Resistance to Anti-Angiogenic Therapy in Cancer—Alterations to Anti-VEGF Pathway


1
Department of Surgery, Graduate School of Medicine, Kyoto University, Kyoto 606-8507, Japan
2
Moores Cancer Center, University of California San Diego, San Diego, CA 92093, USA
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2018, 19(4), 1232; https://doi.org/10.3390/ijms19041232
Submission received: 24 March 2018
/
Revised: 12 April 2018
/
Accepted: 15 April 2018
/
Published: 18 April 2018
(This article belongs to the Special Issue Alterations to Signalling Pathways in Cancer Cells 2018)
{kind=link}
{kind=link}
Abstract
:Anti-angiogenic therapy is one of the promising strategies for many types of solid cancers. Bevacizumab (Avastin), a recombinant humanized monoclonal antibody of vascular endothelial growth factor (VEGF) A, was approved for the first time as an anti-angiogenic drug for the treatment of metastatic colorectal cancer (CRC) by the Food and Drug Administration (FDA) in 2004. In addition, the other VEGF pathway inhibitors including small molecule tyrosine kinase inhibitors (sunitinib, sorafenib, and pazopanib), a soluble VEGF decoy receptor (aflibercept), and a humanized monoclonal antibody of VEGF receptor 2 (VEGFR2) (ramucirumab) have been approved for cancer therapy. Although many types of VEGF pathway inhibitors can improve survival in most cancer patients, some patients have little or no beneficial effect from them. The primary or acquired resistance towards many oncological drugs, including anti-VEGF inhibitors, is a common problem in cancer treatment. This review summarizes the proposed alternative mechanisms of angiogenesis other than the VEGF pathway. These mechanisms are involved in the development of resistance to anti-VEGF therapies in cancer patients.
1. Introduction
In 1971, Judah Folkman for the first time highlighted that angiogenesis is an essential process for the growth and proliferation of solid tumors [1], which resulted in a notion that anti-angiogenesis might be a potential therapeutic approach against various cancers [1,2,3]. Thereafter, several molecules were identified as angiogenic factors, such as acidic and basic fibroblast growth factors (aFGF and bFGF), angiogenin, and transforming growth factor-α and -β (TGF-α and TGF-β). In 1989, vascular endothelial growth factor (VEGF) A was isolated and cloned [4,5], which led to great progress in understanding the angiogenic mechanisms. VEGFA is a growth/survival factor for endothelial cells and binds to two receptor tyrosine kinases (RTKs), VEGF receptor (VEGFR) 1 and 2 [6]. VEGFA has six isoforms—namely, VEGF121, VEGF145, VEGF165, VEGF183, VEGF189, and VEGF206—that are the resultant variants of alternative splicing of a single, 8-exon VEGFA gene [6] (Figure 1). Among these, VEGF121 and VEGF165 are the two major isoforms. VEGF121 binds solely to VEGFR1 and VEGFR2, whereas VEGF165 binds to the co-receptors neuropilin (NRP)-1 and -2 via its basic sequence encoded in exon 7, which enhances the binding of VEGF165 to VEGFR2 and promotes its bioactivity [7]. As for the receptors, VEGFR2 is expressed on endothelial cells whereas VEGFR1 is expressed on endothelial cells and other cell types, such as smooth muscle cells, fibroblasts, myeloid progenitors, macrophages, and various types of cancer cells [8]. Although the angiogenic effect of VEGFA is predominantly mediated by VEGFR2, VEGFR1 signaling plays a role in tumor cell survival and growth [9,10,11].
In 1993, a monoclonal neutralizing antibody against VEGFA was reported to inhibit tumor growth in the in vivo xenograft model [12]. This idea led to the development of bevacizumab (Avastin), a recombinant humanized monoclonal antibody specific to VEGFA. In 2004, bevacizumab was approved by the U.S. Food and Drug Administration (FDA) for the treatment of metastatic colorectal cancer (CRC) [13]. In addition, various other inhibitors of the VEGF signaling pathway have been developed. The RTK inhibitors (RTKIs) sunitinib (Sutent) [14], sorafenib (Nexavar) [15], and pazopanib (Votrient) [16] are currently approved for the treatment of various types of cancers. Aflibercept (Zaltrap), a soluble recombinant fusion protein that consists of the extracellular domains of VEGFR1 and VEGR2 fused to the Fc portion of human IgG1, neutralizes VEGFA, VEGFB, and placental growth factor (PlGF), and was approved in 2012 by the FDA for the treatment of metastatic CRC [17]. Ramucirumab (Cyramza) is also a monoclonal antibody that binds VEGFR2 to block the VEGF signaling pathway and has been approved by the FDA for the treatment of several types of solid cancers [18].
Despite a large amount of promising data from animal experiments, simply blocking the VEGF signaling pathway by an anti-VEGF monotherapy appears to be ineffective for advanced cases in the clinical setting [19]. This primary or de novo treatment resistance is a common problem in the treatment of cancer patients, even with the most recent sophisticated drugs.
Resistance to anti-VEGF therapy often occurs owing to the escape mechanisms of the angiogenic process through the activation of signaling pathways other than the VEGF pathway. Moreover, it has been proposed that the inhibition of VEGFR by RTKI or an antibody promotes tumor invasiveness and metastasis [20,21]. In this review, we summarize the proposed alternative pathways that are involved in the emergence of resistance to anti-VEGF therapy in cancer.
2. Alternative Angiogenic Pathways to the VEGF Pathway That Influence Anti-VEGF Treatment
Although the VEGF pathway induces the most profound angiogenesis during tumor formation, the prediction of the existence of alternative angiogenic pathways is relevant as we observe various anti-VEGF resistant cancers. In this section, we discuss the potential angiogenic factors that are proposed to contribute to the escape from anti-VEGF treatment (Figure 2, right).
2.1. Angiopoietin-2 (Ang2)
Angiopoietin–Tie signaling is a vascular-specific RTK pathway that is essential for blood vessel development, remodeling, and regulation of vascular permeability. Angiopoietin-1 (Ang1) was initially identified as an agonist of the Tie2 receptor, activating this pathway; angiopoietin-2 (Ang2) was identified as an antagonist of the Tie2 receptor [22]. Ang1 affords maturation or stabilization of blood vessels through Tie2, which can be blocked by Ang2, while such inhibition by Ang2 results in the remodeling or initiation of vascular sprouts in the context of VEGF exposure [23]. The Tie2 receptor is expressed on endothelial cells of the blood and lymphatic vessels, the M2 subpopulation of monocytes/macrophages, and hematopoietic stem cells. The Tie2 receptor regulates downstream signaling pathways such as phosphoinositide 3-kinase (PI3K)/Akt and/or mitogen-activated protein kinase (MAPK)/extracellular-related kinase (ERK) (also known as Ras/Raf/MEK/ERK) [24,25]. The Ang/Tie system plays a crucial role in the pathophysiology of the tumor vasculature, as well as normal vasculature, and Ang2 expression is found to be upregulated in many types of cancers [26,27,28,29,30]. Moreover, CRC patients with high serum Ang2 levels exhibited poor response to bevacizumab treatment, suggesting that Ang2 plays an important role in the resistance mechanism against anti-VEGF therapy [31]. In preclinical settings, dual blockade of VEGF and Ang2 suppressed the revascularization and tumor progression of anti-VEGF therapy-resistant cancers [32,33,34,35]. Some clinical trials are underway and their results regarding the efficacy of vanucizumab, a humanized bi-specific monoclonal antibody against two different targets—VEGF-A and Ang2—are now pending [36,37].
2.2. Bombina Variegata Peptide 8 (Bv8)
Bv8, also known as prokineticin-2, was initially purified from the skin secretion of the yellow-bellied toad, Bombina variegata. Bv8 and endocrine gland-derived VEGF (EG-VEGF, also known as prokineticin-1) belong to the same family of proteins and bind to prokineticin receptor (PROKR)-1 and -2 (G-protein coupled receptors) to activate the downstream MAPK/ERK pathway [38,39]. They induce angiogenesis by stimulating proliferation, migration, and survival of vascular endothelial cells [40]. Although Bv8 is mainly expressed in the testis, it is also expressed in bone marrow. Bv8 is highly expressed in the neutrophil population under the control of granulocyte colony-stimulating factor (G-CSF) stimulated by signal transducer and activator of transcription 3 (STAT3) signaling in the bone marrow or inside the tumor microenvironment [41,42]. It is well established that inflammation strongly promotes the initiation of several types of cancers, such as CRC following inflammatory bowel disease, liver cancer following hepatitis, gastric cancer following gastritis, esophageal cancer following esophagitis, and so forth. It was reported that the inflammatory cytokine interleukin-17 (IL-17)-producing T helper type 17 (TH17) cells initiate a paracrine network to confer resistance to anti-VEGF therapy [43]. G-CSF and its upstream cytokine IL-17A are the main players of inflammation; thus, it is conceivable that G-CSF is abundant in tumor tissues [44]. Indeed, serum G-CSF levels in CRC patients are higher than those in healthy volunteers and are associated with the stage of tumor progression [45,46].
It is known that anti-VEGF therapy induces tumor-associated neutrophil (TAN) infiltration into the tumor microenvironment, which would be a predictive biomarker for patients treated with bevacizumab [47,48,49,50,51,52]. These observations led to the discovery that neutrophils provide the resistance mechanisms against anti-VEGF therapy [53]. In brief, tumor cells or tumor tissues secrete G-CSF during the inflammatory conditions and/or in the cytokine-rich environment through nuclear factor-κB (NF-κB) signaling driven by IL-17A [43]. It is also known that the oncogenic Ras pathway promotes G-CSF expression through the activation of its downstream MEK/ERK signaling [54]. Next, G-CSF promotes the recruitment of neutrophils into the tumor site, stimulates them to express Bv8, and promotes angiogenesis, which results in the escape from anti-VEGF therapy [55,56,57]. PKRA7, a small molecule Bv8 antagonist, could suppress tumor formation in vivo by inhibiting angiogenesis and infiltration of myeloid-derived cells [58]. Neutralization of Bv8 and its upstream G-CSF by using monoclonal antibodies was also effective in tumor suppression [41]. To date, effective regimens using Bv8 inhibitors in combination with or without other anti-angiogenic reagents are pending in clinical trials.
2.3. Fibroblast Growth Factor (FGF)
The FGF family consists of 22 members. Among them, 18 are secretory proteins that bind to four types of RTK–FGF receptor (FGFR), i.e., FGFR1–4, whereas four of them are intracellular non-signaling proteins that serve as cofactors of voltage-gated sodium channels [59]. FGF/FGFR signaling is essential in the earliest stage of embryonic development and in the maintenance of adult tissues by stimulating cell proliferation, survival, migration, differentiation, and metabolism of the target cells. Binding of FGF to FGFR tyrosine kinase activates the downstream pathways such as MAPK/ERK, PI3K/Akt, STAT, and/or diacylglycerol (DAG)/protein kinase C (PKC) and the inositol triphosphate (IP3)-Ca2+ signaling cascade via phospholipase-Cγ (PLCγ) activation [60,61,62,63]. In addition to normal tissue development and homeostasis, FGF/FGFR signaling plays crucial roles in cancer development and progression [64,65]. FGFR is expressed on cancer cells as well as several types of stromal cells, such as cancer-associated fibroblasts (CAFs), endothelial cells, and tumor-infiltrating myeloid cells [65]. Angiogenesis is one of the key mechanisms of FGF/FGFR signaling during tumor progression, and the upregulation of FGF2 (also known as bFGF) is observed in anti-VEGF-resistant tumors, especially in tumors that are exposed to a hypoxic environment [66,67,68,69]. Several preclinical studies demonstrated the benefit of dual blockade of VEGF and FGF signaling pathways during cancer treatment [66,70,71,72,73]. These data encouraged us to further study the FGF/FGFR inhibitors against anti-VEGF-resistant tumors. Unfortunately, so far, small molecule tyrosine kinase inhibitors of VEGF, as well as FGF receptors (dovitinib used to inhibit VEGFR and FGFR; and nintedanib used to inhibit VEGFR, FGFR, and platelet-derived growth factor receptor (PDGFR)) were not effective in treating cancer patients after recurrence following anti-VEGF therapy [74,75].
2.4. Interleukin-1 (IL-1)
IL-1 is a family of 11 cytokines that affect tumor progression, as well as inflammatory processes. It includes receptor agonists (IL-1α, IL-1β, IL-18, IL-33, IL-36a, IL-36b, IL-36γ, and IL-37) and antagonists (IL-1Ra, IL-36Ra, and IL-38). Among these 11 cytokines, IL-1α and IL-1β induce the ability of tumor cells to initiate and complete the angiogenic process [76]. IL-1α and IL-1β bind to the same receptor, i.e., type 1 IL-1 receptor (IL-1R1), to activate the downstream signaling, whereas IL-1Ra inhibits IL-1R1 in a competitive binding manner. They initially bind to the extracellular chain of IL-1R1 to recruit its co-receptor IL-1 receptor accessory protein (IL-1RAcP) that is needed to activate the signal transduction of NF-κB, c-Jun N-terminal kinase (JNK), and p38 MAPK [77]. IL-1β was reported to induce the in vivo production of angiogenic factors, such as hypoxia-inducible factor 1α (HIF-1α), VEGF, and C-X-C motif chemokine (CXC) ligand 2 (CXCL2), which results in the rapid growth of tumor cells accompanied by hyperneovascularization [78,79]. IL-1α and IL-1β were upregulated in a mouse model with pancreatic cancer resistant to anti-VEGF therapy, suggesting that they play an important role in the resistance mechanism to anti-VEGF therapy [80]. In vivo experiments demonstrated that the neutralization of IL-1, as well as other candidate molecules such as CXC receptor (CXCR) 1/2 and TGF-β signaling, abrogated resistance to anti-VEGF therapy in a murine model of pancreatic cancer [81]. Clinical trials for dual blockade of IL-1 and VEGF signaling pathways are still pending.
2.5. Platelet-Derived Growth Factor (PDGF)
During embryonic development and tissue repair, the PDGF family members play important roles in cell growth, survival, and motility of mesenchymal cells and other cell types [82]. The PDGF family consists of homodimers of PDGF-AA, -BB, -CC, -DD, and the heterodimer of PDGF-AB. They bind to tyrosine kinase PDGFR, which consists of α and β isoforms, and they dimerize upon binding to the ligand dimers to activate downstream signal transduction, such as PI3K and PLCγ [83]. It is well known that a constitutively active PDGFR mutation contributes to the formation of a gastrointestinal stromal tumor (GIST). Moreover, the overactivation of PDGF signaling in the tumor microenvironment promotes tumor growth through angiogenesis [84]. Among the PDGF family members, PDGF-C was upregulated in CAFs infiltrating into anti-VEGF-resistant tumors in vivo [85]. In this model, a PDGF-C neutralizing antibody suppressed CAF-mediated tumor progression, indicating an additional effect upon anti-VEGF antibody [85].
Sunitinib is a multi-tyrosine kinase inhibitor that blocks VEGFR and PDGFR, as well as c-Kit, fms-like tyrosine kinase 3 (FLT-3), and colony stimulating factor-1 receptor (CSF-1R) [86]. In 2006, sunitinib was first approved by the FDA for the treatment of imatinib-resistant GIST (second-line setting) and metastatic renal cell carcinoma (RCC) (first-line setting); and later, in 2017, its application was expanded to the adjuvant treatment of RCC [87,88]. Despite the success of the dual blockade of VEGFR and PDGFR by sunitinib, a combination strategy using bevacizumab (to block VEGF signaling) and imatinib (to block PDGF signaling) was not effective but was toxic during RCC treatment [89]. Further investigation is required to clearly understand the necessity of dual blockade of these pathways in clinical settings.
2.6. Placental Growth Factor (PlGF)
PlGF is a member of the VEGF subfamily and binds to VEGFR1 and its co-receptors NRP-1 and -2, but not VEGFR2 [90,91]. PlGF stimulates growth, survival, and migration of endothelial cells, macrophages, and bone marrow progenitors, as well as tumor cells via VEGFR1 and its downstream PI3K/Akt and p38 MAPK pathways independent of VEGFA signaling [92,93]. Several observations have reported the upregulation of PlGF in patients treated with anti-VEGF therapy, suggesting that PlGF might be a therapeutic target for anti-angiogenic treatment-resistant tumors [68,94,95,96]. It was reported that PlGF knockout (Pgf−/−) mice exhibited normal embryonic angiogenesis and that Pgf−/− mice subjected to ischemia, wound healing, or tumor burden conditions exhibited impaired pathological angiogenesis [97]. These findings led to the idea that PlGF blockade might inhibit pathological angiogenesis without affecting healthy blood vessels [98]. Unfortunately, so far, anti-PlGF neutralizing antibodies in combination with anti-VEGF antibodies appear to have minimal effect on tumor suppression in vivo [99]. Moreover, its phase I clinical study in combination with bevacizumab demonstrated no improvement in recurrent glioblastoma patients compared to single-agent bevacizumab treatment, although the anti-PlGF antibody toxicity was acceptable and manageable [100].
In 2012, aflibercept (also known as ziv-aflibercept in the U.S.), a soluble VEGF decoy receptor, was approved by the FDA for the treatment of metastatic CRC in combination with 5-fluorouracil, leucovorin, and irinotecan [101]. It is a recombinant protein that consists of the extracellular domain of VEGFR1 and 2 and the Fc portion of human IgG1. Owing to its structure, ziv-aflibercept neutralizes both VEGF and PlGF [102]. In patient-derived xenograft (PDX) models, ziv-aflibercept exhibited higher tumor suppressive activity than bevacizumab [103]. Further evaluation regarding the anti-PlGF activity in addition to anti-VEGF is needed in clinical settings.
2.7. Transforming Growth Factor-β (TGF-β) Signaling
TGF-β signaling is a highly conserved pathway that regulates several cellular processes including growth, differentiation, and apoptosis [104]. The TGF-β superfamily consists of two major branches: TGF-β/Activin and bone morphogenetic protein (BMP). Upon ligand binding, type II receptors activate type I receptors to initiate Smad transcription factors by phosphorylating receptor-regulated Smads (R-Smads, Smad2/3 in the TGF-β/Activin pathway, and Smad1/5 in the BMP pathway), which form a complex with the common partner Smad (Co-Smad, Smad4), and work as transcription factors [105]. TGF-β1 induces angiogenesis either directly or by activating fibroblasts to produce extracellular matrix (ECM) adhesion and stimulating the tube formation of endothelial cells [106,107,108]. Although TGF-β signaling causes tumor suppressive effects during the early stage, it switches toward malignant conversion and tumor progression at later stages [109,110]. Many types of tumor tissues express higher levels of TGF-β compared to the adjacent normal tissues, and its expression levels are correlated with patient survival [111,112,113]. Anti-VEGF therapy-resistant tumors sometimes exhibit high levels of TGF-β1 expression, suggesting that it might play an important role in the acquired resistance to anti-angiogenic therapy [114]. Despite the abundant evidence showing the angiogenic function of TGF-β signaling and the synergistic effect of TGF-β and VEGF signaling in cancer progression, there seem to be few clinical trials showing the combined effect of blocking both TGF-β and VEGF signaling pathways.
3. Phenotypical Changes of Tumor Cells during Anti-Angiogenic Therapy
Anti-angiogenic therapy induces vascular regression, which leads to intratumoral hypoxia and selection of more invasive cancer cells that are resistant to anti-angiogenic therapy. It is well known that hypoxic conditions induce upregulation of VEGF expression at a transcriptional level through its upstream transcription factor HIF-1 [115]. This finding indicates that the anti-angiogenic therapy is ineffective without blocking VEGF signaling. Moreover, HIF-1 was proposed to have several functions that promote cancer cell survival in the hypoxic environment. In this section, we discuss the proposed tumor factors that are altered during the development of resistance to anti-angiogenic therapy in cancers (Figure 2, left).
3.1. Hepatocyte Growth Factor (HGF)/Tyrosine Protein Kinase Met (c-MET) Pathway
c-MET signaling in the presence of its ligand HGF controls tumor growth and invasiveness by activating MAPK/ERK cascades, PI3K/Akt axis, STAT3 pathway, and/or NF-κB inhibitor-α kinase (IKK)—NF-κB complex [116,117,118]. It is one of the most investigated signaling pathways in anti-VEGF therapy-resistant tumors. In glioblastoma patients, c-MET expression was highly upregulated during the recurrence after bevacizumab treatment, which was not observed in patients without bevacizumab treatment [119]. It was reported that c-MET transcription is promoted under hypoxic conditions via the direct regulation of HIF-1 [116]. Moreover, VEGF was reported to negatively regulate c-MET activation, resulting in the direct suppression of tumor invasion in a mouse model of glioblastoma [120]. These findings suggest a hypothesis that the neutralization of VEGF by bevacizumab might promote c-MET protein expression and also remove suppression of c-MET phosphorylation through VEGF, resulting in the dual activation of c-MET signaling. Unfortunately, the c-MET inhibitor onartuzumab did not exhibit any clinical benefit when administered in combination with a bevacizumab regimen to advanced non-small cell lung cancer (NSCLC) patients [121].
3.2. Homeobox B9 (HOXB9)
HOX genes encode highly conserved transcription factors and play crucial roles in embryonic development and oncogenesis, as well as tumor suppression [122]. HOXB9 is one of the HOX superfamily members and is upregulated in many types of cancers [123,124,125,126]. It controls the expression of some angiogenic factors, such as angiopoietin-like 2 (Angptl2), CXCL1, IL8, and TGF-β1, which causes resistance to bevacizumab treatment in mouse xenograft models [123,127]. Importantly, HOXB9 protein expression might be a predictive biomarker for metastatic CRC patients treated with bevacizumab [127]. Although the mechanism of how some of the tumors exhibit high HOXB9 expression remains unidentified, HOXB9 silencing in the bevacizumab-resistant xenograft model significantly decreased the expression of alternative angiogenic factors, causing the model to become sensitive to bevacizumab, and resulting in prolonged survival in vivo [127]. Further clinical studies are needed to validate whether HOXB9 can be a potential therapeutic target for anti-VEGF therapy-resistant tumors.
3.3. Integrin
Integrins are transmembrane receptors that play important roles in cell–cell and cell–ECM adhesion. They are heterodimers formed by the combination of α and β subunits. Upon binding to ECM as their ligand, they induce signal transduction pathways that mediate cytoskeletal organization, cell cycle regulation, cell survival, and proliferation under both normal and pathological states via MAPK/ERK and JNK pathways [128]. Normal host cells in the tumor microenvironment express integrins, which promote angiogenesis and lymphangiogenesis [129,130]. In addition to the host cells in the tumor microenvironment, tumor cells also exhibit high expression of integrins during malignant progression, and their expression levels are correlated with disease progression and poor survival of patients [131,132,133]. Among the members, β1 integrin is implicated in resistance to cancer treatment [134,135,136]. Indeed, in some contexts, β1 integrin is upregulated in the clinical specimens of bevacizumab-resistant glioblastomas [137]. HIF-1α, induced by the hypoxic microenvironment, generated during anti-angiogenic therapy stimulates β1 integrin expression, which interacts with c-MET signaling and results in an enhancement of tumor cell invasiveness [138,139,140]. Preclinical studies of glioblastoma xenograft models in vivo demonstrated the advantage of β1 integrin inhibition in bevacizumab-resistant tumors, as well as non-resistant tumors [138,141].
3.4. Intracellular Cell Adhesion Molecule 1 (ICAM-1)
ICAM-1, also known as CD54, was reported to be overexpressed in bevacizumab-resistant glioblastomas in a mouse xenograft model [142]. ICAM-1 plays a key role as an adhesion molecule by binding to two types of integrins: lymphocyte function-associated antigen-1 (LFA-1, also known as CD11a/CD18) and macrophage antigen-1 (Mac-1, also known as CD11b/CD18) [143]. When the glioma stem cell line GSC11 was subjected to hypoxic conditions, HIF-1-induced phosphorylated STAT3 activated ICAM-1 transcription and promoted macrophage infiltration into the tumor tissues [142]. When ICAM-1 expression in cancer cells was knocked down by shRNA, tumor growth and invasion were significantly suppressed and mice implanted with these cells exhibited improved survival [142].
3.5. Macrophage Migration Inhibitory Factor (MIF)
MIF is classified as an inflammatory cytokine that regulates macrophage function through the suppression of their anti-inflammatory activity. Tumor-associated macrophages (TAMs), mainly M2-polarized macrophages, promote tumor progression by stimulating angiogenesis and tumor cell migration/invasion, as well as suppressing tumor immunity [144]. In the tissue specimens of bevacizumab-resistant glioblastoma patients, MIF expression was decreased and TAM infiltration was increased compared to those in bevacizumab-sensitive ones [145]. As VEGF increases MIF production in a VEGFR-dependent manner, inhibition of the VEGF pathway directly depletes MIF expression, resulting in TAM recruitment and M2 polarization in bevacizumab-resistant glioblastoma patients. Glioblastoma xenograft tumors transduced with MIF expression grew slowly and exhibited low TAM infiltration in vivo [145]. The application of this target in clinical settings is still pending.
4. Discussion
The tumor microenvironment is as important as the tumor cells and is a major component of tumor tissues. It consists of normal host immune cells, bone marrow-derived inflammatory cells, blood vessels, lymphatic vessels, fibroblasts, and ECM. Neutralization of VEGF by using bevacizumab is a pioneering approach to targeting the tumor microenvironment during cancer therapy. The recent success of cancer immunotherapy using immune checkpoint inhibitors is also designed to target the tumor microenvironment. For example, nivolumab, pembrolizumab, and pidilizumab are against programmed cell death protein 1 (PD-1), atezolizumab and avelumab are against programmed death-ligand 1 (PD-L1), and ipilimumab is against cytotoxic T-lymphocyte-associated protein 4 (CTLA-4). As described above, host immune cells such as TANs and TAMs contribute to some of the proposed mechanisms of anti-VEGF therapy resistance [47,48,52,145]. Infiltration of CD8+ cytotoxic T-lymphocytes (CTLs), also known as “immunoscore,” is a good prognostic biomarker for cancer patients [146,147,148]. Similarly, RCC specimens treated with anti-angiogenic therapy exhibited infiltration of CD4+ and forkhead box P3 (FOXP3)+ regulatory T cells (T-reg). A high T-reg infiltration has a significant correlation with poor overall survival [149]. These findings suggest the importance of combination therapy using anti-angiogenic drugs and immune checkpoint inhibitors. Currently, several clinical trials are ongoing, which might lead to a new era of anti-angiogenic therapy [150].
Even after 10 years of approval by the FDA of the first anti-VEGF drug, i.e., bevacizumab, resistance to anti-VEGF therapy remains a challenge in the treatment of cancer patients. So far, the mechanisms of resistance development are not completely unveiled. In addition to the immune checkpoint inhibitors, other potential therapeutic agents appear to be available that need to be clinically validated as treatment strategies for anti-VEGF therapy-resistant tumors.
Acknowledgments
This work was supported by grants from the Ministry of Education, Culture, Sports, Science and Technology of Japan (to K. Kawada), and from Project Mirai Cancer Research Grants (to K. Kawada).
Author Contributions
Yoshiro Itatani and Kenji Kawada wrote the manuscript. Takemasa Yamamoto and Yoshiharu Sakai contributed the conception and the design of the manuscript. All authors reviewed the manuscript for important intellectual content.
Conflicts of Interest
No potential conflicts of interest exist.
Abbreviations
| Ang2 | angiopoietin-2 |
| BMP | bone morphogenetic protein |
| Bv8 | Bombina variegata peptide 8 |
| CRC | colorectal cancer |
| CSF-1R | colony-stimulating factor-1 receptor |
| CTL | cytotoxic T-lymphocyte |
| CTLA-4 | cytotoxic T-lymphocyte-associated protein 4 |
| CXC | C-X-C motif chemokine |
| CXCL | CXC ligand |
| CXCR | CXC receptor |
| DAG | diacylglycerol |
| ECM | extracellular matrix |
| EGFL | epidermal growth factor-like domain |
| EG-VEGF | endocrine gland-derived vascular endothelial growth factor |
| ERK | extracellular-related kinase |
| FDA | Food and Drug Administration |
| FGF | fibroblast growth factor |
| FGFR | FGF receptor |
| FLT-3 | fms-like tyrosine kinase 3 |
| FOXP3 | forkhead box P3 |
| G-CSF | granulocyte colony-stimulating factor |
| GIST | gastrointestinal stromal tumor |
| HIF-1 | hypoxia-inducible factor 1 |
| HOX | homeobox |
| IKK | nuclear factor-κB inhibitor-α kinase |
| IL | interleukin |
| IL-1R1 | type 1 IL-1 receptor |
| IL-1RAcP | IL-1 receptor accessory protein |
| IP3 | inositol triphosphate |
| LFA-1 | lymphocyte function-associated antigen-1 |
| JNK | c-Jun N-terminal kinase |
| MAPK | mitogen-activated protein kinase |
| Mac-1 | macrophage antigen-1 |
| MIF | macrophage migration inhibitory factor |
| NF-κB | nuclear factor-κB |
| NRP | neuropilin |
| NSCLC | non-small cell lung cancer |
| PD-1 | programmed cell death protein 1 |
| PD-L1 | programmed death-ligand 1 |
| PDGF | platelet-derived growth factor |
| PDGFR | platelet-derived growth factor receptor |
| PI3K | phosphoinositide 3-kinase |
| PKC | protein kinase C |
| PLC γ | phospholipase C γ |
| PlGF | placental growth factor |
| PROKR | prokineticin receptor |
| RCC | renal cell carcinoma |
| RTK | receptor tyrosine kinase |
| RTKI | RTK inhibitor |
| STAT | signal transducer and activator of transcription |
| TAN | tumor-associated neutrophil |
| T-reg | regulatory T cells |
| TAM | tumor-associated macrophage |
| TGF | transforming growth factor |
| TH17 | T helper type 17 |
| VEGF | vascular endothelial growth factor |
| VEGFR | VEGF receptor |
References
- Folkman, J. Tumor angiogenesis: Therapeutic implications. N. Engl. J. Med. 1971, 285, 1182–1186. [Google Scholar] [PubMed]
- Folkman, J.; Klagsbrun, M. Angiogenic factors. Science 1987, 235, 442–447. [Google Scholar] [CrossRef] [PubMed]
- Folkman, J. Anti-angiogenesis: New concept for therapy of solid tumors. Ann. Surg. 1972, 175, 409–416. [Google Scholar] [CrossRef] [PubMed]
- Keck, P.J.; Hauser, S.D.; Krivi, G.; Sanzo, K.; Warren, T.; Feder, J.; Connolly, D.T. Vascular permeability factor, an endothelial cell mitogen related to PDGF. Science 1989, 246, 1309–1312. [Google Scholar] [CrossRef] [PubMed]
- Leung, D.W.; Cachianes, G.; Kuang, W.J.; Goeddel, D.V.; Ferrara, N. Vascular endothelial growth factor is a secreted angiogenic mitogen. Science 1989, 246, 1306–1309. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, N.; Gerber, H.P.; LeCouter, J. The biology of VEGF and its receptors. Nat. Med. 2003, 9, 669–676. [Google Scholar] [CrossRef] [PubMed]
- Soker, S.; Takashima, S.; Miao, H.Q.; Neufeld, G.; Klagsbrun, M. Neuropilin-1 is expressed by endothelial and tumor cells as an isoform-specific receptor for vascular endothelial growth factor. Cell 1998, 92, 735–745. [Google Scholar] [CrossRef]
- Fischer, C.; Mazzone, M.; Jonckx, B.; Carmeliet, P. FLT1 and its ligands VEGFB and PlGF: Drug targets for anti-angiogenic therapy? Nat. Rev. Cancer 2008, 8, 942–956. [Google Scholar] [CrossRef] [PubMed]
- Kerbel, R.S. Tumor angiogenesis. N. Engl. J. Med. 2008, 358, 2039–2049. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Hooper, A.T.; Zhong, Z.; Witte, L.; Bohlen, P.; Rafii, S.; Hicklin, D.J. The vascular endothelial growth factor receptor (VEGFR-1) supports growth and survival of human breast carcinoma. Int. J. Cancer 2006, 119, 1519–1529. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Zhong, Z.; Huber, J.; Bassi, R.; Finnerty, B.; Corcoran, E.; Li, H.; Navarro, E.; Balderes, P.; Jimenez, X.; et al. Anti-Vascular Endothelial Growth Factor Receptor-1 Antagonist Antibody as a Therapeutic Agent for Cancer. Clin. Cancer Res. 2006, 12, 6573–6584. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.J.; Li, B.; Winer, J.; Armanini, M.; Gillett, N.; Phillips, H.S.; Ferrara, N. Inhibition of vascular endothelial growth factor-induced angiogenesis suppresses tumour growth in vivo. Nature 1993, 362, 841–844. [Google Scholar] [CrossRef] [PubMed]
- Hurwitz, H.; Fehrenbacher, L.; Novotny, W.; Cartwright, T.; Hainsworth, J.; Heim, W.; Berlin, J.; Baron, A.; Griffing, S.; Holmgren, E.; et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N. Engl. J. Med. 2004, 350, 2335–2342. [Google Scholar] [CrossRef] [PubMed]
- Motzer, R.J.; Michaelson, M.D.; Redman, B.G.; Hudes, G.R.; Wilding, G.; Figlin, R.A.; Ginsberg, M.S.; Kim, S.T.; Baum, C.M.; DePrimo, S.E.; et al. Activity of SU11248, a multitargeted inhibitor of vascular endothelial growth factor receptor and platelet-derived growth factor receptor, in patients with metastatic renal cell carcinoma. J. Clin. Oncol. 2006, 24, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Escudier, B.; Eisen, T.; Stadler, W.M.; Szczylik, C.; Oudard, S.; Staehler, M.; Negrier, S.; Chevreau, C.; Desai, A.A.; Rolland, F.; et al. Sorafenib for treatment of renal cell carcinoma: Final efficacy and safety results of the phase III treatment approaches in renal cancer global evaluation trial. J. Clin. Oncol. 2009, 27, 3312–3318. [Google Scholar] [CrossRef] [PubMed]
- Sternberg, C.N.; Davis, I.D.; Mardiak, J.; Szczylik, C.; Lee, E.; Wagstaff, J.; Barrios, C.H.; Salman, P.; Gladkov, O.A.; Kavina, A.; et al. Pazopanib in locally advanced or metastatic renal cell carcinoma: Results of a randomized phase III trial. J. Clin. Oncol. 2010, 28, 1061–1068. [Google Scholar] [CrossRef] [PubMed]
- Van Cutsem, E.; Tabernero, J.; Lakomy, R.; Prenen, H.; Prausova, J.; Macarulla, T.; Ruff, P.; van Hazel, G.A.; Moiseyenko, V.; Ferry, D.; et al. Addition of aflibercept to fluorouracil, leucovorin, and irinotecan improves survival in a phase III randomized trial in patients with metastatic colorectal cancer previously treated with an oxaliplatin-based regimen. J. Clin. Oncol. 2012, 30, 3499–3506. [Google Scholar] [CrossRef] [PubMed]
- Wilke, H.; Muro, K.; Van Cutsem, E.; Oh, S.C.; Bodoky, G.; Shimada, Y.; Hironaka, S.; Sugimoto, N.; Lipatov, O.; Kim, T.Y.; et al. Ramucirumab plus paclitaxel versus placebo plus paclitaxel in patients with previously treated advanced gastric or gastro-oesophageal junction adenocarcinoma (RAINBOW): A double-blind, randomised phase 3 trial. Lancet Oncol. 2014, 15, 1224–1235. [Google Scholar] [CrossRef]
- Giantonio, B.J.; Catalano, P.J.; Meropol, N.J.; O’Dwyer, P.J.; Mitchell, E.P.; Alberts, S.R.; Schwartz, M.A.; Benson, A.B., 3rd. Bevacizumab in combination with oxaliplatin, fluorouracil, and leucovorin (FOLFOX4) for previously treated metastatic colorectal cancer: Results from the Eastern Cooperative Oncology Group Study E3200. J. Clin. Oncol. 2007, 25, 1539–1544. [Google Scholar] [CrossRef] [PubMed]
- Ebos, J.M.; Lee, C.R.; Cruz-Munoz, W.; Bjarnason, G.A.; Christensen, J.G.; Kerbel, R.S. Accelerated metastasis after short-term treatment with a potent inhibitor of tumor angiogenesis. Cancer Cell 2009, 15, 232–239. [Google Scholar] [CrossRef] [PubMed]
- Paez-Ribes, M.; Allen, E.; Hudock, J.; Takeda, T.; Okuyama, H.; Vinals, F.; Inoue, M.; Bergers, G.; Hanahan, D.; Casanovas, O. Antiangiogenic therapy elicits malignant progression of tumors to increased local invasion and distant metastasis. Cancer Cell 2009, 15, 220–231. [Google Scholar] [CrossRef] [PubMed]
- Maisonpierre, P.C.; Suri, C.; Jones, P.F.; Bartunkova, S.; Wiegand, S.J.; Radziejewski, C.; Compton, D.; McClain, J.; Aldrich, T.H.; Papadopoulos, N.; et al. Angiopoietin-2, a natural antagonist for Tie2 that disrupts in vivo angiogenesis. Science 1997, 277, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D. Signaling vascular morphogenesis and maintenance. Science 1997, 277, 48–50. [Google Scholar] [CrossRef] [PubMed]
- Eklund, L.; Kangas, J.; Saharinen, P. Angiopoietin–Tie signalling in the cardiovascular and lymphatic systems. Clin. Sci. 2017, 131, 87–103. [Google Scholar] [CrossRef] [PubMed]
- Brindle, N.P.; Saharinen, P.; Alitalo, K. Signaling and functions of angiopoietin-1 in vascular protection. Circ. Res. 2006, 98, 1014–1023. [Google Scholar] [CrossRef] [PubMed]
- Eklund, L.; Saharinen, P. Angiopoietin signaling in the vasculature. Exp. Cell Res. 2013, 319, 1271–1280. [Google Scholar] [CrossRef] [PubMed]
- Helfrich, I.; Edler, L.; Sucker, A.; Thomas, M.; Christian, S.; Schadendorf, D.; Augustin, H.G. Angiopoietin-2 levels are associated with disease progression in metastatic malignant melanoma. Clin. Cancer Res. 2009, 15, 1384–1392. [Google Scholar] [CrossRef] [PubMed]
- Sfiligoi, C.; de Luca, A.; Cascone, I.; Sorbello, V.; Fuso, L.; Ponzone, R.; Biglia, N.; Audero, E.; Arisio, R.; Bussolino, F.; et al. Angiopoietin-2 expression in breast cancer correlates with lymph node invasion and short survival. Int. J. Cancer 2003, 103, 466–474. [Google Scholar] [CrossRef] [PubMed]
- Moon, W.S.; Rhyu, K.H.; Kang, M.J.; Lee, D.G.; Yu, H.C.; Yeum, J.H.; Koh, G.Y.; Tarnawski, A.S. Overexpression of VEGF and angiopoietin 2: A key to high vascularity of hepatocellular carcinoma? Mod. Pathol. 2003, 16, 552–557. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, M.; Yamamoto, H.; Nagano, H.; Miyake, Y.; Sugita, Y.; Hata, T.; Kim, B.N.; Ngan, C.Y.; Damdinsuren, B.; Ikenaga, M.; et al. Hepatic expression of ANG2 RNA in metastatic colorectal cancer. Hepatology 2004, 39, 528–539. [Google Scholar] [CrossRef] [PubMed]
- Goede, V.; Coutelle, O.; Neuneier, J.; Reinacher-Schick, A.; Schnell, R.; Koslowsky, T.C.; Weihrauch, M.R.; Cremer, B.; Kashkar, H.; Odenthal, M.; et al. Identification of serum angiopoietin-2 as a biomarker for clinical outcome of colorectal cancer patients treated with bevacizumab-containing therapy. Br. J. Cancer 2010, 103, 1407–1414. [Google Scholar] [CrossRef] [PubMed]
- Rigamonti, N.; Kadioglu, E.; Keklikoglou, I.; Wyser Rmili, C.; Leow, C.C.; De Palma, M. Role of angiopoietin-2 in adaptive tumor resistance to VEGF signaling blockade. Cell Rep. 2014, 8, 696–706. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.L.; Cao, Z.A.; Pinzon-Ortiz, M.; Kendrew, J.; Reimer, C.; Wen, S.; Zhou, J.Q.; Tabrizi, M.; Emery, S.; McDermott, B.; et al. A human monoclonal anti-ANG2 antibody leads to broad antitumor activity in combination with VEGF inhibitors and chemotherapy agents in preclinical models. Mol. Cancer Ther. 2010, 9, 145–156. [Google Scholar] [CrossRef] [PubMed]
- Kienast, Y.; Klein, C.; Scheuer, W.; Raemsch, R.; Lorenzon, E.; Bernicke, D.; Herting, F.; Yu, S.; The, H.H.; Martarello, L.; et al. Ang-2-VEGF-A CrossMab, a novel bispecific human IgG1 antibody blocking VEGF-A and Ang-2 functions simultaneously, mediates potent antitumor, antiangiogenic, and antimetastatic efficacy. Clin. Cancer Res. 2013, 19, 6730–6740. [Google Scholar] [CrossRef] [PubMed]
- Scholz, A.; Harter, P.N.; Cremer, S.; Yalcin, B.H.; Gurnik, S.; Yamaji, M.; Di Tacchio, M.; Sommer, K.; Baumgarten, P.; Bahr, O.; et al. Endothelial cell-derived angiopoietin-2 is a therapeutic target in treatment-naive and bevacizumab-resistant glioblastoma. EMBO Mol. Med. 2016, 8, 39–57. [Google Scholar] [CrossRef] [PubMed]
- A Study of Vanucizumab (RO5520985) alone or in Combination with Atezolizumab in Participants with Locally Advanced or Metastatic Solid Tumors. Available online: https://clinicaltrials.gov/ct2/show/NCT01688206 (accessed on 5 April 2018).
- A Study Comparing the Efficacy and Safety of Vanucizumab and FOLFOX with Bevacizmab and FOLFOX in Participants with Untreated Metastatic Colorectal Cancer (McCAVE). Available online: https://clinicaltrials.gov/ct2/show/NCT02141295 (accessed on 5 April 2018).
- Evans, J.; Catalano, R.D.; Morgan, K.; Critchley, H.O.; Millar, R.P.; Jabbour, H.N. Prokineticin 1 signaling and gene regulation in early human pregnancy. Endocrinology 2008, 149, 2877–2887. [Google Scholar] [CrossRef] [PubMed]
- Martin, C.; Balasubramanian, R.; Dwyer, A.A.; Au, M.G.; Sidis, Y.; Kaiser, U.B.; Seminara, S.B.; Pitteloud, N.; Zhou, Q.Y.; Crowley, W.F. The Role of the Prokineticin 2 Pathway in Human Reproduction: Evidence from the Study of Human and Murine Gene Mutations. Endocr. Rev. 2011, 32, 225–246. [Google Scholar] [CrossRef] [PubMed]
- LeCouter, J.; Lin, R.; Tejada, M.; Frantz, G.; Peale, F.; Hillan, K.J.; Ferrara, N. The endocrine-gland-derived VEGF homologue Bv8 promotes angiogenesis in the testis: Localization of Bv8 receptors to endothelial cells. Proc. Natl. Acad. Sci. USA 2003, 100, 2685–2690. [Google Scholar] [CrossRef] [PubMed]
- Shojaei, F.; Wu, X.; Zhong, C.; Yu, L.; Liang, X.H.; Yao, J.; Blanchard, D.; Bais, C.; Peale, F.V.; van Bruggen, N.; et al. Bv8 regulates myeloid-cell-dependent tumour angiogenesis. Nature 2007, 450, 825–831. [Google Scholar] [CrossRef] [PubMed]
- Zhong, C.; Qu, X.; Tan, M.; Meng, Y.G.; Ferrara, N. Characterization and regulation of bv8 in human blood cells. Clin. Cancer Res. 2009, 15, 2675–2684. [Google Scholar] [CrossRef] [PubMed]
- Chung, A.S.; Wu, X.; Zhuang, G.; Ngu, H.; Kasman, I.; Zhang, J.; Vernes, J.M.; Jiang, Z.; Meng, Y.G.; Peale, F.V.; et al. An interleukin-17-mediated paracrine network promotes tumor resistance to anti-angiogenic therapy. Nat. Med. 2013, 19, 1114–1123. [Google Scholar] [CrossRef] [PubMed]
- Morris, K.T.; Khan, H.; Ahmad, A.; Weston, L.L.; Nofchissey, R.A.; Pinchuk, I.V.; Beswick, E.J. G-CSF and G-CSFR are highly expressed in human gastric and colon cancers and promote carcinoma cell proliferation and migration. Br. J. Cancer 2014, 110, 1211–1220. [Google Scholar] [CrossRef] [PubMed]
- Mroczko, B.; Groblewska, M.; Wereszczynska-Siemiatkowska, U.; Kedra, B.; Konopko, M.; Szmitkowski, M. The diagnostic value of G-CSF measurement in the sera of colorectal cancer and adenoma patients. Clin. Chim. Acta 2006, 371, 143–147. [Google Scholar] [CrossRef] [PubMed]
- Krzystek-Korpacka, M.; Diakowska, D.; Kapturkiewicz, B.; Bebenek, M.; Gamian, A. Profiles of circulating inflammatory cytokines in colorectal cancer (CRC), high cancer risk conditions, and health are distinct. Possible implications for CRC screening and surveillance. Cancer Lett. 2013, 337, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Roland, C.L.; Dineen, S.P.; Lynn, K.D.; Sullivan, L.A.; Dellinger, M.T.; Sadegh, L.; Sullivan, J.P.; Shames, D.S.; Brekken, R.A. Inhibition of vascular endothelial growth factor reduces angiogenesis and modulates immune cell infiltration of orthotopic breast cancer xenografts. Mol. Cancer Ther. 2009, 8, 1761–1771. [Google Scholar] [CrossRef] [PubMed]
- Piao, Y.; Liang, J.; Holmes, L.; Zurita, A.J.; Henry, V.; Heymach, J.V.; de Groot, J.F. Glioblastoma resistance to anti-VEGF therapy is associated with myeloid cell infiltration, stem cell accumulation, and a mesenchymal phenotype. Neuro Oncol. 2012, 14, 1379–1392. [Google Scholar] [CrossRef] [PubMed]
- Cetin, B.; Kaplan, M.A.; Berk, V.; Ozturk, S.C.; Benekli, M.; Isikdogan, A.; Ozkan, M.; Coskun, U.; Buyukberber, S. Prognostic factors for overall survival in patients with metastatic colorectal carcinoma treated with vascular endothelial growth factor-targeting agents. Asian Pac. J. Cancer Prev. 2012, 13, 1059–1063. [Google Scholar] [CrossRef] [PubMed]
- Botta, C.; Barbieri, V.; Ciliberto, D.; Rossi, A.; Rocco, D.; Addeo, R.; Staropoli, N.; Pastina, P.; Marvaso, G.; Martellucci, I.; et al. Systemic inflammatory status at baseline predicts bevacizumab benefit in advanced non-small cell lung cancer patients. Cancer Biol. Ther. 2013, 14, 469–475. [Google Scholar] [CrossRef] [PubMed]
- Dirican, A.; Varol, U.; Kucukzeybek, Y.; Alacacioglu, A.; Erten, C.; Somali, I.; Can, A.; Demir, L.; Bayoglu, I.V.; Akyol, M.; et al. Treatment of metastatic colorectal cancer with or without bevacizumab: Can the neutrophil/lymphocyte ratio predict the efficiency of bevacizumab? Asian Pac. J. Cancer Prev. 2014, 15, 4781–4786. [Google Scholar] [CrossRef] [PubMed]
- Passardi, A.; Scarpi, E.; Cavanna, L.; Dall’Agata, M.; Tassinari, D.; Leo, S.; Bernardini, I.; Gelsomino, F.; Tamberi, S.; Brandes, A.A.; et al. Inflammatory indexes as predictors of prognosis and bevacizumab efficacy in patients with metastatic colorectal cancer. Oncotarget 2016, 7, 33210–33219. [Google Scholar] [CrossRef] [PubMed]
- Shojaei, F.; Wu, X.; Malik, A.K.; Zhong, C.; Baldwin, M.E.; Schanz, S.; Fuh, G.; Gerber, H.P.; Ferrara, N. Tumor refractoriness to anti-VEGF treatment is mediated by CD11b+Gr1+ myeloid cells. Nat. Biotechnol. 2007, 25, 911–920. [Google Scholar] [CrossRef] [PubMed]
- Phan, V.T.; Wu, X.; Cheng, J.H.; Sheng, R.X.; Chung, A.S.; Zhuang, G.; Tran, C.; Song, Q.; Kowanetz, M.; Sambrone, A.; et al. Oncogenic RAS pathway activation promotes resistance to anti-VEGF therapy through G-CSF-induced neutrophil recruitment. Proc. Natl. Acad. Sci. USA 2013, 110, 6079–6084. [Google Scholar] [CrossRef] [PubMed]
- Qu, X.; Zhuang, G.; Yu, L.; Meng, G.; Ferrara, N. Induction of Bv8 expression by granulocyte colony-stimulating factor in CD11b+Gr1+ cells: Key role of Stat3 signaling. J. Biol. Chem. 2012, 287, 19574–19584. [Google Scholar] [CrossRef] [PubMed]
- Kowanetz, M.; Wu, X.; Lee, J.; Tan, M.; Hagenbeek, T.; Qu, X.; Yu, L.; Ross, J.; Korsisaari, N.; Cao, T.; et al. Granulocyte-colony stimulating factor promotes lung metastasis through mobilization of Ly6G+Ly6C+ granulocytes. Proc. Natl. Acad. Sci. USA 2010, 107, 21248–21255. [Google Scholar] [CrossRef] [PubMed]
- Shojaei, F.; Wu, X.; Qu, X.; Kowanetz, M.; Yu, L.; Tan, M.; Meng, Y.G.; Ferrara, N. G-CSF-initiated myeloid cell mobilization and angiogenesis mediate tumor refractoriness to anti-VEGF therapy in mouse models. Proc. Natl. Acad. Sci. USA 2009, 106, 6742–6747. [Google Scholar] [CrossRef] [PubMed]
- Curtis, V.F.; Wang, H.; Yang, P.; McLendon, R.E.; Li, X.; Zhou, Q.Y.; Wang, X.F. A PK2/Bv8/PROK2 antagonist suppresses tumorigenic processes by inhibiting angiogenesis in glioma and blocking myeloid cell infiltration in pancreatic cancer. PLoS ONE 2013, 8, e54916. [Google Scholar] [CrossRef] [PubMed]
- Ornitz, D.M.; Itoh, N. The Fibroblast Growth Factor signaling pathway. Wiley Interdiscip. Rev. Dev. Biol. 2015, 4, 215–266. [Google Scholar] [CrossRef] [PubMed]
- LaVallee, T.M.; Prudovsky, I.A.; McMahon, G.A.; Hu, X.; Maciag, T. Activation of the MAP Kinase Pathway by FGF-1 Correlates with Cell Proliferation Induction While Activation of the Src Pathway Correlates with Migration. J. Cell Biol. 1998, 141, 1647–1658. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Li, X.; Eswarakumar, V.P.; Seger, R.; Lonai, P. Fibroblast growth factor (FGF) signaling through PI 3-kinase and Akt/PKB is required for embryoid body differentiation. Oncogene 2000, 19, 3750–3756. [Google Scholar] [CrossRef] [PubMed]
- Hart, K.C.; Robertson, S.C.; Kanemitsu, M.Y.; Meyer, A.N.; Tynan, J.A.; Donoghue, D.J. Transformation and Stat activation by derivatives of FGFR1, FGFR3, and FGFR4. Oncogene 2000, 19, 3309–3320. [Google Scholar] [CrossRef] [PubMed]
- Chae, Y.K.; Ranganath, K.; Hammerman, P.S.; Vaklavas, C.; Mohindra, N.; Kalyan, A.; Matsangou, M.; Costa, R.; Carneiro, B.; Villaflor, V.M.; et al. Inhibition of the fibroblast growth factor receptor (FGFR) pathway: The current landscape and barriers to clinical application. Oncotarget 2017, 8, 16052–16074. [Google Scholar] [CrossRef] [PubMed]
- Turner, N.; Grose, R. Fibroblast growth factor signalling: From development to cancer. Nat. Rev. Cancer 2010, 10, 116–129. [Google Scholar] [CrossRef] [PubMed]
- Poon, R.T.; Fan, S.T.; Wong, J. Clinical implications of circulating angiogenic factors in cancer patients. J. Clin. Oncol. 2001, 19, 1207–1225. [Google Scholar] [CrossRef] [PubMed]
- Casanovas, O.; Hicklin, D.J.; Bergers, G.; Hanahan, D. Drug resistance by evasion of antiangiogenic targeting of VEGF signaling in late-stage pancreatic islet tumors. Cancer Cell 2005, 8, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Batchelor, T.T.; Sorensen, A.G.; di Tomaso, E.; Zhang, W.T.; Duda, D.G.; Cohen, K.S.; Kozak, K.R.; Cahill, D.P.; Chen, P.J.; Zhu, M.; et al. AZD2171, a pan-VEGF receptor tyrosine kinase inhibitor, normalizes tumor vasculature and alleviates edema in glioblastoma patients. Cancer Cell 2007, 11, 83–95. [Google Scholar] [CrossRef] [PubMed]
- Kopetz, S.; Hoff, P.M.; Morris, J.S.; Wolff, R.A.; Eng, C.; Glover, K.Y.; Adinin, R.; Overman, M.J.; Valero, V.; Wen, S.; et al. Phase II trial of infusional fluorouracil, irinotecan, and bevacizumab for metastatic colorectal cancer: Efficacy and circulating angiogenic biomarkers associated with therapeutic resistance. J. Clin. Oncol. 2010, 28, 453–459. [Google Scholar] [CrossRef] [PubMed]
- Mitsuhashi, A.; Goto, H.; Saijo, A.; Trung, V.T.; Aono, Y.; Ogino, H.; Kuramoto, T.; Tabata, S.; Uehara, H.; Izumi, K.; et al. Fibrocyte-like cells mediate acquired resistance to anti-angiogenic therapy with bevacizumab. Nat. Commun. 2015, 6, 8792. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.K.; Lee, M.E.; Lee, W.S.; Kim, J.M.; Park, K.H.; Kim, T.S.; Lee, K.Y.; Ahn, J.B.; Chung, H.C.; Rha, S.Y. Dovitinib (TKI258), a multi-target angiokinase inhibitor, is effective regardless of KRAS or BRAF mutation status in colorectal cancer. Am. J. Cancer Res. 2015, 5, 72–86. [Google Scholar] [PubMed]
- Andre, F.; Bachelot, T.; Campone, M.; Dalenc, F.; Perez-Garcia, J.M.; Hurvitz, S.A.; Turner, N.; Rugo, H.; Smith, J.W.; Deudon, S.; et al. Targeting FGFR with dovitinib (TKI258): Preclinical and clinical data in breast cancer. Clin. Cancer Res. 2013, 19, 3693–3702. [Google Scholar] [CrossRef] [PubMed]
- Allen, E.; Walters, I.B.; Hanahan, D. Brivanib, a dual FGF/VEGF inhibitor, is active both first and second line against mouse pancreatic neuroendocrine tumors developing adaptive/evasive resistance to VEGF inhibition. Clin. Cancer Res. 2011, 17, 5299–5310. [Google Scholar] [CrossRef] [PubMed]
- Burbridge, M.F.; Bossard, C.J.; Saunier, C.; Fejes, I.; Bruno, A.; Leonce, S.; Ferry, G.; Da Violante, G.; Bouzom, F.; Cattan, V.; et al. S49076 is a novel kinase inhibitor of MET, AXL, and FGFR with strong preclinical activity alone and in association with bevacizumab. Mol. Cancer Ther. 2013, 12, 1749–1762. [Google Scholar] [CrossRef] [PubMed]
- Semrad, T.J.; Kim, E.J.; Tanaka, M.S.; Sands, J.; Roberts, C.; Burich, R.A.; Li, Y.; Gandara, D.R.; Lara, P.; Mack, P.C. Phase II study of dovitinib in patients progressing on anti-vascular endothelial growth factor therapy. Cancer Treat. Res. Commun. 2017, 10, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Norden, A.D.; Schiff, D.; Ahluwalia, M.S.; Lesser, G.J.; Nayak, L.; Lee, E.Q.; Rinne, M.L.; Muzikansky, A.; Dietrich, J.; Purow, B.; et al. Phase II trial of triple tyrosine kinase receptor inhibitor nintedanib in recurrent high-grade gliomas. J. Neurooncol. 2015, 121, 297–302. [Google Scholar] [CrossRef] [PubMed]
- Voronov, E.; Shouval, D.S.; Krelin, Y.; Cagnano, E.; Benharroch, D.; Iwakura, Y.; Dinarello, C.A.; Apte, R.N. IL-1 is required for tumor invasiveness and angiogenesis. Proc. Natl. Acad. Sci. USA 2003, 100, 2645–2650. [Google Scholar] [CrossRef] [PubMed]
- Weber, A.; Wasiliew, P.; Kracht, M. Interleukin-1 (IL-1) pathway. Sci. Signal. 2010, 3. [Google Scholar] [CrossRef] [PubMed]
- Saijo, Y.; Tanaka, M.; Miki, M.; Usui, K.; Suzuki, T.; Maemondo, M.; Hong, X.; Tazawa, R.; Kikuchi, T.; Matsushima, K.; et al. Proinflammatory cytokine IL-1 beta promotes tumor growth of Lewis lung carcinoma by induction of angiogenic factors: In vivo analysis of tumor-stromal interaction. J. Immunol. 2002, 169, 469–475. [Google Scholar] [CrossRef] [PubMed]
- Thornton, R.D.; Lane, P.; Borghaei, R.C.; Pease, E.A.; Caro, J.; Mochan, E. Interleukin 1 induces hypoxia-inducible factor 1 in human gingival and synovial fibroblasts. Biochem. J. 2000, 350 Pt 1, 307–312. [Google Scholar] [CrossRef] [PubMed]
- Carbone, C.; Moccia, T.; Zhu, C.; Paradiso, G.; Budillon, A.; Chiao, P.J.; Abbruzzese, J.L.; Melisi, D. Anti-VEGF treatment-resistant pancreatic cancers secrete proinflammatory factors that contribute to malignant progression by inducing an EMT cell phenotype. Clin. Cancer Res. 2011, 17, 5822–5832. [Google Scholar] [CrossRef] [PubMed]
- Carbone, C.; Tamburrino, A.; Piro, G.; Boschi, F.; Cataldo, I.; Zanotto, M.; Mina, M.M.; Zanini, S.; Sbarbati, A.; Scarpa, A.; et al. Combined inhibition of IL1, CXCR1/2, and TGFbeta signaling pathways modulates in-vivo resistance to anti-VEGF treatment. Anticancer Drugs 2016, 27, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Heldin, C.-H. Targeting the PDGF signaling pathway in tumor treatment. Cell Commun. Signal. 2013, 11, 97. [Google Scholar] [CrossRef] [PubMed]
- Montmayeur, J.P.; Valius, M.; Vandenheede, J.; Kazlauskas, A. The platelet-derived growth factor beta receptor triggers multiple cytoplasmic signaling cascades that arrive at the nucleus as distinguishable inputs. J. Biol. Chem. 1997, 272, 32670–32678. [Google Scholar] [CrossRef] [PubMed]
- Francia, G.; Emmenegger, U.; Kerbel, R.S. Tumor-associated fibroblasts as “Trojan Horse” mediators of resistance to anti-VEGF therapy. Cancer Cell 2009, 15, 3–5. [Google Scholar] [CrossRef] [PubMed]
- Crawford, Y.; Kasman, I.; Yu, L.; Zhong, C.; Wu, X.; Modrusan, Z.; Kaminker, J.; Ferrara, N. PDGF-C mediates the angiogenic and tumorigenic properties of fibroblasts associated with tumors refractory to anti-VEGF treatment. Cancer Cell 2009, 15, 21–34. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R., Jr. Sunitinib: A VEGF and PDGF receptor protein kinase and angiogenesis inhibitor. Biochem. Biophys. Res. Commun. 2007, 356, 323–328. [Google Scholar] [CrossRef] [PubMed]
- Rock, E.P.; Goodman, V.; Jiang, J.X.; Mahjoob, K.; Verbois, S.L.; Morse, D.; Dagher, R.; Justice, R.; Pazdur, R. Food and Drug Administration drug approval summary: Sunitinib malate for the treatment of gastrointestinal stromal tumor and advanced renal cell carcinoma. Oncologist 2007, 12, 107–113. [Google Scholar] [CrossRef] [PubMed]
- FDA Approves Sunitinib Malate for Adjuvant Treatment of Renal Cell Carcinoma. Available online: https://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm585686.htm (accessed on 16 November 2017).
- Hainsworth, J.D.; Spigel, D.R.; Sosman, J.A.; Burris, H.A., 3rd; Farley, C.; Cucullu, H.; Yost, K.; Hart, L.L.; Sylvester, L.; Waterhouse, D.M.; et al. Treatment of advanced renal cell carcinoma with the combination bevacizumab/erlotinib/imatinib: A phase I/II trial. Clin. Genitourin. Cancer 2007, 5, 427–432. [Google Scholar] [CrossRef] [PubMed]
- Maglione, D.; Guerriero, V.; Viglietto, G.; Delli-Bovi, P.; Persico, M.G. Isolation of a human placenta cDNA coding for a protein related to the vascular permeability factor. Proc. Natl. Acad. Sci. USA 1991, 88, 9267–9271. [Google Scholar] [CrossRef] [PubMed]
- Park, J.E.; Chen, H.H.; Winer, J.; Houck, K.A.; Ferrara, N. Placenta growth factor. Potentiation of vascular endothelial growth factor bioactivity, in vitro and in vivo, and high affinity binding to Flt-1 but not to Flk-1/KDR. J. Biol. Chem. 1994, 269, 25646–25654. [Google Scholar] [PubMed]
- Kaplan, R.N.; Riba, R.D.; Zacharoulis, S.; Bramley, A.H.; Vincent, L.; Costa, C.; MacDonald, D.D.; Jin, D.K.; Shido, K.; Kerns, S.A.; et al. VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature 2005, 438, 820–827. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Tu, C.; Zhao, Y.; Liu, H.; Zhang, S. Placental growth factor enhances angiogenesis in human intestinal microvascular endothelial cells via PI3K/Akt pathway: Potential implications of inflammation bowel disease. Biochem. Biophys. Res. Commun. 2016, 470, 967–974. [Google Scholar] [CrossRef] [PubMed]
- Bagley, R.G.; Ren, Y.; Weber, W.; Yao, M.; Kurtzberg, L.; Pinckney, J.; Bangari, D.; Nguyen, C.; Brondyk, W.; Kaplan, J.; et al. Placental growth factor upregulation is a host response to antiangiogenic therapy. Clin. Cancer Res. 2011, 17, 976–988. [Google Scholar] [CrossRef] [PubMed]
- Willett, C.G.; Duda, D.G.; di Tomaso, E.; Boucher, Y.; Ancukiewicz, M.; Sahani, D.V.; Lahdenranta, J.; Chung, D.C.; Fischman, A.J.; Lauwers, G.Y.; et al. Efficacy, safety, and biomarkers of neoadjuvant bevacizumab, radiation therapy, and fluorouracil in rectal cancer: A multidisciplinary phase II study. J. Clin. Oncol. 2009, 27, 3020–3026. [Google Scholar] [CrossRef] [PubMed]
- Rini, B.I.; Michaelson, M.D.; Rosenberg, J.E.; Bukowski, R.M.; Sosman, J.A.; Stadler, W.M.; Hutson, T.E.; Margolin, K.; Harmon, C.S.; DePrimo, S.E.; et al. Antitumor activity and biomarker analysis of sunitinib in patients with bevacizumab-refractory metastatic renal cell carcinoma. J. Clin. Oncol. 2008, 26, 3743–3748. [Google Scholar] [CrossRef] [PubMed]
- Carmeliet, P.; Moons, L.; Luttun, A.; Vincenti, V.; Compernolle, V.; De Mol, M.; Wu, Y.; Bono, F.; Devy, L.; Beck, H.; et al. Synergism between vascular endothelial growth factor and placental growth factor contributes to angiogenesis and plasma extravasation in pathological conditions. Nat. Med. 2001, 7, 575–583. [Google Scholar] [CrossRef] [PubMed]
- Fischer, C.; Jonckx, B.; Mazzone, M.; Zacchigna, S.; Loges, S.; Pattarini, L.; Chorianopoulos, E.; Liesenborghs, L.; Koch, M.; De Mol, M.; et al. Anti-PlGF inhibits growth of VEGF(R)-inhibitor-resistant tumors without affecting healthy vessels. Cell 2007, 131, 463–475. [Google Scholar] [CrossRef] [PubMed]
- Bais, C.; Wu, X.; Yao, J.; Yang, S.; Crawford, Y.; McCutcheon, K.; Tan, C.; Kolumam, G.; Vernes, J.M.; Eastham-Anderson, J.; et al. PlGF blockade does not inhibit angiogenesis during primary tumor growth. Cell 2010, 141, 166–177. [Google Scholar] [CrossRef] [PubMed]
- Lassen, U.; Chinot, O.L.; McBain, C.; Mau-Sorensen, M.; Larsen, V.A.; Barrie, M.; Roth, P.; Krieter, O.; Wang, K.; Habben, K.; et al. Phase 1 dose-escalation study of the antiplacental growth factor monoclonal antibody RO5323441 combined with bevacizumab in patients with recurrent glioblastoma. Neuro Oncol. 2015, 17, 1007–1015. [Google Scholar] [CrossRef] [PubMed]
- FDA Approval for Ziv-Aflibercept. Available online: https://www.cancer.gov/about-cancer/treatment/drugs/fda-ziv-aflibercept (accessed on 3 August 2012).
- Ciombor, K.K.; Berlin, J.; Chan, E. Aflibercept. Clin. Cancer Res. 2013, 19, 1920–1925. [Google Scholar] [CrossRef] [PubMed]
- Chiron, M.; Bagley, R.G.; Pollard, J.; Mankoo, P.K.; Henry, C.; Vincent, L.; Geslin, C.; Baltes, N.; Bergstrom, D.A. Differential antitumor activity of aflibercept and bevacizumab in patient-derived xenograft models of colorectal cancer. Mol. Cancer Ther. 2014, 13, 1636–1644. [Google Scholar] [CrossRef] [PubMed]
- Massague, J.; Blain, S.W.; Lo, R.S. TGFbeta signaling in growth control, cancer, and heritable disorders. Cell 2000, 103, 295–309. [Google Scholar] [CrossRef]
- Massague, J. TGF-beta signal transduction. Annu. Rev. Biochem. 1998, 67, 753–791. [Google Scholar] [CrossRef] [PubMed]
- Roberts, A.B.; Sporn, M.B.; Assoian, R.K.; Smith, J.M.; Roche, N.S.; Wakefield, L.M.; Heine, U.I.; Liotta, L.A.; Falanga, V.; Kehrl, J.H.; et al. Transforming growth factor type beta: Rapid induction of fibrosis and angiogenesis in vivo and stimulation of collagen formation in vitro. Proc. Natl. Acad. Sci. USA 1986, 83, 4167–4171. [Google Scholar] [CrossRef] [PubMed]
- Madri, J.A.; Pratt, B.M.; Tucker, A.M. Phenotypic modulation of endothelial cells by transforming growth factor-beta depends upon the composition and organization of the extracellular matrix. J. Cell Biol. 1988, 106, 1375–1384. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, G.; Cook, B.D.; Terushkin, V.; Pintucci, G.; Mignatti, P. Transforming growth factor-beta 1 (TGF-β1) induces angiogenesis through vascular endothelial growth factor (VEGF)-mediated apoptosis. J. Cell. Physiol. 2009, 219, 449–458. [Google Scholar] [CrossRef] [PubMed]
- Barcellos-Hoff, M.H.; Akhurst, R.J. Transforming growth factor-beta in breast cancer: Too much, too late. Breast Cancer Res. 2009, 11, 202. [Google Scholar] [CrossRef] [PubMed]
- Derynck, R.; Akhurst, R.J.; Balmain, A. TGF-beta signaling in tumor suppression and cancer progression. Nat. Genet. 2001, 29, 117–129. [Google Scholar] [CrossRef] [PubMed]
- Derynck, R.; Goeddel, D.V.; Ullrich, A.; Gutterman, J.U.; Williams, R.D.; Bringman, T.S.; Berger, W.H. Synthesis of messenger RNAs for transforming growth factors alpha and beta and the epidermal growth factor receptor by human tumors. Cancer Res. 1987, 47, 707–712. [Google Scholar] [PubMed]
- Chen, X.; Chen, Z.; Zhu, S.; Liu, T.; Wen, Y.; Su, Y.; Xi, X.; Hu, Y.; Lian, L.; Liu, F. Prognostic value of transforming growth factor-beta in patients with colorectal cancer who undergo surgery: A meta-analysis. BMC Cancer 2017, 17, 240. [Google Scholar] [CrossRef] [PubMed]
- Dave, H.; Shah, M.; Trivedi, S.; Shukla, S. Prognostic utility of circulating transforming growth factor beta 1 in breast cancer patients. Int. J. Biol. Markers 2012, 27, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Park, S.Y.; Piao, Y.; Jeong, K.J.; Dong, J.; de Groot, J.F. Periostin (POSTN) Regulates Tumor Resistance to Antiangiogenic Therapy in Glioma Models. Mol. Cancer Ther. 2016, 15, 2187–2197. [Google Scholar] [CrossRef] [PubMed]
- Forsythe, J.A.; Jiang, B.H.; Iyer, N.V.; Agani, F.; Leung, S.W.; Koos, R.D.; Semenza, G.L. Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol. Cell. Biol. 1996, 16, 4604–4613. [Google Scholar] [CrossRef] [PubMed]
- Pennacchietti, S.; Michieli, P.; Galluzzo, M.; Mazzone, M.; Giordano, S.; Comoglio, P.M. Hypoxia promotes invasive growth by transcriptional activation of the met protooncogene. Cancer Cell 2003, 3, 347–361. [Google Scholar] [CrossRef]
- Birchmeier, C.; Birchmeier, W.; Gherardi, E.; Vande Woude, G.F. Met, metastasis, motility and more. Nat. Rev. Mol. Cell Biol. 2003, 4, 915–925. [Google Scholar] [CrossRef] [PubMed]
- Trusolino, L.; Bertotti, A.; Comoglio, P.M. MET signalling: Principles and functions in development, organ regeneration and cancer. Nat. Rev. Mol. Cell Biol. 2010, 11, 834–848. [Google Scholar] [CrossRef] [PubMed]
- Jahangiri, A.; De Lay, M.; Miller, L.M.; Carbonell, W.S.; Hu, Y.L.; Lu, K.; Tom, M.W.; Paquette, J.; Tokuyasu, T.A.; Tsao, S.; et al. Gene expression profile identifies tyrosine kinase c-Met as a targetable mediator of antiangiogenic therapy resistance. Clin. Cancer Res. 2013, 19, 1773–1783. [Google Scholar] [CrossRef] [PubMed]
- Lu, K.V.; Chang, J.P.; Parachoniak, C.A.; Pandika, M.M.; Aghi, M.K.; Meyronet, D.; Isachenko, N.; Fouse, S.D.; Phillips, J.J.; Cheresh, D.A.; et al. VEGF inhibits tumor cell invasion and mesenchymal transition through a MET/VEGFR2 complex. Cancer Cell 2012, 22, 21–35. [Google Scholar] [CrossRef] [PubMed]
- Wakelee, H.; Zvirbule, Z.; De Braud, F.; Kingsley, C.D.; Mekhail, T.; Lowe, T.; Schutte, W.; Lena, H.; Lawler, W.; Braiteh, F.; et al. Efficacy and Safety of Onartuzumab in Combination With First-Line Bevacizumab- or Pemetrexed-Based Chemotherapy Regimens in Advanced Non-Squamous Non-Small-Cell Lung Cancer. Clin. Lung Cancer 2017, 18, 50–59. [Google Scholar] [CrossRef] [PubMed]
- Shah, N.; Sukumar, S. The Hox genes and their roles in oncogenesis. Nat. Rev. Cancer 2010, 10, 361–371. [Google Scholar] [CrossRef] [PubMed]
- Hayashida, T.; Takahashi, F.; Chiba, N.; Brachtel, E.; Takahashi, M.; Godin-Heymann, N.; Gross, K.W.; Vivanco, M.; Wijendran, V.; Shioda, T.; et al. HOXB9, a gene overexpressed in breast cancer, promotes tumorigenicity and lung metastasis. Proc. Natl. Acad. Sci. USA 2010, 107, 1100–1105. [Google Scholar] [CrossRef] [PubMed]
- Zhan, J.; Wang, P.; Niu, M.; Wang, Y.; Zhu, X.; Guo, Y.; Zhang, H. High expression of transcriptional factor HoxB9 predicts poor prognosis in patients with lung adenocarcinoma. Histopathology 2015, 66, 955–965. [Google Scholar] [CrossRef] [PubMed]
- Hoshino, Y.; Hayashida, T.; Hirata, A.; Takahashi, H.; Chiba, N.; Ohmura, M.; Wakui, M.; Jinno, H.; Hasegawa, H.; Maheswaran, S.; et al. Bevacizumab terminates homeobox B9-induced tumor proliferation by silencing microenvironmental communication. Mol. Cancer 2014, 13, 102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiba, N.; Ozawa, Y.; Hikita, K.; Okihara, M.; Sano, T.; Tomita, K.; Takano, K.; Kawachi, S. Increased expression of HOXB9 in hepatocellular carcinoma predicts poor overall survival but a beneficial response to sorafenib. Oncol. Rep. 2017, 37, 2270–2276. [Google Scholar] [CrossRef] [PubMed]
- Carbone, C.; Piro, G.; Simionato, F.; Ligorio, F.; Cremolini, C.; Loupakis, F.; Ali, G.; Rossini, D.; Merz, V.; Santoro, R.; et al. Homeobox B9 Mediates Resistance to Anti-VEGF Therapy in Colorectal Cancer Patients. Clin. Cancer Res. 2017, 23, 4312–4322. [Google Scholar] [CrossRef] [PubMed]
- Giancotti, F.G.; Ruoslahti, E. Integrin signaling. Science 1999, 285, 1028–1032. [Google Scholar] [CrossRef] [PubMed]
- Avraamides, C.J.; Garmy-Susini, B.; Varner, J.A. Integrins in angiogenesis and lymphangiogenesis. Nat. Rev. Cancer 2008, 8, 604–617. [Google Scholar] [CrossRef] [PubMed]
- Desgrosellier, J.S.; Cheresh, D.A. Integrins in cancer: Biological implications and therapeutic opportunities. Nat. Rev. Cancer 2010, 10, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Gotzmann, J.; Mikula, M.; Eger, A.; Schulte-Hermann, R.; Foisner, R.; Beug, H.; Mikulits, W. Molecular aspects of epithelial cell plasticity: Implications for local tumor invasion and metastasis. Mutat. Res. 2004, 566, 9–20. [Google Scholar] [CrossRef]
- Foubert, P.; Varner, J.A. Integrins in tumor angiogenesis and lymphangiogenesis. Methods Mol. Biol. 2012, 757, 471–486. [Google Scholar] [PubMed]
- Tchaicha, J.H.; Reyes, S.B.; Shin, J.; Hossain, M.G.; Lang, F.F.; McCarty, J.H. Glioblastoma angiogenesis and tumor cell invasiveness are differentially regulated by beta8 integrin. Cancer Res. 2011, 71, 6371–6381. [Google Scholar] [CrossRef] [PubMed]
- Cordes, N.; Park, C.C. beta1 integrin as a molecular therapeutic target. Int. J. Radiat. Biol. 2007, 83, 753–760. [Google Scholar] [CrossRef] [PubMed]
- Sethi, T.; Rintoul, R.C.; Moore, S.M.; MacKinnon, A.C.; Salter, D.; Choo, C.; Chilvers, E.R.; Dransfield, I.; Donnelly, S.C.; Strieter, R.; et al. Extracellular matrix proteins protect small cell lung cancer cells against apoptosis: A mechanism for small cell lung cancer growth and drug resistance in vivo. Nat. Med. 1999, 5, 662–668. [Google Scholar] [CrossRef] [PubMed]
- Meads, M.B.; Gatenby, R.A.; Dalton, W.S. Environment-mediated drug resistance: A major contributor to minimal residual disease. Nat. Rev. Cancer 2009, 9, 665–674. [Google Scholar] [CrossRef] [PubMed]
- DeLay, M.; Jahangiri, A.; Carbonell, W.S.; Hu, Y.L.; Tsao, S.; Tom, M.W.; Paquette, J.; Tokuyasu, T.A.; Aghi, M.K. Microarray analysis verifies two distinct phenotypes of glioblastomas resistant to antiangiogenic therapy. Clin. Cancer Res. 2012, 18, 2930–2942. [Google Scholar] [CrossRef] [PubMed]
- Carbonell, W.S.; DeLay, M.; Jahangiri, A.; Park, C.C.; Aghi, M.K. beta1 integrin targeting potentiates antiangiogenic therapy and inhibits the growth of bevacizumab-resistant glioblastoma. Cancer Res. 2013, 73, 3145–3154. [Google Scholar] [CrossRef] [PubMed]
- Sidorov, M.; Jahangiri, A.; Han, S.W.; De Lay, M.; Wagner, J.; Castro, B.; Flanigan, P.M.; Imber, B.S.; Weiss, W.A.; Aghi, M.K. 340 c-Met/beta1 Integrin: A Receptor Complex Driving Invasive Glioblastoma Resistance to Antiangiogenic Therapy. Neurosurgery 2016, 63 (Suppl. S1), 199–200. [Google Scholar] [CrossRef] [PubMed]
- Jahangiri, A.; Nguyen, A.; Chandra, A.; Sidorov, M.K.; Yagnik, G.; Rick, J.; Han, S.W.; Chen, W.; Flanigan, P.M.; Schneidman-Duhovny, D.; et al. Cross-activating c-Met/beta1 integrin complex drives metastasis and invasive resistance in cancer. Proc. Natl. Acad. Sci. USA 2017, 114, E8685–E8694. [Google Scholar] [PubMed]
- Jahangiri, A.; Aghi, M.K.; Carbonell, W.S. beta1 integrin: Critical path to antiangiogenic therapy resistance and beyond. Cancer Res 2014, 74, 3–7. [Google Scholar] [CrossRef] [PubMed]
- Piao, Y.; Henry, V.; Tiao, N.; Park, S.Y.; Martinez-Ledesma, J.; Dong, J.W.; Balasubramaniyan, V.; de Groot, J.F. Targeting intercellular adhesion molecule-1 prolongs survival in mice bearing bevacizumab-resistant glioblastoma. Oncotarget 2017, 8, 96970–96983. [Google Scholar] [CrossRef] [PubMed]
- Van de Stolpe, A.; van der Saag, P.T. Intercellular adhesion molecule-1. J. Mol. Med. 1996, 74, 13–33. [Google Scholar] [CrossRef] [PubMed]
- Qian, B.Z.; Pollard, J.W. Macrophage diversity enhances tumor progression and metastasis. Cell 2010, 141, 39–51. [Google Scholar] [CrossRef] [PubMed]
- Castro, B.A.; Flanigan, P.; Jahangiri, A.; Hoffman, D.; Chen, W.; Kuang, R.; De Lay, M.; Yagnik, G.; Wagner, J.R.; Mascharak, S.; et al. Macrophage migration inhibitory factor downregulation: A novel mechanism of resistance to anti-angiogenic therapy. Oncogene 2017, 36, 3749–3759. [Google Scholar] [CrossRef] [PubMed]
- Galon, J.; Pages, F.; Marincola, F.M.; Angell, H.K.; Thurin, M.; Lugli, A.; Zlobec, I.; Berger, A.; Bifulco, C.; Botti, G.; et al. Cancer classification using the Immunoscore: A worldwide task force. J. Transl. Med. 2012, 10, 205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galon, J.; Costes, A.; Sanchez-Cabo, F.; Kirilovsky, A.; Mlecnik, B.; Lagorce-Pages, C.; Tosolini, M.; Camus, M.; Berger, A.; Wind, P.; et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science 2006, 313, 1960–1964. [Google Scholar] [CrossRef] [PubMed]
- Pagès, F.; Berger, A.; Camus, M.; Sanchez-Cabo, F.; Costes, A.; Molidor, R.; Mlecnik, B.; Kirilovsky, A.; Nilsson, M.; Damotte, D.; et al. Effector Memory T Cells, Early Metastasis, and Survival in Colorectal Cancer. N. Engl. J. Med. 2005, 353, 2654–2666. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.D.; Hoang, A.; Zhou, L.; Kalra, S.; Yetil, A.; Sun, M.; Ding, Z.; Zhang, X.; Bai, S.; German, P.; et al. Resistance to Antiangiogenic Therapy Is Associated with an Immunosuppressive Tumor Microenvironment in Metastatic Renal Cell Carcinoma. Cancer Immunol. Res. 2015, 3, 1017–1029. [Google Scholar] [CrossRef] [PubMed]
- Kuusk, T.; Albiges, L.; Escudier, B.; Grivas, N.; Haanen, J.; Powles, T.; Bex, A. Antiangiogenic therapy combined with immune checkpoint blockade in renal cancer. Angiogenesis 2017, 20, 205–215. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Schematic representation of the VEGFA isoforms. Each number indicates the exon composition and the isoforms consist of splicing variants of these exons from the VEGFA gene.
Figure 1.
Schematic representation of the VEGFA isoforms. Each number indicates the exon composition and the isoforms consist of splicing variants of these exons from the VEGFA gene.
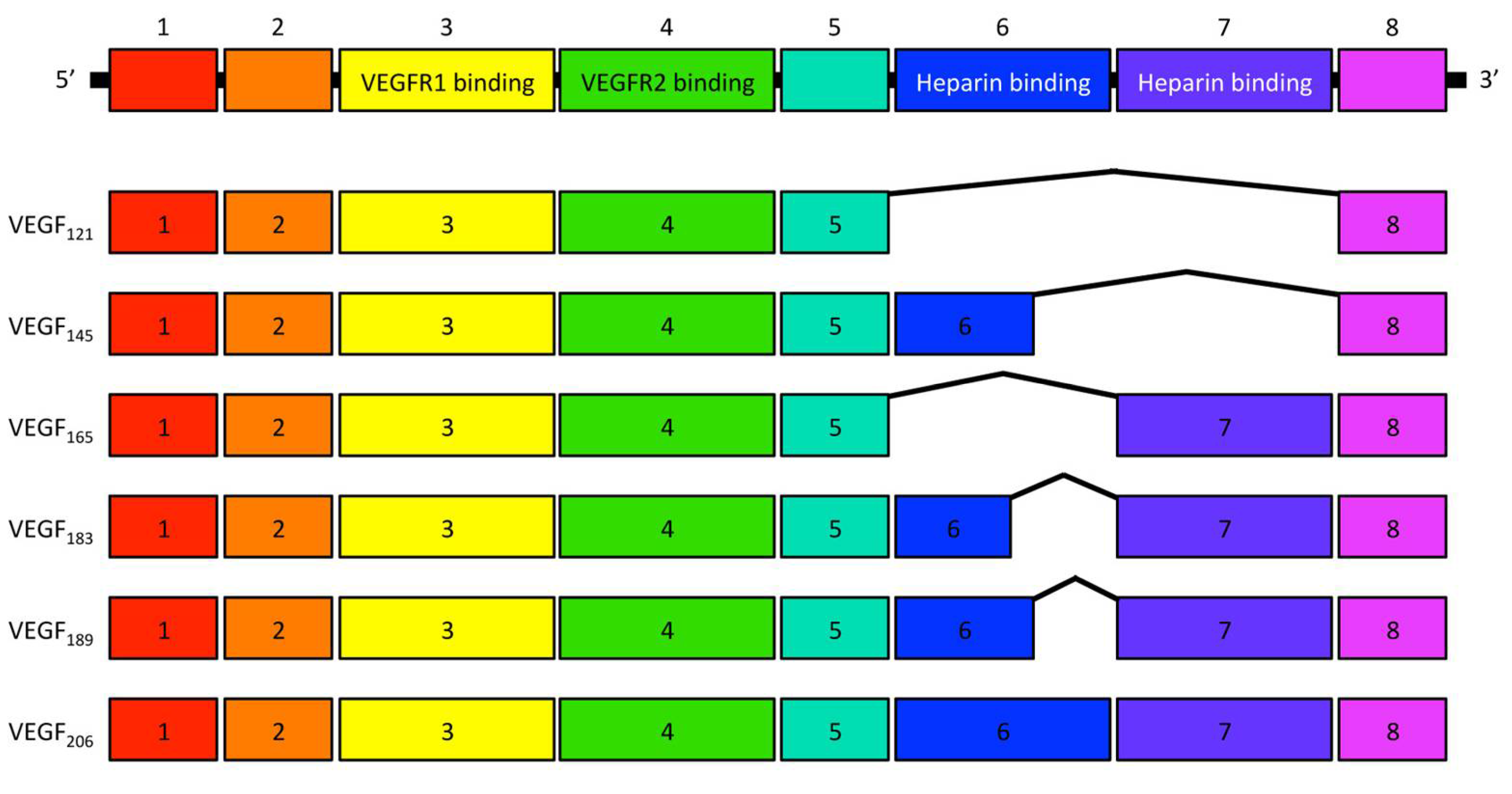
Figure 2.
Alternative angiogenic factors are listed on the right side and phenotypical tumor changes are listed on the left side.
Figure 2.
Alternative angiogenic factors are listed on the right side and phenotypical tumor changes are listed on the left side.

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Itatani, Y.; Kawada, K.; Yamamoto, T.; Sakai, Y. Resistance to Anti-Angiogenic Therapy in Cancer—Alterations to Anti-VEGF Pathway. Int. J. Mol. Sci. 2018, 19, 1232. https://doi.org/10.3390/ijms19041232
AMA Style
Itatani Y, Kawada K, Yamamoto T, Sakai Y. Resistance to Anti-Angiogenic Therapy in Cancer—Alterations to Anti-VEGF Pathway. International Journal of Molecular Sciences. 2018; 19(4):1232. https://doi.org/10.3390/ijms19041232
Chicago/Turabian StyleItatani, Yoshiro, Kenji Kawada, Takamasa Yamamoto, and Yoshiharu Sakai. 2018. "Resistance to Anti-Angiogenic Therapy in Cancer—Alterations to Anti-VEGF Pathway" International Journal of Molecular Sciences 19, no. 4: 1232. https://doi.org/10.3390/ijms19041232
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.