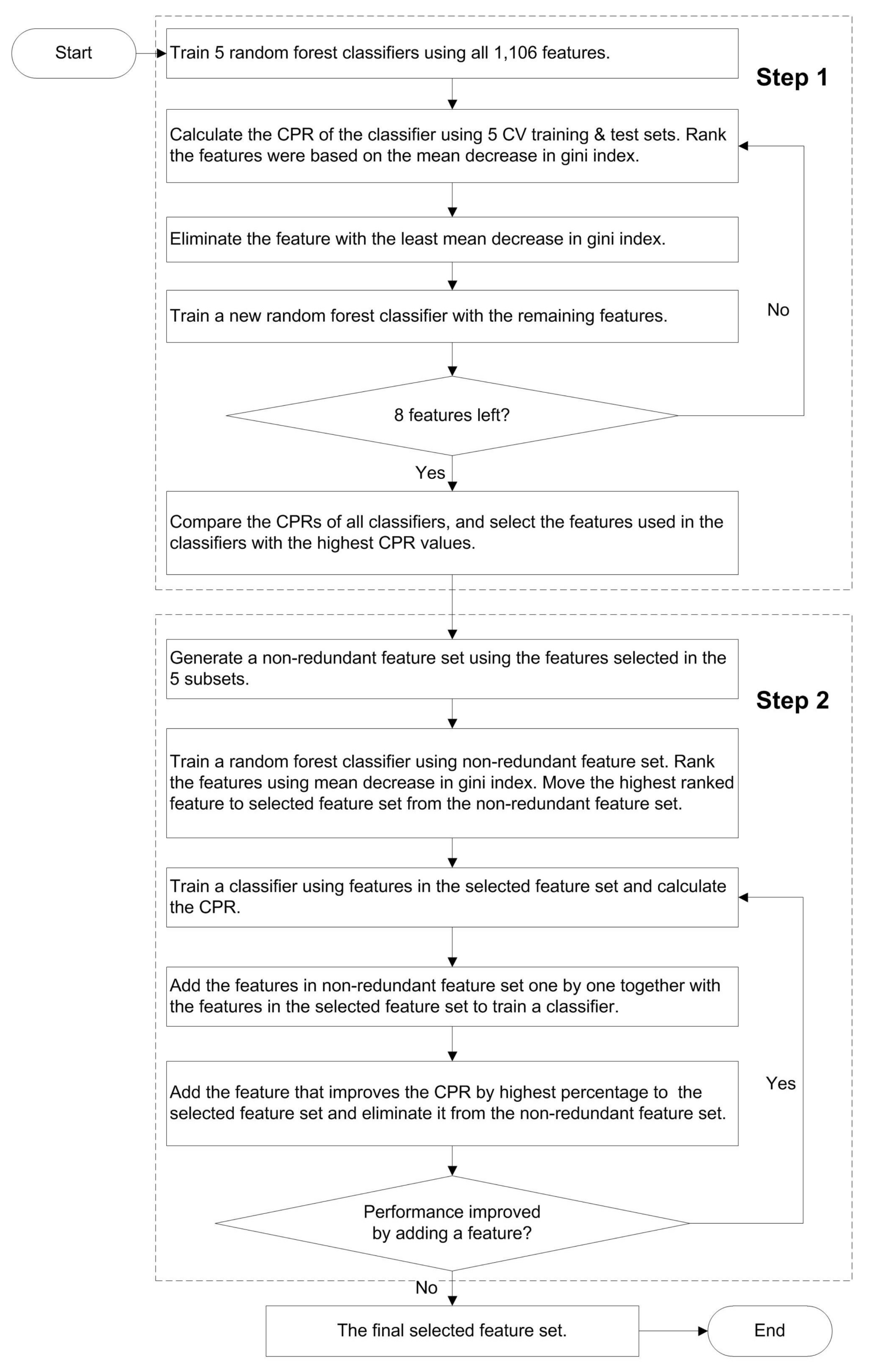{kind=link}
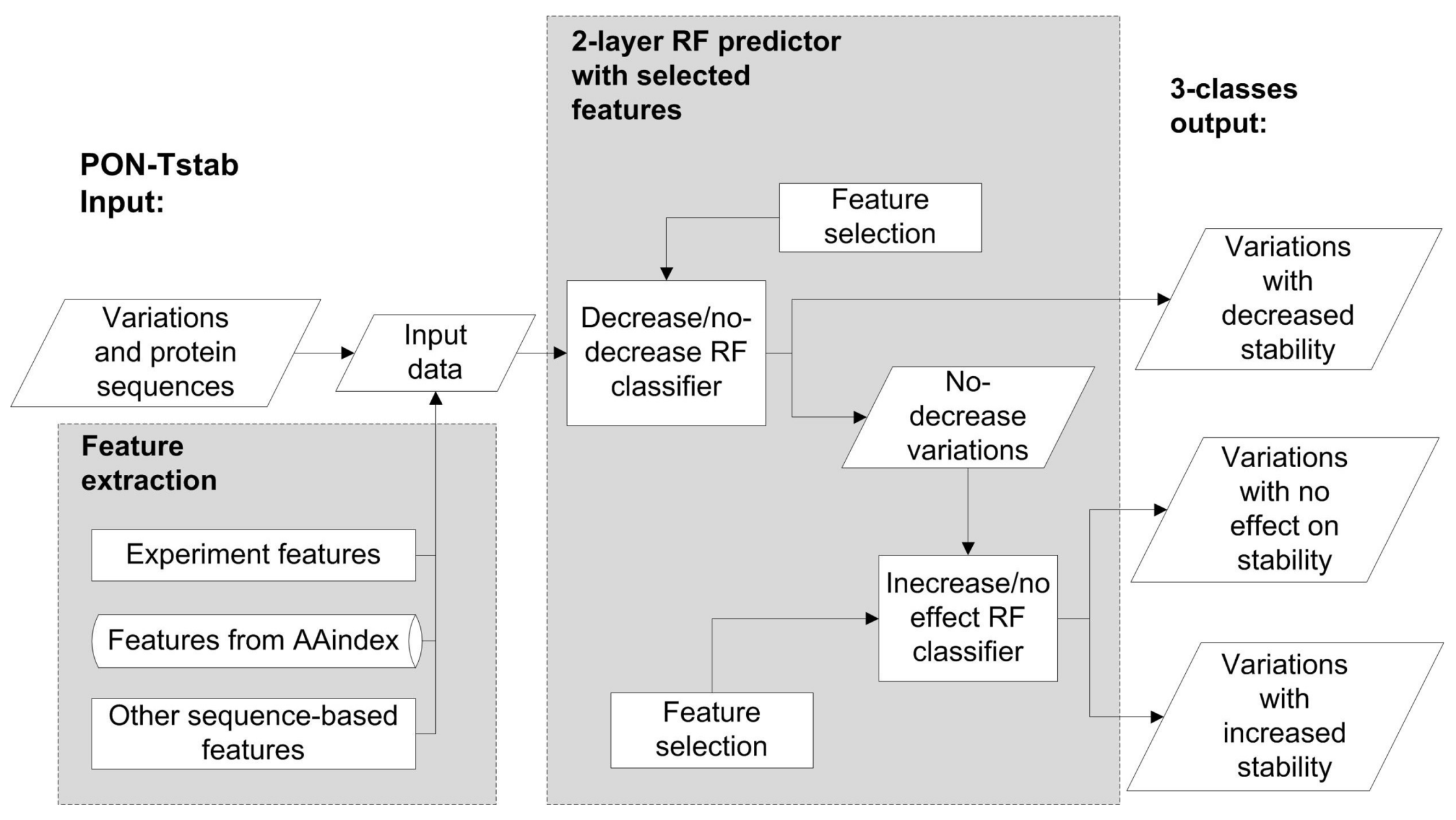{kind=link}
| Performance Measures | Predictors Trained and Test on Original Dataset | Feature Selection on Original Dataset and Training/Testing on Balanced Original and Reversed Dataset | |||||||
|---|---|---|---|---|---|---|---|---|---|
| 3-Class RF with All Features a | 3-Class RF with 8 Selected Features | 2-Layer Predictor with All Features | 2-Layer Predictor with 10 Selected Features (PON-tstab) | 3-Class RF with All Features | 3-Class RF with 5 Selected Features | 2-Layer Predictor on 3 Classifiers with All Features | 2-Layer Predictor on 3 Classifiers with Selected Features | ||
| TP b | + | 21.6/43.5 | 9.6/19.0 | 19/38.3 | 23.2/46.3 | 40.8 | 44.8 | 42.8 | 43.4 |
| − | 74.8/40.2 | 124.4/67.2 | 90.6/48.1 | 91.4/49.0 | 43.4 | 42 | 41.6 | 37.4 | |
| no | 34 | 26.4 | 28.8 | 30.8 | 36.2 | 37.2 | 37.8 | 40 | |
| TN | + | 180.6/123.6 | 32.2/64.4 | 183.4/125.7 | 186.2/127.9 | 125.4 | 122.2 | 125.4 | 123.8 |
| − | 95/130.4 | 30.2/16.2 | 85.8/118.1 | 88.2/122.7 | 125.4 | 129.2 | 126.4 | 131.4 | |
| no | 134.6/113.9 | 57 | 149/121.6 | 150.8/125.6 | 121.4 | 123.4 | 121.2 | 116.4 | |
| FP | + | 57.4/43.2 | 10.6/8.0 | 54.6/41.1 | 51.8/38.9 | 41.8 | 45 | 41.8 | 43.4 |
| − | 30.2/36.4 | 71.8/91.6 | 39.4/48.7 | 37/44.1 | 42.8 | 38 | 40.8 | 35.8 | |
| no | 61.8/52.9 | 37/38.0 | 47.4/45.2 | 45.6/41.2 | 45.8 | 43.8 | 46 | 50.8 | |
| FN | + | 20.2/39.9 | 227.4/158.7 | 22.8/45.1 | 18.6/37.1 | 42.8 | 38.8 | 40.8 | 40.2 |
| − | 79.8/43.2 | 53.4/75.2 | 64/35.3 | 63.2/34.4 | 40.2 | 41.6 | 42 | 46.2 | |
| no | 49.4 | 159.4/128.8 | 54.6 | 52.6 | 47.4 | 46.4 | 45.8 | 43.6 | |
| Sensitivity | + | 0.516 | 0.228 | 0.455 | 0.554 | 0.488 | 0.537 | 0.511 | 0.518 |
| − | 0.483 | 0.805 | 0.583 | 0.59 | 0.52 | 0.504 | 0.498 | 0.449 | |
| no | 0.406 | 0.318 | 0.339 | 0.367 | 0.432 | 0.445 | 0.451 | 0.478 | |
| Specificity | + | 0.759/0.744 | 0.955/0.953 | 0.771/0.756 | 0.783/0.769 | 0.75 | 0.732 | 0.75 | 0.741 |
| − | 0.755/0.776 | 0.427/0.451 | 0.68/0.700 | 0.701/0.730 | 0.743 | 0.772 | 0.755 | 0.785 | |
| no | 0.686/0.683 | 0.812/0.772 | 0.757/0.732 | 0.767/0.756 | 0.727 | 0.739 | 0.725 | 0.697 | |
| PPV | + | 0.271/0.498 | 0.463/0.691 | 0.258/0.481 | 0.318/0.551 | 0.492 | 0.505 | 0.505 | 0.502 |
| − | 0.714/0.527 | 0.635/0.424 | 0.701/0.503 | 0.715/0.530 | 0.505 | 0.528 | 0.548 | 0.513 | |
| no | 0.354/0.388 | 0.421/0.413 | 0.371/0.386 | 0.399/0.428 | 0.445 | 0.462 | 0.452 | 0.444 | |
| NPV | + | 0.9/0.757 | 0.876/0.712 | 0.89/0.763 | 0.909/0.774 | 0.746 | 0.76 | 0.755 | 0.756 |
| − | 0.543/0.75 | 0.643/0.824 | 0.57/0.771 | 0.58/0.781 | 0.756 | 0.758 | 0.75 | 0.741 | |
| no | 0.732/0.698 | 0.737/0.694 | 0.731/0.690 | 0.741/0.705 | 0.719 | 0.727 | 0.726 | 0.728 | |
| GC2 | 0.172/0.078 | 0.101/0.281 | 0.121/0.085 | 0.162/0.112 | 0.063 | 0.08 | 0.068 | 0.071 | |
| CPR | 0.466/0.469 | 0.573/0.450 | 0.495/0.459 | 0.520/0.503 | 0.48 | 0.495 | 0.487 | 0.481 | |
| Performance Measures | Predictors | ||||
|---|---|---|---|---|---|
| 3-Class RF with All 1106 Features a | 3-Class RF with 8 Selected Features | 2-Layer Predictor with All 1106 Features | 2-Layer Predictor with 10 Selected Features (PON-tstab) | ||
| TP | + | 2/4.4 | 1/2.2 | 4/8.7 | 3/6.5 |
| − | 66/35.9 | 69/37.5 | 62/33.7 | 66/35.9 | |
| no | 18 | 16 | 20 | 22 | |
| TN | + | 135/94.8 | 134/94.7 | 120/83.5 | 126/88.1 |
| − | 28/36.2 | 20/22.3 | 37/45.2 | 37/46.4 | |
| no | 88/77.2 | 97/88.6 | 94/83.7 | 93/79.9 | |
| FP | + | 7/5.2 | 8/5.3 | 22/16.5 | 16/11.9 |
| − | 45/63.8 | 53/77.7 | 36/54.8 | 36/53.6 | |
| no | 27/22.8 | 18/11.4 | 21/16.3 | 22/20.1 | |
| FN | + | 21/45.7 | 22/47.8 | 19/41.3 | 20/43.5 |
| − | 26/14.1 | 23/12.5 | 30/16.3 | 26/14.1 | |
| no | 32 | 34 | 30 | 28 | |
| Sensitivity | + | 0.087 | 0.043 | 0.174 | 0.130 |
| − | 0.717 | 0.750 | 0.674 | 0.717 | |
| no | 0.360 | 0.320 | 0.400 | 0.440 | |
| Specificity | + | 0.951/0.948 | 0.944/0.947 | 0.845/0.835 | 0.887/0.881 |
| − | 0.384/0.362 | 0.274/0.223 | 0.507/0.452 | 0.507/0.464 | |
| no | 0.765/0.772 | 0.843/0.886 | 0.817/0.837 | 0.809/0.799 | |
| PPV | + | 0.222/0.457 | 0.111/0.292 | 0.154/0.345 | 0.158/0.354 |
| − | 0.595/0.36 | 0.566/0.326 | 0.633/0.381 | 0.647/0.401 | |
| no | 0,4/0.441 | 0.471/0.584 | 0.488/0.551 | 0.5/0.522 | |
| NPV | + | 0.865/0.675 | 0.859/0.665 | 0.863/0.669 | 0.863/0.670 |
| − | 0.519/0.719 | 0.465/0.641 | 0.552/0.735 | 0.587/0.767 | |
| no | 0.733/0.707 | 0.74/0.723 | 0.758/0.736 | 0.769/0.740 | |
| GC2 | 0.049/0.291 | 0.091/0.476 | 0.043/0.200 | 0.046/0.219 | |
| CPR | 0.521/0.388 | 0.521/0.371 | 0.521/0.416 | 0.552/0.429 | |
| Predictors | |||||
|---|---|---|---|---|---|
| Performance Measures | EASE-MM | I-Mutant | INPS | PON-tstab | |
| Variants Predicted | 40 | 40 | 15 | 165 | |
| TP | + | 0 | 0 | 0 | 3/6.5 |
| − | 22/6.8 | 22/6.8 | 9 | 66/35.9 | |
| no | 2 | 2 | 0 | 22 | |
| TN | + | 34/16 | 33/15.7 | 15 | 126/88.1 |
| − | 2 | 3/3.3 | 0 | 37/46.4 | |
| no | 28/14.77 | 28/13.7 | 9 | 93/79.9 | |
| FP | + | 0 | 1/0.3 | 0 | 16/11.9 |
| − | 12/14 | 11/12.7 | 2 | 36/53.6 | |
| no | 4/1.23 | 4/2.3 | 4 | 22/20.1 | |
| FN | + | 6/8 | 6/8 | 0 | 20/43.5 |
| − | 4/1.2 | 4/1.2 | 4 | 26/14.1 | |
| no | 6 | 6 | 2 | 28 | |
| Sensitivity | + | 0 | 0 | NA a | 0.13 |
| − | 0.85 | 0.85 | 0.69 | 0.717 | |
| no | 0.25 | 0.25 | 0 | 0.44 | |
| Specificity | + | 1 | 0.97/0.98 | 1 | 0.89/0.88 |
| − | 0.14/0.13 | 0.21 | 0 | 0.51/0.46 | |
| no | 0.88/0.92 | 0.88/0.86 | 0.69 | 0.81/0.80 | |
| PPV | + | 0 | 0 | NA | 0.16/0.35 |
| − | 0.65/0.33 | 0.67/0.35 | 0.82 | 0.65/0.40 | |
| no | 0.33/0.62 | 0.33/0.47 | 0 | 0.5/0.52 | |
| NPV | + | 0.85/0.67 | 0.85/0.66 | 1 | 0.86/0.67 |
| − | 0.33/0.62 | 0.43/0.73 | 0 | 0.59/0.77 | |
| no | 0.82/0.71 | 0.83/0.70 | 0.82 | 0.77/0.74 | |
| GC2 | 0.13/0.68 | 0.09/0.54 | NA | 0.05/0.22 | |
| CPR | 0.6/0.36 | 0.6/0.37 | 0.6 | 0.55/0.43 | |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, Y.; Urolagin, S.; Niroula, A.; Ding, X.; Shen, B.; Vihinen, M. PON-tstab: Protein Variant Stability Predictor. Importance of Training Data Quality. Int. J. Mol. Sci. 2018, 19, 1009. https://doi.org/10.3390/ijms19041009
Yang Y, Urolagin S, Niroula A, Ding X, Shen B, Vihinen M. PON-tstab: Protein Variant Stability Predictor. Importance of Training Data Quality. International Journal of Molecular Sciences. 2018; 19(4):1009. https://doi.org/10.3390/ijms19041009
Chicago/Turabian StyleYang, Yang, Siddhaling Urolagin, Abhishek Niroula, Xuesong Ding, Bairong Shen, and Mauno Vihinen. 2018. "PON-tstab: Protein Variant Stability Predictor. Importance of Training Data Quality" International Journal of Molecular Sciences 19, no. 4: 1009. https://doi.org/10.3390/ijms19041009