Pivotal Roles of Peroxisome Proliferator-Activated Receptors (PPARs) and Their Signal Cascade for Cellular and Whole-Body Energy Homeostasis

Abstract
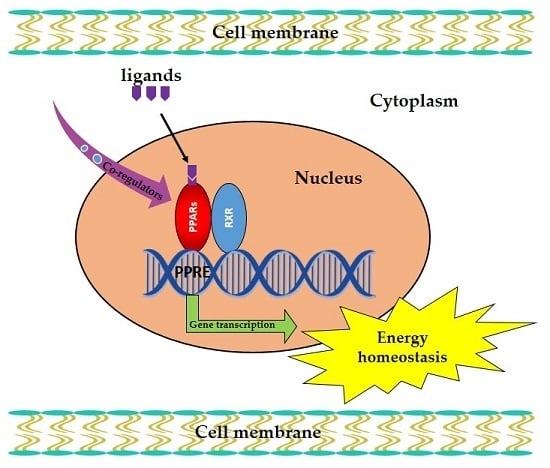
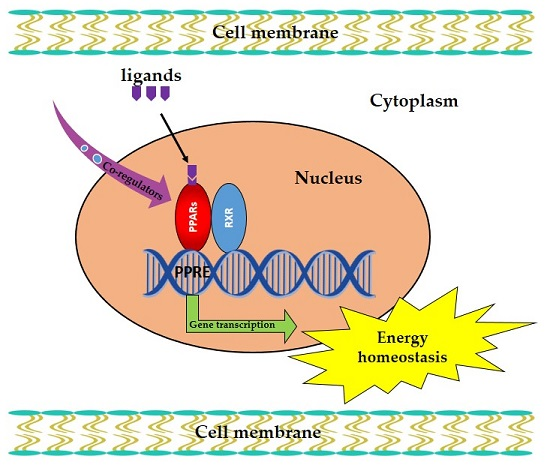
:
{kind=link}
{kind=link}
{kind=link}
1. Introduction
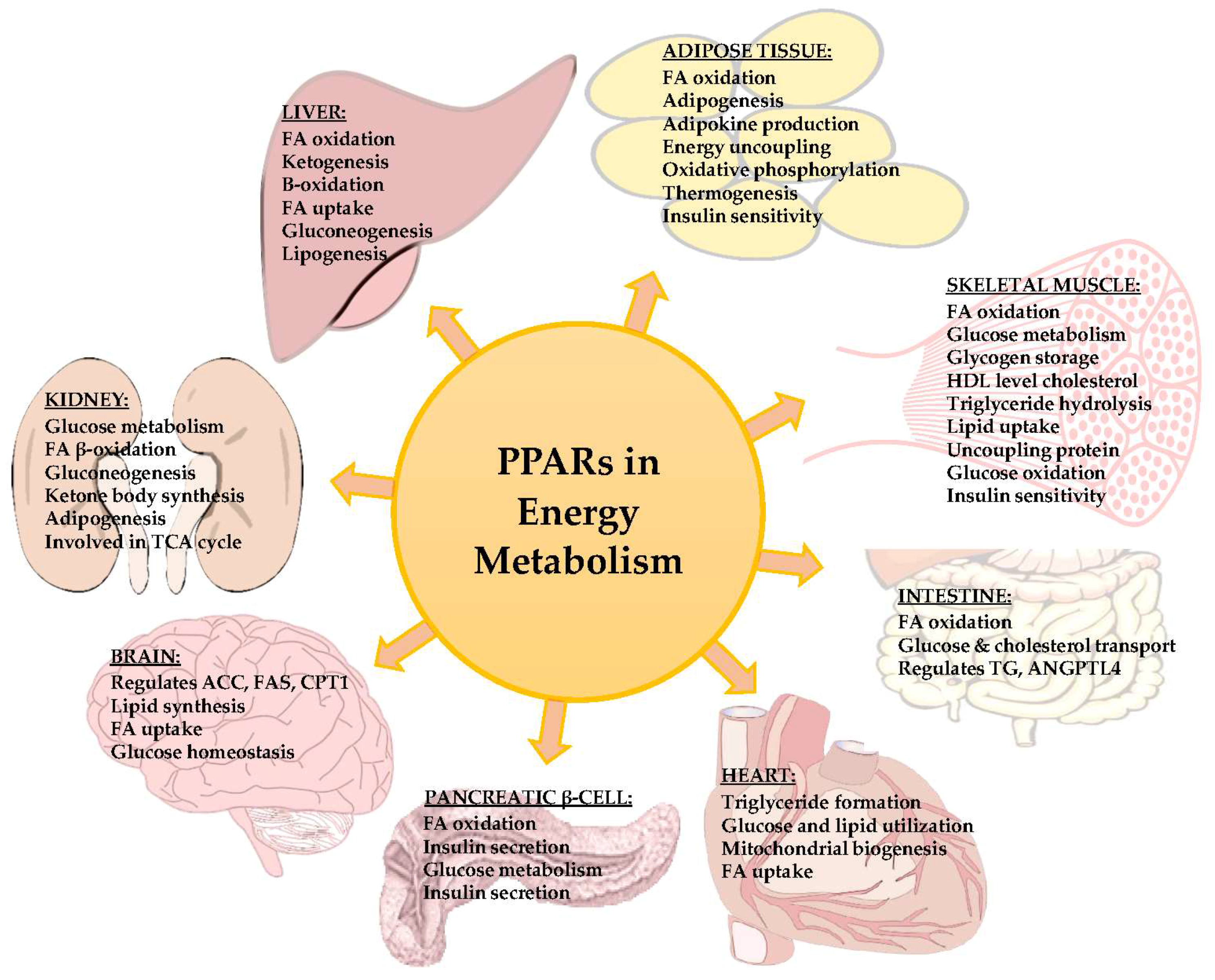
2. PPAR Signals in Liver
3. PPAR Signals in Adipose Tissue
4. PPAR Signals in Skeletal Muscle
5. PPAR Signals in Kidney
6. PPAR Signals in Heart
7. PPAR Signals in Brain
8. PPAR Signals in Pancreatic β-Cells
9. PPAR Signals in Intestine
10. Co-Regulators of PPAR in Energy Homeostasis
11. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| PPARs | Peroxisome proliferator-activated receptors |
| FAs | Fatty acids |
| PPREs | PPAR response elements |
| FAO | Fatty acid oxidation |
| CPT1 | Carnitine palmitoyltransferase 1 |
| HDL | High-density lipoprotein |
| WAT | White adipose tissue |
| BAT | Brown adipose tissue |
| GPDH | Glycerol-3-phosphate dehydrogenase |
| OXPHOS | Oxidative phosphorylation |
| PDK4 | Pyruvate dehydrogenase kinase 4 |
| LDHB | Lactate dehydrogenase B |
| PGC-1α | PPARγ coactivator-1α |
| UCP1 | Uncoupling protein 1 |
| ATP | Adenosine triphosphate |
| TFEB | Transcription factor EB |
| TCA | Tricarboxylic acid |
| GIT1 | G-protein-coupled receptor kinase interacting protein-1 |
| ACC | Acetyl-coenzyme A carboxylase |
| FAS | Fatty acid synthase |
| VMN | Ventromedial nucleus |
| ARC | Arcuate nucleus of the hypothalamus |
| NPY | Neuropeptide Y |
| POMC | Pro-opiomelanocortin |
| CNS | Central nervous system |
| DIO | Diet induced obesity |
| LCAD | Long chain acyl-CoA dehydrogenase |
| NCOR1 | Nuclear co-repressor 1 |
| HNFα | Hepatocyte nuclear factor α |
| C/EBPα | CCAAT/enhancer-binding protein α |
| MED1 | Mediator complex subunit 1 |
| ANGPTL4 | Angiopoietin-like protein 4 |
| TG | Triglyceride |
| HMG-CoAS2 | Mitochondrial 3-hydroxy-3-methylglutaryl-CoA synthase |
| SCFAs | Short chain fatty acids |
| EFA-CLA | Fatty acids from conjugated linoleic acid-enriched egg yolks |
| CLA | Conjugated linoleic acid |
| NAFLD | Non-alcoholic fatty liver disease |
| NASH | Non-alcoholic steatohepatitis |
| TZD | Thiazolidinediones |
| ERK1/2 | Extracellular signal-regulated kinase type 1 and 2 |
| P38-MAPK | Mitogen-activated protein kinase p38 |
| PKC | Protein kinase C |
| AMPK | 5’Adenosine monophosphate-activated protein kinase |
| GSK3 | Glycogen synthase kinase 3 |
Appendix A

References
- Fan, W.; Evans, R. PPARs and ERRs: Molecular mediators of mitochondrial metabolism. Curr. Opin. Cell Biol. 2015, 33, 49–54. [Google Scholar] [CrossRef] [PubMed]
- Kota, B.P.; Huang, T.H.-W.; Roufogalis, B.D. An overview on biological mechanisms of PPARs. Pharmacol. Res. 2005, 51, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, W.; Ziouzenkova, O.; Brown, J.; Devchand, P.; Francis, S.; Kadakia, M.; Kanda, T.; Orasanu, G.; Sharlach, M.; Zandbergen, F.; et al. PPARs and their metabolic modulation: New mechanisms for transcriptional regulation? J. Intern. Med. 2007, 262, 184–198. [Google Scholar] [CrossRef] [PubMed]
- Kersten, S.; Desvergne, B.; Wahli, W. Roles of PPARs in health and disease. Nature 2000, 405, 421–424. [Google Scholar] [CrossRef] [PubMed]
- Pyper, S.R.; Viswakarma, N.; Yu, S.; Reddy, J.K. PPARα: Energy combustion, hypolipidemia, inflammation and cancer. Nucl. Recept. Signal. 2010, 8, e002. [Google Scholar] [CrossRef] [PubMed]
- Tyagi, S.; Gupta, P.; Saini, A.S.; Kaushal, C.; Sharma, S. The peroxisome proliferator-activated receptor: A family of nuclear receptors role in various diseases. J. Adv. Pharm. Technol. Res. 2011, 2, 236–240. [Google Scholar] [CrossRef] [PubMed]
- Ferré, P. The Biology of Peroxisome Proliferator-Activated Receptors: Relationship with Lipid Metabolism and Insulin Sensitivity. Diabetes 2004, 53, S43–S50. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.X. PPARs: Diverse regulators in energy metabolism and metabolic diseases. Cell Res. 2010, 20, 124–137. [Google Scholar] [CrossRef] [PubMed]
- Tachibana, K.; Yamasaki, D.; Ishimoto, K.; Doi, T. The Role of PPARs in Cancer. PPAR Res. 2008, 2008. [Google Scholar] [CrossRef] [PubMed]
- La Paglia, L.; Listi, A.; Caruso, S.; Amodeo, V.; Passiglia, F.; Bazan, V.; Fanale, D. Potential Role of ANGPTL4 in the Cross Talk between Metabolism and Cancer through PPAR Signaling Pathway. PPAR Res. 2017, 2017. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Chen, L.; Zhang, D.; Huo, M.; Zhang, X.; Pu, D.; Guan, Y. Peroxisome proliferator-activated receptors and renal diseases. Front. Biosci. (Landmark Ed.) 2009, 14, 995–1009. [Google Scholar] [CrossRef] [PubMed]
- Dubois, V.; Eeckhoute, J.; Lefebvre, P.; Staels, B. Distinct but complementary contributions of PPAR isotypes to energy homeostasis. J. Clin. Investig. 2017, 127, 1202–1214. [Google Scholar] [CrossRef] [PubMed]
- Feingold, K.R.; Wang, Y.; Moser, A.; Shigenaga, J.K.; Grunfeld, C. LPS decreases fatty acid oxidation and nuclear hormone receptors in the kidney. J. Lipid Res. 2008, 49, 2179–2187. [Google Scholar] [CrossRef] [PubMed]
- Dressel, U.; Allen, T.L.; Pippal, J.B.; Rohde, P.R.; Lau, P.; Muscat, G.E. The peroxisome proliferator-activated receptor beta/delta agonist, GW501516, regulates the expression of genes involved in lipid catabolism and energy uncoupling in skeletal muscle cells. Mol. Endocrinol. 2003, 17, 2477–2493. [Google Scholar] [CrossRef] [PubMed]
- Lefebvre, P.; Chinetti, G.; Fruchart, J.C.; Staels, B. Sorting out the roles of PPARα in energy metabolism and vascular homeostasis. J. Clin. Investig. 2006, 116, 571–580. [Google Scholar] [CrossRef] [PubMed]
- Kondo, Y.; Uno, K.; Machida, K.; Terajima, M. PPARs and liver disease. PPAR Res. 2013, 2013. [Google Scholar] [CrossRef] [PubMed]
- Choe, S.S.; Huh, J.Y.; Hwang, I.J.; Kim, J.I.; Kim, J.B. Adipose Tissue Remodeling: Its Role in Energy Metabolism and Metabolic Disorders. Front. Endocrinol. (Lausanne) 2016, 7, 30. [Google Scholar] [CrossRef] [PubMed]
- Birsoy, K.; Festuccia, W.T.; Laplante, M. A comparative perspective on lipid storage in animals. J. Cell Sci. 2013, 126, 1541–1552. [Google Scholar] [CrossRef] [PubMed]
- Rosen, E.D.; Spiegelman, B.M. Adipocytes as regulators of energy balance and glucose homeostasis. Nature 2006, 444, 847–853. [Google Scholar] [CrossRef] [PubMed]
- Tontonoz, P.; Hu, E.; Graves, R.A.; Budavari, A.I.; Spiegelman, B.M. mPPAR gamma 2: Tissue-specific regulator of an adipocyte enhancer. Genes Dev. 1994, 8, 1224–1234. [Google Scholar] [CrossRef] [PubMed]
- Tontonoz, P.; Hu, E.; Spiegelman, B.M. Stimulation of adipogenesis in fibroblasts by PPAR gamma 2, a lipid-activated transcription factor. Cell 1994, 79, 1147–1156. [Google Scholar] [CrossRef]
- Rosen, E.D.; Hsu, C.H.; Wang, X.; Sakai, S.; Freeman, M.W.; Gonzalez, F.J.; Spiegelman, B.M. C/EBPα induces adipogenesis through PPARgamma: A unified pathway. Genes Dev. 2002, 16, 22–26. [Google Scholar] [CrossRef] [PubMed]
- Mullican, S.E.; Dispirito, J.R.; Lazar, M.A. The orphan nuclear receptors at their 25-year reunion. J. Mol. Endocrinol. 2013, 51, T115–T140. [Google Scholar] [CrossRef] [PubMed]
- Manickam, R.; Wahli, W. Roles of Peroxisome Proliferator-Activated Receptor beta/delta in skeletal muscle physiology. Biochimie 2017, 136, 42–48. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.Y.; Jeong, H.W.; Sohn, J.H.; Seo, D.B.; Kim, W.G.; Lee, S.J. An ethanol extract of Artemisia iwayomogi activates PPARdelta leading to activation of fatty acid oxidation in skeletal muscle. PLoS ONE 2012, 7, e33815. [Google Scholar] [CrossRef] [PubMed]
- Periasamy, M.; Herrera, J.L.; Reis, F.C.G. Skeletal Muscle Thermogenesis and Its Role in Whole Body Energy Metabolism. Diabetes Metab. J. 2017, 41, 327–336. [Google Scholar] [CrossRef] [PubMed]
- Gan, Z.; Burkart-Hartman, E.M.; Han, D.H.; Finck, B.; Leone, T.C.; Smith, E.Y.; Ayala, J.E.; Holloszy, J.; Kelly, D.P. The nuclear receptor PPARβ/δ programs muscle glucose metabolism in cooperation with AMPK and MEF2. Genes Dev. 2011, 25, 2619–2630. [Google Scholar] [CrossRef] [PubMed]
- Schnuck, J.K.; Sunderland, K.L.; Gannon, N.P.; Kuennen, M.R.; Vaughan, R.A. Leucine stimulates PPARbeta/delta-dependent mitochondrial biogenesis and oxidative metabolism with enhanced GLUT4 content and glucose uptake in myotubes. Biochimie 2016, 128, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Perez-Schindler, J.; Svensson, K.; Vargas-Fernandez, E.; Santos, G.; Wahli, W.; Handschin, C. The coactivator PGC-1α regulates skeletal muscle oxidative metabolism independently of the nuclear receptor PPARbeta/delta in sedentary mice fed a regular chow diet. Diabetologia 2014, 57, 2405–2412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thach, T.T.; Lee, C.K.; Park, H.W.; Lee, S.J.; Lee, S.J. Syringaresinol induces mitochondrial biogenesis through activation of PPARbeta pathway in skeletal muscle cells. Bioorg. Med. Chem. Lett. 2016, 26, 3978–3983. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.; Hazen, B.C.; Russell, A.P.; Kralli, A. Peroxisome proliferator-activated receptor gamma coactivator 1 (PGC-1)- and estrogen-related receptor (ERR)-induced regulator in muscle 1 (Perm1) is a tissue-specific regulator of oxidative capacity in skeletal muscle cells. J. Biol. Chem. 2013, 288, 25207–25218. [Google Scholar] [CrossRef] [PubMed]
- Kong, X.Y.; Feng, Y.Z.; Eftestol, E.; Kase, E.T.; Haugum, H.; Eskild, W.; Rustan, A.C.; Thoresen, G.H. Increased glucose utilization and decreased fatty acid metabolism in myotubes from Glmp(gt/gt) mice. Arch. Physiol. Biochem. 2016, 122, 36–45. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Nagothu, K.K.; Desai, V.; Lee, T.; Branham, W.; Moland, C.; Megyesi, J.K.; Crew, M.D.; Portilla, D. Transgenic expression of proximal tubule peroxisome proliferator-activated receptor-α in mice confers protection during acute kidney injury. Kidney Int. 2009, 76, 1049–1062. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Zhou, Y.; Guan, Y. PPARγ as a therapeutic target in diabetic nephropathy and other renal diseases. Curr. Opin. Nephrol. Hypertens. 2012, 21, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.; Nakamura, F.; Taguchi, E.; Nakata, M.; Wada, F.; Takihi, M.; Inoue, T.; Ohta, S.; Kawachi, H. 4′,6-Dimethoxyisoflavone-7-O-β-d-glucopyranoside (wistin) is a peroxisome proliferator-activated receptor α (PPARα) agonist in mouse hepatocytes. Mol. Cell. Biochem. 2018, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Negishi, K.; Noiri, E.; Maeda, R.; Portilla, D.; Sugaya, T.; Fujita, T. Renal l-type fatty acid-binding protein mediates the bezafibrate reduction of cisplatin-induced acute kidney injury. Kidney Int. 2008, 73, 1374–1384. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Hernandez, F.J.; Lopez-Novoa, J.M. Potential utility of PPARα activation in the prevention of ischemic and drug-induced acute renal damage. Kidney Int. 2009, 76, 1022–1024. [Google Scholar] [CrossRef] [PubMed]
- Teng, C.T.; Li, Y.; Stockton, P.; Foley, J. Fasting induces the expression of PGC-1α and ERR isoforms in the outer stripe of the outer medulla (OSOM) of the mouse kidney. PLoS ONE 2011, 6, e26961. [Google Scholar] [CrossRef] [PubMed]
- Guan, Y.; Breyer, M.D. Peroxisome proliferator-activated receptors (PPARs): Novel therapeutic targets in renal disease. Kidney Int. 2001, 60, 14–30. [Google Scholar] [CrossRef] [PubMed]
- Roe, N.D.; Standage, S.W.; Tian, R. The Relationship Between KLF5 and PPARα in the Heart: It’s Complicated. Circ. Res. 2016, 118, 193–195. [Google Scholar] [CrossRef] [PubMed]
- Ravingerova, T.; Adameova, A.; Carnicka, S.; Nemcekova, M.; Kelly, T.; Matejikova, J.; Galatou, E.; Barlaka, E.; Lazou, A. The role of PPAR in myocardial response to ischemia in normal and diseased heart. Gen. Physiol. Biophys. 2011, 30, 329–341. [Google Scholar] [CrossRef] [PubMed]
- Drosatos, K.; Pollak, N.M.; Pol, C.J.; Ntziachristos, P.; Willecke, F.; Valenti, M.C.; Trent, C.M.; Hu, Y.; Guo, S.; Aifantis, I.; et al. Cardiac Myocyte KLF5 Regulates Ppara Expression and Cardiac Function. Circ. Res. 2016, 118, 241–253. [Google Scholar] [CrossRef] [PubMed]
- Mora, C.; Pintado, C.; Rubio, B.; Mazuecos, L.; Lopez, V.; Fernandez, A.; Salamanca, A.; Barcena, B.; Fernandez-Agullo, T.; Arribas, C.; et al. Central leptin regulates heart lipid content by selectively increasing PPAR beta/delta expression. J. Endocrinol. 2018, 236, 43–56. [Google Scholar] [CrossRef] [PubMed]
- Pang, J.; Xu, X.; Getman, M.R.; Shi, X.; Belmonte, S.L.; Michaloski, H.; Mohan, A.; Blaxall, B.C.; Berk, B.C. G protein coupled receptor kinase 2 interacting protein 1 (GIT1) is a novel regulator of mitochondrial biogenesis in heart. J. Mol. Cell Cardiol. 2011, 51, 769–776. [Google Scholar] [CrossRef] [PubMed]
- Arumugam, S.; Sreedhar, R.; Thandavarayan, R.A.; Karuppagounder, V.; Watanabe, K. Targeting fatty acid metabolism in heart failure: Is it a suitable therapeutic approach? Drug Discov. Today 2016, 21, 1003–1008. [Google Scholar] [CrossRef] [PubMed]
- Di Giacomo, E.; Benedetti, E.; Cristiano, L.; Antonosante, A.; d’Angelo, M.; Fidoamore, A.; Barone, D.; Moreno, S.; Ippoliti, R.; Ceru, M.P.; et al. Roles of PPAR transcription factors in the energetic metabolic switch occurring during adult neurogenesis. Cell Cycle 2017, 16, 59–72. [Google Scholar] [CrossRef] [PubMed]
- Kocalis, H.E.; Turney, M.K.; Printz, R.L.; Laryea, G.N.; Muglia, L.J.; Davies, S.S.; Stanwood, G.D.; McGuinness, O.P.; Niswender, K.D. Neuron-specific deletion of peroxisome proliferator-activated receptor delta (PPARdelta) in mice leads to increased susceptibility to diet-induced obesity. PLoS ONE 2012, 7, e42981. [Google Scholar] [CrossRef] [PubMed]
- Kouidhi, S.; Seugnet, I.; Decherf, S.; Guissouma, H.; Elgaaied, A.B.; Demeneix, B.; Clerget-Froidevaux, M.S. Peroxisome proliferator-activated receptor-gamma (PPARγ) modulates hypothalamic Trh regulation in vivo. Mol. Cell. Endocrinol. 2010, 317, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Stump, M.; Guo, D.F.; Lu, K.T.; Mukohda, M.; Cassell, M.D.; Norris, A.W.; Rahmouni, K.; Sigmund, C.D. Nervous System Expression of PPARgamma and Mutant PPARgamma Has Profound Effects on Metabolic Regulation and Brain Development. Endocrinology 2016, 157, 4266–4275. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Sarruf, D.A.; Talukdar, S.; Sharma, S.; Li, P.; Bandyopadhyay, G.; Nalbandian, S.; Fan, W.; Gayen, J.R.; Mahata, S.K.; et al. Brain PPAR-γ promotes obesity and is required for the insulin-sensitizing effect of thiazolidinediones. Nat. Med. 2011, 17, 618–622. [Google Scholar] [CrossRef] [PubMed]
- Rijnsburger, M.; Belegri, E.; Eggels, L.; Unmehopa, U.A.; Boelen, A.; Serlie, M.J.; la Fleur, S.E. The effect of diet interventions on hypothalamic nutrient sensing pathways in rodents. Physiol. Behav. 2016, 162, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Ryan, K.K.; Li, B.; Grayson, B.E.; Matter, E.K.; Woods, S.C.; Seeley, R.J. A role for central nervous system PPAR-γ in the regulation of energy balance. Nat. Med. 2011, 17, 623–626. [Google Scholar] [CrossRef] [PubMed]
- Sarruf, D.A.; Yu, F.; Nguyen, H.T.; Williams, D.L.; Printz, R.L.; Niswender, K.D.; Schwartz, M.W. Expression of peroxisome proliferator-activated receptor-γ in key neuronal subsets regulating glucose metabolism and energy homeostasis. Endocrinology 2009, 150, 707–712. [Google Scholar] [CrossRef] [PubMed]
- Wan, J.; Jiang, L.; Lu, Q.; Ke, L.; Li, X.; Tong, N. Activation of PPARδ up-regulates fatty acid oxidation and energy uncoupling genes of mitochondria and reduces palmitate-induced apoptosis in pancreatic β-cells. Biochem. Biophys. Res. Commun. 2010, 391, 1567–1572. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Wan, J.; Ke, L.Q.; Lu, Q.G.; Tong, N.W. Activation of PPARδ promotes mitochondrial energy metabolism and decreases basal insulin secretion in palmitate-treated β-cells. Mol. Cell. Biochem. 2010, 343, 249–256. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Li, T.; Zhang, Y.; Pan, Z.; Wu, B.; Huang, X.; Zhang, Y.; Mei, Y.; Ge, L.; Shen, G.; et al. Peroxisome proliferator-activated receptorβ/δ activation is essential for modulating p-Foxo1/Foxo1 status in functional insulin-positive cell differentiation. Cell Death Dis. 2015, 6, e1715. [Google Scholar] [CrossRef] [PubMed]
- Bendlova, B.; Vejrazkova, D.; Vcelak, J.; Lukasova, P.; Burkonova, D.; Kunesova, M.; Vrbikova, J.; Dvorakova, K.; Vondra, K.; Vankova, M. PPARγ2 Pro12Ala polymorphism in relation to free fatty acids concentration and composition in lean healthy Czech individuals with and without family history of diabetes type 2. Physiol. Res. 2008, 57 (Suppl S1), S77–S90. [Google Scholar] [PubMed]
- Roduit, R.; Morin, J.; Masse, F.; Segall, L.; Roche, E.; Newgard, C.B.; Assimacopoulos-Jeannet, F.; Prentki, M. Glucose down-regulates the expression of the peroxisome proliferator-activated receptor-α gene in the pancreatic β-cell. J. Biol. Chem. 2000, 275, 35799–35806. [Google Scholar] [CrossRef] [PubMed]
- Cohen, G.; Riahi, Y.; Shamni, O.; Guichardant, M.; Chatgilialoglu, C.; Ferreri, C.; Kaiser, N.; Sasson, S. Role of lipid peroxidation and PPAR-δ in amplifying glucose-stimulated insulin secretion. Diabetes 2011, 60, 2830–2842. [Google Scholar] [CrossRef] [PubMed]
- Vrins, C.L.; van der Velde, A.E.; van den Oever, K.; Levels, J.H.; Huet, S.; Oude Elferink, R.P.; Kuipers, F.; Groen, A.K. Peroxisome proliferator-activated receptor delta activation leads to increased transintestinal cholesterol efflux. J. Lipid Res. 2009, 50, 2046–2054. [Google Scholar] [CrossRef] [PubMed]
- Higashimura, Y.; Naito, Y.; Takagi, T.; Uchiyama, K.; Mizushima, K.; Yoshikawa, T. Propionate Promotes Fatty Acid Oxidation through the Up-Regulation of Peroxisome Proliferator-Activated Receptor α in Intestinal Epithelial Cells. J. Nutr. Sci. Vitaminol. (Tokyo) 2015, 61, 511–515. [Google Scholar] [CrossRef] [PubMed]
- Korecka, A.; de Wouters, T.; Cultrone, A.; Lapaque, N.; Pettersson, S.; Dore, J.; Blottiere, H.M.; Arulampalam, V. ANGPTL4 expression induced by butyrate and rosiglitazone in human intestinal epithelial cells utilizes independent pathways. Am. J. Physiol. Gastrointest. Liver Physiol. 2013, 304, G1025–G1037. [Google Scholar] [CrossRef] [PubMed]
- Karimian Azari, E.; Leitner, C.; Jaggi, T.; Langhans, W.; Mansouri, A. Possible role of intestinal fatty acid oxidation in the eating-inhibitory effect of the PPAR-α agonist Wy-14643 in high-fat diet fed rats. PLoS ONE 2013, 8, e74869. [Google Scholar] [CrossRef] [PubMed]
- de Vogel-van den Bosch, H.M.; Bunger, M.; de Groot, P.J.; Bosch-Vermeulen, H.; Hooiveld, G.J.; Muller, M. PPARα-mediated effects of dietary lipids on intestinal barrier gene expression. BMC Genom. 2008, 9, 231. [Google Scholar] [CrossRef] [PubMed]
- van den Bosch, H.M.; Bunger, M.; de Groot, P.J.; van der Meijde, J.; Hooiveld, G.J.; Muller, M. Gene expression of transporters and phase I/II metabolic enzymes in murine small intestine during fasting. BMC Genom. 2007, 8, 267. [Google Scholar] [CrossRef] [PubMed]
- Takei, K.; Nakagawa, Y.; Wang, Y.; Han, S.I.; Satoh, A.; Sekiya, M.; Matsuzaka, T.; Shimano, H. Effects of K-877, a novel selective PPARα modulator, on small intestine contribute to the amelioration of hyperlipidemia in low-density lipoprotein receptor knockout mice. J. Pharmacol. Sci. 2017, 133, 214–222. [Google Scholar] [CrossRef] [PubMed]
- Lempradl, A.; Pospisilik, J.A.; Penninger, J.M. Exploring the emerging complexity in transcriptional regulation of energy homeostasis. Nat. Rev. Genet. 2015, 16, 665–681. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, H.; Williams, E.G.; Mouchiroud, L.; Canto, C.; Fan, W.; Downes, M.; Heligon, C.; Barish, G.D.; Desvergne, B.; Evans, R.M.; et al. NCoR1 is a conserved physiological modulator of muscle mass and oxidative function. Cell 2011, 147, 827–839. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Jimenez, C.P.; Kyrmizi, I.; Cardot, P.; Gonzalez, F.J.; Talianidis, I. Hepatocyte nuclear factor 4α coordinates a transcription factor network regulating hepatic fatty acid metabolism. Mol. Cell. Biol. 2010, 30, 565–577. [Google Scholar] [CrossRef] [PubMed]
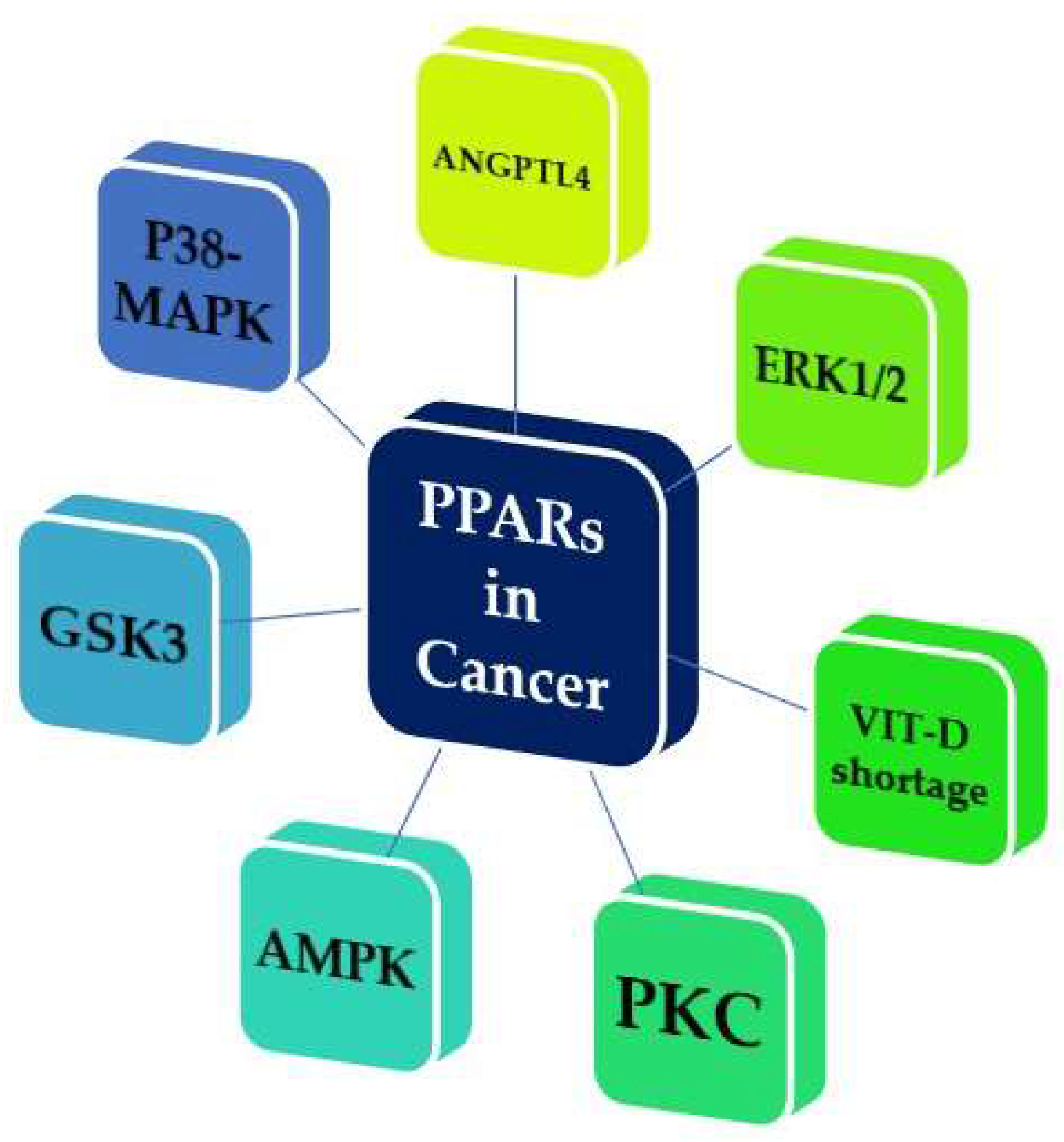
- Mello, T.; Materozzi, M.; Galli, A. PPARs and Mitochondrial Metabolism: From NAFLD to HCC. PPAR Res. 2016, 2016. [Google Scholar] [CrossRef] [PubMed]
- Scatena, R.; Bottoni, P.; Giardina, B. Mitochondria, PPARs, and Cancer: Is Receptor-Independent Action of PPAR Agonists a Key? PPAR Res. 2008, 2008. [Google Scholar] [CrossRef] [PubMed]
- Vitale, S.G.; Lagana, A.S.; Nigro, A.; La Rosa, V.L.; Rossetti, P.; Rapisarda, A.M.; La Vignera, S.; Condorelli, R.A.; Corrado, F.; Buscema, M.; et al. Peroxisome Proliferator-Activated Receptor Modulation during Metabolic Diseases and Cancers: Master and Minions. PPAR Res. 2016, 2016. [Google Scholar] [CrossRef] [PubMed]
- Fanale, D.; Amodeo, V.; Caruso, S. The Interplay between Metabolism, PPAR Signaling Pathway, and Cancer. PPAR Res. 2017, 2017. [Google Scholar] [CrossRef] [PubMed]
- Bandera Merchan, B.; Tinahones, F.J.; Macias-Gonzalez, M. Commonalities in the Association between PPARG and Vitamin D Related with Obesity and Carcinogenesis. PPAR Res. 2016, 2016. [Google Scholar] [CrossRef] [PubMed]
- Glazer, R.I. PPARδ as a Metabolic Initiator of Mammary Neoplasia and Immune Tolerance. PPAR Res. 2016, 2016. [Google Scholar] [CrossRef] [PubMed]
- Lakshmi, S.P.; Reddy, A.T.; Banno, A.; Reddy, R.C. PPAR Agonists for the Prevention and Treatment of Lung Cancer. PPAR Res. 2017, 2017. [Google Scholar] [CrossRef] [PubMed]
- Chiu, M.; McBeth, L.; Sindhwani, P.; Hinds, T.D. Deciphering the Roles of Thiazolidinediones and PPARγ in Bladder Cancer. PPAR Res. 2017, 2017. [Google Scholar] [CrossRef] [PubMed]
- Koronowicz, A.A.; Banks, P.; Master, A.; Domagala, D.; Piasna-Slupecka, E.; Drozdowska, M.; Sikora, E.; Laidler, P. Fatty Acids of CLA-Enriched Egg Yolks Can Induce Transcriptional Activation of Peroxisome Proliferator-Activated Receptors in MCF-7 Breast Cancer Cells. PPAR Res. 2017, 2017. [Google Scholar] [CrossRef] [PubMed]
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lamichane, S.; Dahal Lamichane, B.; Kwon, S.-M. Pivotal Roles of Peroxisome Proliferator-Activated Receptors (PPARs) and Their Signal Cascade for Cellular and Whole-Body Energy Homeostasis. Int. J. Mol. Sci. 2018, 19, 949. https://doi.org/10.3390/ijms19040949
Lamichane S, Dahal Lamichane B, Kwon S-M. Pivotal Roles of Peroxisome Proliferator-Activated Receptors (PPARs) and Their Signal Cascade for Cellular and Whole-Body Energy Homeostasis. International Journal of Molecular Sciences. 2018; 19(4):949. https://doi.org/10.3390/ijms19040949
Chicago/Turabian StyleLamichane, Shreekrishna, Babita Dahal Lamichane, and Sang-Mo Kwon. 2018. "Pivotal Roles of Peroxisome Proliferator-Activated Receptors (PPARs) and Their Signal Cascade for Cellular and Whole-Body Energy Homeostasis" International Journal of Molecular Sciences 19, no. 4: 949. https://doi.org/10.3390/ijms19040949