TGF-β and the Tissue Microenvironment: Relevance in Fibrosis and Cancer

 , , ,
, , , 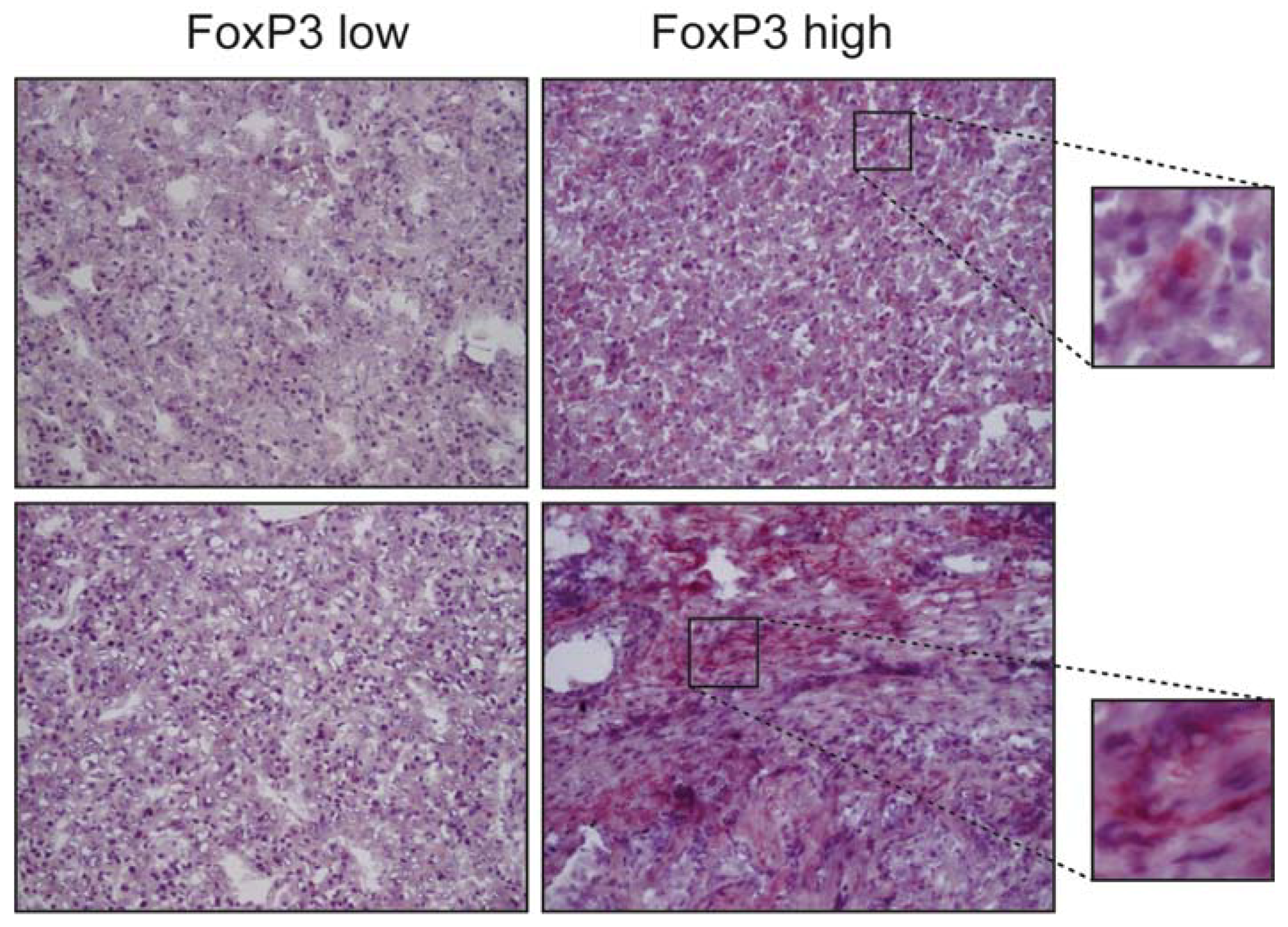{kind=link}
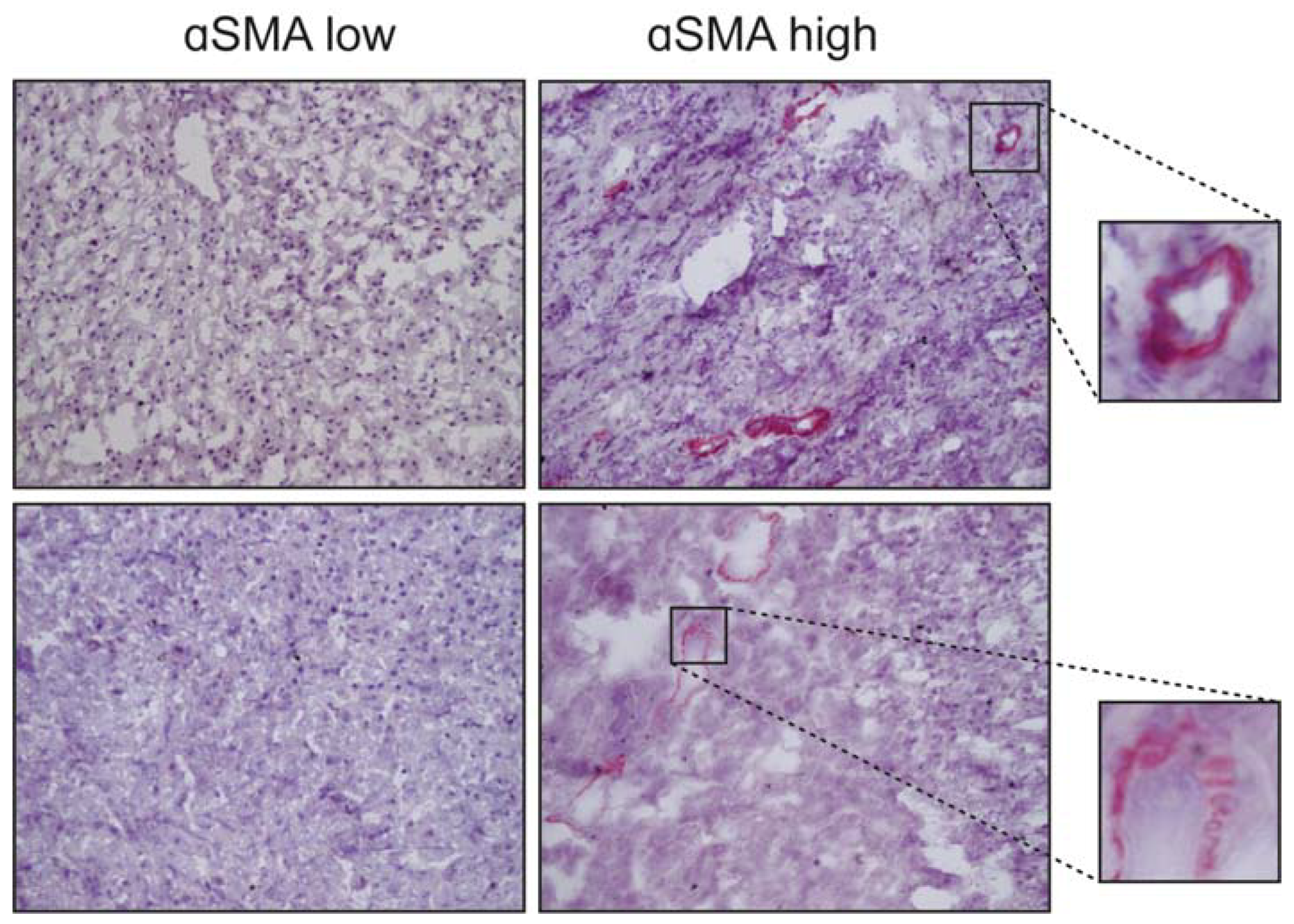{kind=link}
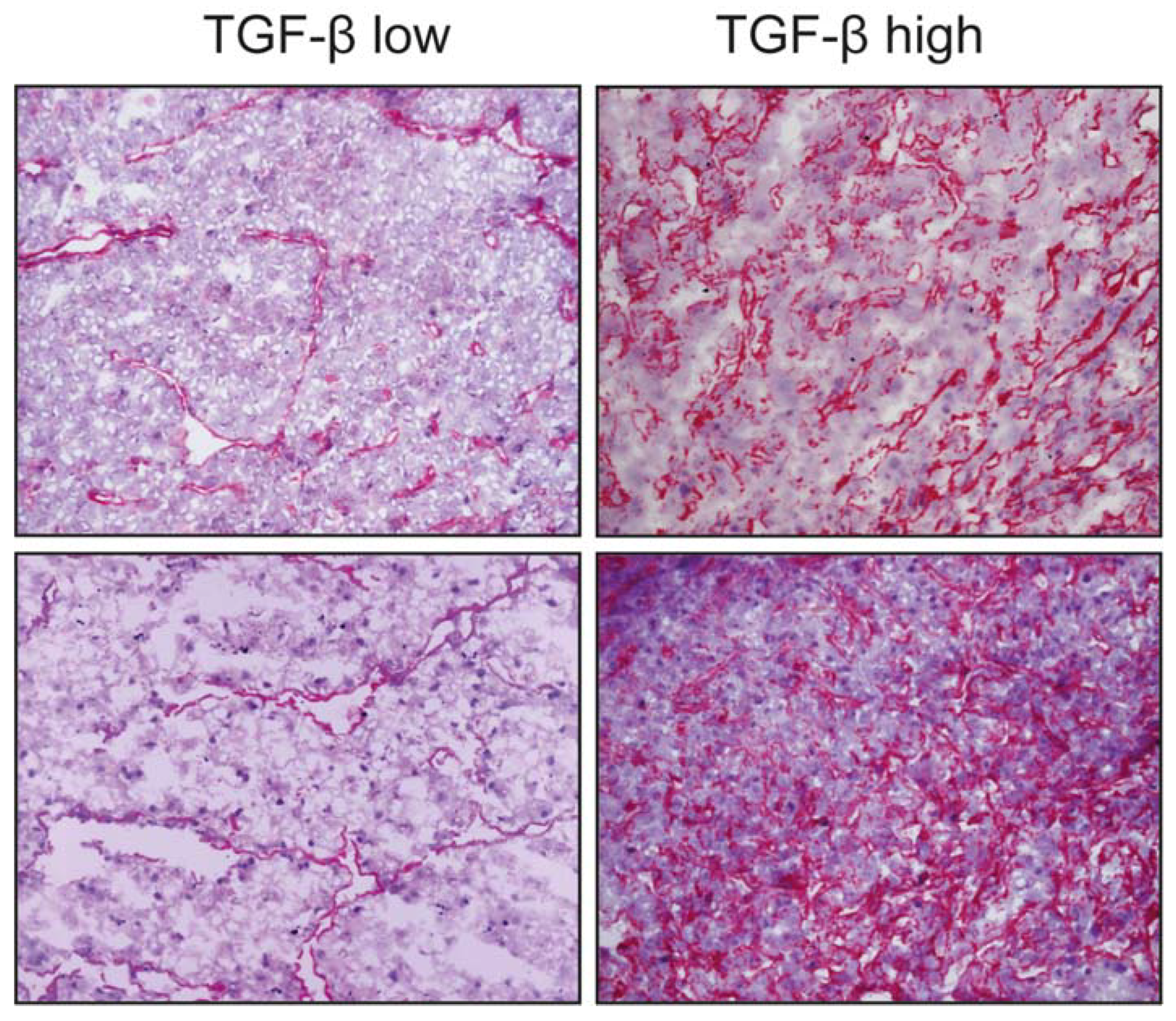{kind=link}
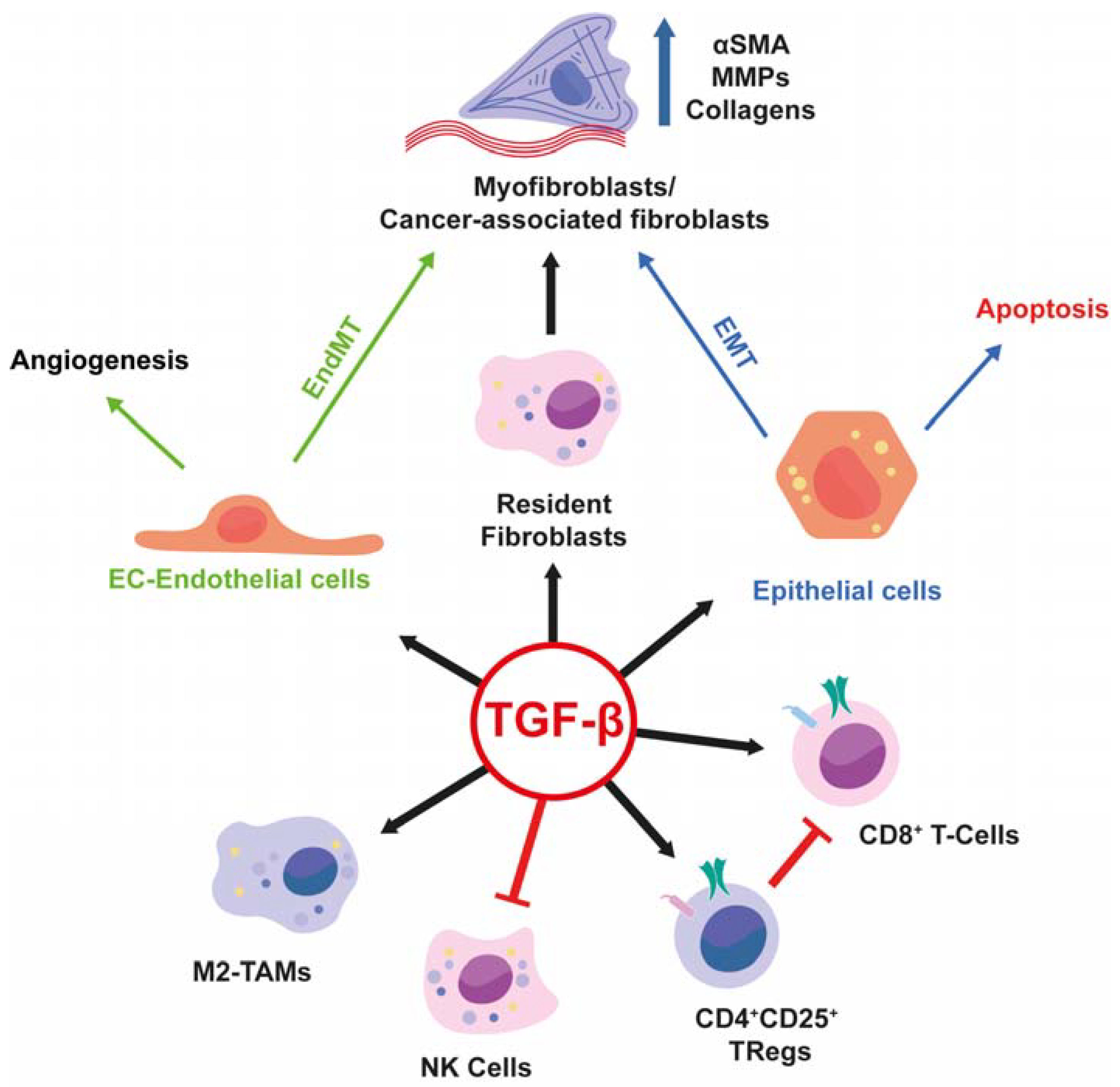{kind=link}
Abstract
:1. Introduction: Transforming Growth Factor-β (TGF-β), Fibrosis and Cancer
2. Stromal Activation of Latent TGF-β: Role of Integrins
3. TGF-β as a Master Regulator of Extracellular Matrix Remodelling
4. TGF-β Signalling in Fibrosis and Cancer-Related Invasiveness
5. Cancer-Associated Fibroblasts (CAFs): Their Origin and Links to the Epithelial-Mesenchymal Transition (EMT) in Fibrosis and Cancer
6. TGF-β Signalling in Key Cell Types Involved in the Pathogenic Microenvironment
7. Stromal Derived TGF-β Modulates the Cancer Immune Response
8. TGF-β as a Link between Fibrosis and Hepatocellular Carcinoma Progression
9. Systemic Therapy Based on TGF-β Pathway Inhibition
10. Concluding Remarks
Acknowledgments
Conflicts of Interest
References
- Nanthakumar, C.B.; Hatley, R.J.; Lemma, S.; Gauldie, J.; Marshall, R.P.; Macdonald, S.J. Dissecting fibrosis: Therapeutic insights from the small-molecule toolbox. Nat. Rev. Drug Discov. 2015, 14, 693–720. [Google Scholar] [CrossRef] [PubMed]
- Travers, J.G.; Kamal, F.A.; Robbins, J.; Yutzey, K.E.; Blaxall, B.C. Cardiac fibrosis: The Fibroblast awakens. Circ. Res. 2016, 118, 1021–1040. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.M.; Nikolic-Paterson, D.J.; Lan, H.Y. Inflammatory processes in renal fibrosis. Nat. Rev. Nephrol. 2014, 10, 493–503. [Google Scholar] [CrossRef] [PubMed]
- Hammerich, L.; Bangen, J.M.; Govaere, O.; Zimmermann, H.W.; Gassler, N.; Huss, S.; Liedtke, C.; Prinz, I.; Lira, S.A.; Luedde, T.; et al. Chemokine receptor CCR6-dependent accumulation of gammadelta T cells in injured liver restricts hepatic inflammation and fibrosis. Hepatology 2014, 59, 630–642. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Coussens, L.M. Accessories to the crime: Functions of cells recruited to the tumor microenvironment. Cancer Cell 2012, 21, 309–322. [Google Scholar] [CrossRef] [PubMed]
- Xavier, S.; Sahu, R.K.; Landes, S.G.; Yu, J.; Taylor, R.P.; Ayyadevara, S.; Megyesi, J.; Stallcup, W.B.; Duffield, J.S.; Reis, E.S.; et al. Pericytes and immune cells contribute to complement activation in tubulointerstitial fibrosis. Am. J. Physiol. Ren. Physiol. 2017, 312, F516–F532. [Google Scholar] [CrossRef] [PubMed]
- Pickup, M.W.; Owens, P.; Moses, H.L. TGF-β, bone morphogenetic protein, and activin signaling and the tumor microenvironment. Cold Spring Harb. Perspect. Biol. 2017, 9, a022285. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Kim, T.J.; Peng, D.H.; Duan, D.; Gibbons, D.L.; Yamauchi, M.; Jackson, J.R.; Le Saux, C.J.; Calhoun, C.; Peters, J.; et al. Fibroblast-specific inhibition of TGF-β1 signaling attenuates lung and tumor fibrosis. J. Clin. Investig. 2017, 127, 3675–3688. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.K.; Sheppard, D.; Chapman, H.A. TGF-β1 signaling and tissue fibrosis. Cold Spring Harb. Perspect. Biol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Okabe, H.; Hayashi, H.; Nakagawa, S.; Imai, K.; Nitta, H.; Arima, K.; Hashimoto, D.; Chikamoto, A.; Ishiko, T.; Beppu, T.; et al. Inducible factors for cancer-associated fibroblasts in liver cancer versus myofibroblasts in inflammatory liver disease. Histol. Histopathol. 2016, 31, 141–148. [Google Scholar] [PubMed]
- Yu, J.; Zhang, L.; Chen, A.; Xiang, G.; Wang, Y.; Wu, J.; Mitchelson, K.; Cheng, J.; Zhou, Y. Identification of the gene transcription and apoptosis mediated by TGF-β-Smad2/3-Smad4 signaling. J. Cell. Physiol. 2008, 215, 422–433. [Google Scholar] [CrossRef] [PubMed]
- Pardali, K.; Moustakas, A. Actions of TGF-β as tumor suppressor and pro-metastatic factor in human cancer. Biochim. Biophys. Acta 2007, 1775, 21–62. [Google Scholar] [CrossRef] [PubMed]
- Sundar, R.; Gudey, S.K.; Heldin, C.H.; Landstrom, M. TRAF6 promotes TGFβ-induced invasion and cell-cycle regulation via Lys63-linked polyubiquitination of Lys178 in TGFβ type I receptor. Cell Cycle 2015, 14, 554–565. [Google Scholar] [CrossRef] [PubMed]
- Hong, M.; Wilkes, M.C.; Penheiter, S.G.; Gupta, S.K.; Edens, M.; Leof, E.B. Non-Smad transforming growth factor-β signaling regulated by focal adhesion kinase binding the p85 subunit of phosphatidylinositol 3-kinase. J. Biol. Chem. 2011, 286, 17841–17850. [Google Scholar] [CrossRef] [PubMed]
- Horiguchi, M.; Ota, M.; Rifkin, D.B. Matrix control of transforming growth factor-β function. J. Biochem. 2012, 152, 321–329. [Google Scholar] [CrossRef] [PubMed]
- Robertson, I.B.; Horiguchi, M.; Zilberberg, L.; Dabovic, B.; Hadjiolova, K.; Rifkin, D.B. Latent TGF-β-binding proteins. Matrix Biol. J. Int. Soc. Matrix Biol. 2015, 47, 44–53. [Google Scholar] [CrossRef] [PubMed]
- Zilberberg, L.; Todorovic, V.; Dabovic, B.; Horiguchi, M.; Courousse, T.; Sakai, L.Y.; Rifkin, D.B. Specificity of latent TGF-β binding protein (LTBP) incorporation into matrix: Role of fibrillins and fibronectin. J. Cell. Physiol. 2012, 227, 3828–3836. [Google Scholar] [CrossRef] [PubMed]
- Ono, R.N.; Sengle, G.; Charbonneau, N.L.; Carlberg, V.; Bachinger, H.P.; Sasaki, T.; Lee-Arteaga, S.; Zilberberg, L.; Rifkin, D.B.; Ramirez, F.; et al. Latent transforming growth factor β-binding proteins and fibulins compete for fibrillin-1 and exhibit exquisite specificities in binding sites. J. Biol. Chem. 2009, 284, 16872–16881. [Google Scholar] [CrossRef] [PubMed]
- Troilo, H.; Steer, R.; Collins, R.F.; Kielty, C.M.; Baldock, C. Independent multimerization of Latent TGFβ Binding Protein-1 stabilized by cross-linking and enhanced by heparan sulfate. Sci. Rep. 2016, 6, 34347. [Google Scholar] [CrossRef] [PubMed]
- Khan, Z.; Marshall, J.F. The role of integrins in TGFβ activation in the tumour stroma. Cell Tissue Res. 2016, 365, 657–673. [Google Scholar] [CrossRef] [PubMed]
- Thomas, G.J.; Hart, I.R.; Speight, P.M.; Marshall, J.F. Binding of TGF-β1 latency-associated peptide (LAP) to αvβ6 integrin modulates behaviour of squamous carcinoma cells. Br. J. Cancer 2002, 87, 859–867. [Google Scholar] [CrossRef] [PubMed]
- Hinz, B. The extracellular matrix and transforming growth factor-β1: Tale of a strained relationship. Matrix Biol. J. Int. Soc. Matrix Biol. 2015, 47, 54–65. [Google Scholar] [CrossRef] [PubMed]
- Klingberg, F.; Chow, M.L.; Koehler, A.; Boo, S.; Buscemi, L.; Quinn, T.M.; Costell, M.; Alman, B.A.; Genot, E.; Hinz, B. Prestress in the extracellular matrix sensitizes latent TGF-β1 for activation. J. Cell Biol. 2014, 207, 283–297. [Google Scholar] [CrossRef] [PubMed]
- Wipff, P.J.; Rifkin, D.B.; Meister, J.J.; Hinz, B. Myofibroblast contraction activates latent TGF-β1 from the extracellular matrix. J. Cell Biol. 2007, 179, 1311–1323. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Mu, Z.; Dabovic, B.; Jurukovski, V.; Yu, D.; Sung, J.; Xiong, X.; Munger, J.S. Absence of integrin-mediated TGFβ1 activation in vivo recapitulates the phenotype of TGFβ1-null mice. J. Cell Biol. 2007, 176, 787–793. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Zhu, J.; Wang, R.; Chen, X.; Mi, L.; Walz, T.; Springer, T.A. Latent TGF-β structure and activation. Nature 2011, 474, 343–349. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.; Hudson, N.E.; Lu, C.; Springer, T.A. Structural determinants of integrin β-subunit specificity for latent TGF-β. Nat. Struct. Mol. Biol. 2014, 21, 1091–1096. [Google Scholar] [CrossRef] [PubMed]
- Henderson, N.C.; Arnold, T.D.; Katamura, Y.; Giacomini, M.M.; Rodriguez, J.D.; McCarty, J.H.; Pellicoro, A.; Raschperger, E.; Betsholtz, C.; Ruminski, P.G.; et al. Targeting of αv integrin identifies a core molecular pathway that regulates fibrosis in several organs. Nat. Med. 2013, 19, 1617–1624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, Z.W.; Ikenaga, N.; Liu, S.B.; Sverdlov, D.Y.; Vaid, K.A.; Dixit, R.; Weinreb, P.H.; Violette, S.; Sheppard, D.; Schuppan, D.; et al. Integrin αvβ6 critically regulates hepatic progenitor cell function and promotes ductular reaction, fibrosis, and tumorigenesis. Hepatology 2016, 63, 217–232. [Google Scholar] [CrossRef] [PubMed]
- Griggs, L.A.; Hassan, N.T.; Malik, R.S.; Griffin, B.P.; Martinez, B.A.; Elmore, L.W.; Lemmon, C.A. Fibronectin fibrils regulate TGF-β1-induced Epithelial-Mesenchymal Transition. Matrix Biol. J. Int. Soc. Matrix Biol. 2017, 60–61, 157–175. [Google Scholar] [CrossRef] [PubMed]
- Ignotz, R.A.; Massague, J. Transforming growth factor-β stimulates the expression of fibronectin and collagen and their incorporation into the extracellular matrix. J. Biol. Chem. 1986, 261, 4337–4345. [Google Scholar] [PubMed]
- Heino, J.; Ignotz, R.A.; Hemler, M.E.; Crouse, C.; Massague, J. Regulation of cell adhesion receptors by transforming growth factor-β. Concomitant regulation of integrins that share a common β 1 subunit. J. Biol. Chem. 1989, 264, 380–388. [Google Scholar] [PubMed]
- Edwards, D.R.; Murphy, G.; Reynolds, J.J.; Whitham, S.E.; Docherty, A.J.; Angel, P.; Heath, J.K. Transforming growth factor β modulates the expression of collagenase and metalloproteinase inhibitor. EMBO J. 1987, 6, 1899–1904. [Google Scholar] [PubMed]
- Nakatsukasa, H.; Nagy, P.; Evarts, R.P.; Hsia, C.C.; Marsden, E.; Thorgeirsson, S.S. Cellular distribution of transforming growth factor-β 1 and procollagen types I, III, and IV transcripts in carbon tetrachloride-induced rat liver fibrosis. J. Clin. Investig. 1990, 85, 1833–1843. [Google Scholar] [CrossRef] [PubMed]
- Hecker, L.; Vittal, R.; Jones, T.; Jagirdar, R.; Luckhardt, T.R.; Horowitz, J.C.; Pennathur, S.; Martinez, F.J.; Thannickal, V.J. NADPH oxidase-4 mediates myofibroblast activation and fibrogenic responses to lung injury. Nat. Med. 2009, 15, 1077. [Google Scholar] [CrossRef] [PubMed]
- Sancho, P.; Mainez, J.; Crosas-Molist, E.; Roncero, C.; Fernandez-Rodriguez, C.M.; Pinedo, F.; Huber, H.; Eferl, R.; Mikulits, W.; Fabregat, I. NADPH oxidase NOX4 mediates stellate cell activation and hepatocyte cell death during liver fibrosis development. PLoS ONE 2012, 7, e45285. [Google Scholar] [CrossRef] [PubMed]
- Fabregat, I.; Moreno-Caceres, J.; Sanchez, A.; Dooley, S.; Dewidar, B.; Giannelli, G.; ten Dijke, P.; Consortium, I.-L. TGF-β signalling and liver disease. FEBS J. 2016, 283, 2219–2232. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.M.; Nikolic-Paterson, D.J.; Lan, H.Y. TGF-β: The master regulator of fibrosis. Nat. Rev. Nephrol. 2016, 12, 325–338. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, T.; O’Reilly, P.; Antony, V.B.; Gaggar, A.; Thannickal, V.J. Matrix remodeling in pulmonary fibrosis and emphysema. Am. J. Respir. Cell Mol. Biol. 2016, 54, 751–760. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Zhen, G.; Chai, Y.; Xie, L.; Crane, J.L.; Farber, E.; Farber, C.R.; Luo, X.; Gao, P.; Cao, X.; et al. RhoA determines lineage fate of mesenchymal stem cells by modulating CTGF-VEGF complex in extracellular matrix. Nat. Commun. 2016, 7, 11455. [Google Scholar] [CrossRef] [PubMed]
- Crosas-Molist, E.; Meirelles, T.; Lopez-Luque, J.; Serra-Peinado, C.; Selva, J.; Caja, L.; Gorbenko del Blanco, D.; Uriarte, J.J.; Bertran, E.; Mendizabal, Y.; et al. Vascular smooth muscle cell phenotypic changes in patients with Marfan syndrome. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 960–972. [Google Scholar] [CrossRef] [PubMed]
- Busnadiego, O.; Gonzalez-Santamaria, J.; Lagares, D.; Guinea-Viniegra, J.; Pichol-Thievend, C.; Muller, L.; Rodriguez-Pascual, F. LOXL4 is induced by transforming growth factor β1 through Smad and JunB/Fra2 and contributes to vascular matrix remodeling. Mol. Cell. Biol. 2013, 33, 2388–2401. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.C.; Tse, A.P.; Huang, Y.P.; Zhu, Y.T.; Chiu, D.K.; Lai, R.K.; Au, S.L.; Kai, A.K.; Lee, J.M.; Wei, L.L.; et al. Lysyl oxidase-like 2 is critical to tumor microenvironment and metastatic niche formation in hepatocellular carcinoma. Hepatology 2014, 60, 1645–1658. [Google Scholar] [CrossRef] [PubMed]
- Varadaraj, A.; Jenkins, L.M.; Singh, P.; Chanda, A.; Snider, J.; Lee, N.Y.; Amsalem-Zafran, A.R.; Ehrlich, M.; Henis, Y.I.; Mythreye, K. TGF-β triggers rapid fibrillogenesis via a novel TβRII-dependent fibronectin-trafficking mechanism. Mol. Biol. Cell 2017, 28, 1195–1207. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.K.; Bhattacharyya, S.; Varga, J. The tumor suppressor p53 abrogates Smad-dependent collagen gene induction in mesenchymal cells. J. Biol. Chem. 2004, 279, 47455–47463. [Google Scholar] [CrossRef] [PubMed]
- Samarakoon, R.; Overstreet, J.M.; Higgins, P.J. TGF-β signaling in tissue fibrosis: Redox controls, target genes and therapeutic opportunities. Cell. Signal. 2013, 25, 264–268. [Google Scholar] [CrossRef] [PubMed]
- Phanish, M.K.; Wahab, N.A.; Colville-Nash, P.; Hendry, B.M.; Dockrell, M.E. The differential role of Smad2 and Smad3 in the regulation of pro-fibrotic TGFβ1 responses in human proximal-tubule epithelial cells. Biochem. J. 2006, 393, 601–607. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Liu, H.; Meyer, C.; Li, J.; Nadalin, S.; Konigsrainer, A.; Weng, H.; Dooley, S.; ten Dijke, P. Transforming growth factor-β (TGF-β)-mediated connective tissue growth factor (CTGF) expression in hepatic stellate cells requires Stat3 signaling activation. J. Biol. Chem. 2013, 288, 30708–30719. [Google Scholar] [CrossRef] [PubMed]
- Tang, L.Y.; Heller, M.; Meng, Z.; Yu, L.R.; Tang, Y.; Zhou, M.; Zhang, Y.E. Transforming growth factor-β (TGF-β) directly activates the JAK1-STAT3 axis to induce hepatic fibrosis in coordination with the SMAD pathway. J. Biol. Chem. 2017, 292, 4302–4312. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Wang, H.; Liao, H.J.; Hu, W.; Gewin, L.; Mernaugh, G.; Zhang, S.; Zhang, Z.Y.; Vega-Montoto, L.; Vanacore, R.M.; et al. Integrin-mediated type II TGF-β receptor tyrosine dephosphorylation controls SMAD-dependent profibrotic signaling. J. Clin. Investig. 2014, 124, 3295–3310. [Google Scholar] [CrossRef] [PubMed]
- Ding, Q.; Subramanian, I.; Luckhardt, T.R.; Che, P.; Waghray, M.; Zhao, X.K.; Bone, N.; Kurundkar, A.R.; Hecker, L.; Hu, M.; et al. Focal adhesion kinase signaling determines the fate of lung epithelial cells in response to TGF-β. Am. J. Physiol. Lung Cell. Mol. Physiol. 2017, 312, L926–L935. [Google Scholar] [CrossRef] [PubMed]
- Dufton, N.P.; Peghaire, C.R.; Osuna-Almagro, L.; Raimondi, C.; Kalna, V.; Chuahan, A.; Webb, G.; Yang, Y.; Birdsey, G.M.; Lalor, P.; et al. Dynamic regulation of canonical TGFβ signalling by endothelial transcription factor ERG protects from liver fibrogenesis. Nat. Commun. 2017, 8, 895. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, M.; Matsushita, Y.; Horikawa, M.; Higashi, K.; Tomigahara, Y.; Kaneko, H.; Shirasaki, F.; Fujimoto, M.; Takehara, K.; Sato, S. A novel inhibitor of Smad-dependent transcriptional activation suppresses tissue fibrosis in mouse models of systemic sclerosis. Arthritis Rheum. 2009, 60, 3465–3475. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.H.; Jung, M.Y.; Yin, X.; Andrianifahanana, M.; Hernandez, D.M.; Leof, E.B. Cell-penetrating peptides selectively targeting SMAD3 inhibit profibrotic TGF-β signaling. J. Clin. Investig. 2017, 127, 2541–2554. [Google Scholar] [CrossRef] [PubMed]
- Sorrentino, A.; Thakur, N.; Grimsby, S.; Marcusson, A.; von Bulow, V.; Schuster, N.; Zhang, S.; Heldin, C.H.; Landstrom, M. The type I TGF-β receptor engages TRAF6 to activate TAK1 in a receptor kinase-independent manner. Nat. Cell Biol. 2008, 10, 1199–1207. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhou, F.; Garcia de Vinuesa, A.; de Kruijf, E.M.; Mesker, W.E.; Hui, L.; Drabsch, Y.; Li, Y.; Bauer, A.; Rousseau, A.; et al. TRAF4 promotes TGF-β receptor signaling and drives breast cancer metastasis. Mol. Cell 2013, 51, 559–572. [Google Scholar] [CrossRef] [PubMed]
- Hamidi, A.; Song, J.; Thakur, N.; Itoh, S.; Marcusson, A.; Bergh, A.; Heldin, C.H.; Landstrom, M. TGF-β promotes PI3K-AKT signaling and prostate cancer cell migration through the TRAF6-mediated ubiquitylation of p85α. Sci. Signal. 2017, 10, eaal4186. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Wang, X.; Wang, Q.; Deng, Y.; Li, K.; Zhang, M.; Zhang, Q.; Zhou, J.; Wang, H.Y.; Bai, P.; et al. USP2a Supports Metastasis by Tuning TGF-β Signaling. Cell Rep. 2018, 22, 2442–2454. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.K.; Pardoux, C.; Hall, M.C.; Lee, P.S.; Warburton, D.; Qing, J.; Smith, S.M.; Derynck, R. TGF-β activates Erk MAP kinase signalling through direct phosphorylation of ShcA. EMBO J. 2007, 26, 3957–3967. [Google Scholar] [CrossRef] [PubMed]
- Niculescu-Duvaz, I.; Phanish, M.K.; Colville-Nash, P.; Dockrell, M.E. The TGFβ1-induced fibronectin in human renal proximal tubular epithelial cells is p38 MAP kinase dependent and Smad independent. Nephron Exp. Nephrol. 2007, 105, e108–e116. [Google Scholar] [CrossRef] [PubMed]
- Ding, Z.Y.; Jin, G.N.; Liang, H.F.; Wang, W.; Chen, W.X.; Datta, P.K.; Zhang, M.Z.; Zhang, B.; Chen, X.P. Transforming growth factor β induces expression of connective tissue growth factor in hepatic progenitor cells through Smad independent signaling. Cell. Signal. 2013, 25, 1981–1992. [Google Scholar] [CrossRef] [PubMed]
- Neef, M.; Ledermann, M.; Saegesser, H.; Schneider, V.; Reichen, J. Low-dose oral rapamycin treatment reduces fibrogenesis, improves liver function, and prolongs survival in rats with established liver cirrhosis. J. Hepatol. 2006, 45, 786–796. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Wilkes, M.C.; Leof, E.B.; Hirschberg, R. Noncanonical TGF-β pathways, mTORC1 and Abl, in renal interstitial fibrogenesis. Am. J. Physiol. Ren. Physiol. 2010, 298, F142–F149. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Ren, J.; Liu, X.; Jiang, L.; He, W.; Yuan, W.; Yang, J.; Dai, C. Rictor/mTORC2 signaling mediates TGFβ1-induced fibroblast activation and kidney fibrosis. Kidney Int. 2015, 88, 515–527. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Chen, H.; Wang, C.; Peng, Y.; Sun, L.; Liu, H.; Liu, F. Rapamycin ameliorates kidney fibrosis by inhibiting the activation of mTOR signaling in interstitial macrophages and myofibroblasts. PLoS ONE 2012, 7, e33626. [Google Scholar] [CrossRef] [PubMed]
- Lamouille, S.; Connolly, E.; Smyth, J.W.; Akhurst, R.J.; Derynck, R. TGF-β-induced activation of mTOR complex 2 drives epithelial-mesenchymal transition and cell invasion. J. Cell Sci. 2012, 125 Pt 5, 1259–1273. [Google Scholar] [CrossRef] [PubMed]
- Chang, W.; Wei, K.; Ho, L.; Berry, G.J.; Jacobs, S.S.; Chang, C.H.; Rosen, G.D. A critical role for the mTORC2 pathway in lung fibrosis. PLoS ONE 2014, 9, e106155. [Google Scholar] [CrossRef] [PubMed]
- Tsapara, A.; Luthert, P.; Greenwood, J.; Hill, C.S.; Matter, K.; Balda, M.S. The RhoA activator GEF-H1/Lfc is a transforming growth factor-β target gene and effector that regulates α-smooth muscle actin expression and cell migration. Mol. Biol. Cell 2010, 21, 860–870. [Google Scholar] [CrossRef] [PubMed]
- Jain, M.; Rivera, S.; Monclus, E.A.; Synenki, L.; Zirk, A.; Eisenbart, J.; Feghali-Bostwick, C.; Mutlu, G.M.; Budinger, G.R.; Chandel, N.S. Mitochondrial reactive oxygen species regulate transforming growth factor-β signaling. J. Biol. Chem. 2013, 288, 770–777. [Google Scholar] [CrossRef] [PubMed]
- Cencetti, F.; Bernacchioni, C.; Nincheri, P.; Donati, C.; Bruni, P. Transforming growth factor-β1 induces transdifferentiation of myoblasts into myofibroblasts via up-regulation of sphingosine kinase-1/S1P3 axis. Mol. Biol. Cell 2010, 21, 1111–1124. [Google Scholar] [CrossRef] [PubMed]
- Kisseleva, T. The origin of fibrogenic myofibroblasts in fibrotic liver. Hepatology 2017, 65, 1039–1043. [Google Scholar] [CrossRef] [PubMed]
- LeBleu, V.S.; Taduri, G.; O’Connell, J.; Teng, Y.; Cooke, V.G.; Woda, C.; Sugimoto, H.; Kalluri, R. Origin and function of myofibroblasts in kidney fibrosis. Nat. Med. 2013, 19, 1047–1053. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.K.; Kugler, M.C.; Wolters, P.J.; Robillard, L.; Galvez, M.G.; Brumwell, A.N.; Sheppard, D.; Chapman, H.A. Alveolar epithelial cell mesenchymal transition develops in vivo during pulmonary fibrosis and is regulated by the extracellular matrix. Proc. Natl. Acad. Sci. USA 2006, 103, 13180–13185. [Google Scholar] [CrossRef] [PubMed]
- Scholten, D.; Osterreicher, C.H.; Scholten, A.; Iwaisako, K.; Gu, G.; Brenner, D.A.; Kisseleva, T. Genetic labeling does not detect epithelial-to-mesenchymal transition of cholangiocytes in liver fibrosis in mice. Gastroenterology 2010, 139, 987–998. [Google Scholar] [CrossRef] [PubMed]
- Nieto, M.A.; Huang, R.Y.; Jackson, R.A.; Thiery, J.P. EMT: 2016. Cell 2016, 166, 21–45. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [PubMed]
- Willis, B.C.; Liebler, J.M.; Luby-Phelps, K.; Nicholson, A.G.; Crandall, E.D.; du Bois, R.M.; Borok, Z. Induction of epithelial-mesenchymal transition in alveolar epithelial cells by transforming growth factor-β1: Potential role in idiopathic pulmonary fibrosis. Am. J. Pathol. 2005, 166, 1321–1332. [Google Scholar] [CrossRef]
- Balli, D.; Ustiyan, V.; Zhang, Y.; Wang, I.C.; Masino, A.J.; Ren, X.; Whitsett, J.A.; Kalinichenko, V.V.; Kalin, T.V. Foxm1 transcription factor is required for lung fibrosis and epithelial-to-mesenchymal transition. EMBO J. 2013, 32, 231–244. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.J.; Ye, H.; Zhang, Q.; Li, F.Z.; Song, L.J.; Yang, J.; Mu, Q.; Rao, S.S.; Cai, P.C.; Xiang, F.; et al. Bleomycin induced epithelial-mesenchymal transition (EMT) in pleural mesothelial cells. Toxicol. Appl. Pharmacol. 2015, 283, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Bakin, A.V.; Tomlinson, A.K.; Bhowmick, N.A.; Moses, H.L.; Arteaga, C.L. Phosphatidylinositol 3-kinase function is required for transforming growth factor β-mediated epithelial to mesenchymal transition and cell migration. J. Biol. Chem. 2000, 275, 36803–36810. [Google Scholar] [CrossRef] [PubMed]
- Lamouille, S.; Derynck, R. Cell size and invasion in TGF-β-induced epithelial to mesenchymal transition is regulated by activation of the mTOR pathway. J. Cell Biol. 2007, 178, 437–451. [Google Scholar] [CrossRef] [PubMed]
- Gal, A.; Sjoblom, T.; Fedorova, L.; Imreh, S.; Beug, H.; Moustakas, A. Sustained TGF β exposure suppresses Smad and non-Smad signalling in mammary epithelial cells, leading to EMT and inhibition of growth arrest and apoptosis. Oncogene 2008, 27, 1218–1230. [Google Scholar] [CrossRef] [PubMed]
- Del Castillo, G.; Murillo, M.M.; Alvarez-Barrientos, A.; Bertran, E.; Fernandez, M.; Sanchez, A.; Fabregat, I. Autocrine production of TGF-β confers resistance to apoptosis after an epithelial-mesenchymal transition process in hepatocytes: Role of EGF receptor ligands. Exp. Cell Res. 2006, 312, 2860–2871. [Google Scholar] [CrossRef] [PubMed]
- Caja, L.; Bertran, E.; Campbell, J.; Fausto, N.; Fabregat, I. The transforming growth factor-β (TGF-β) mediates acquisition of a mesenchymal stem cell-like phenotype in human liver cells. J. Cell. Physiol. 2011, 226, 1214–1223. [Google Scholar] [CrossRef] [PubMed]
- Franco, D.L.; Mainez, J.; Vega, S.; Sancho, P.; Murillo, M.M.; de Frutos, C.A.; del Castillo, G.; Lopez-Blau, C.; Fabregat, I.; Nieto, M.A. Snail1 suppresses TGF-β-induced apoptosis and is sufficient to trigger EMT in hepatocytes. J. Cell Sci. 2010, 123, 3467–3477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nitta, T.; Kim, J.S.; Mohuczy, D.; Behrns, K.E. Murine cirrhosis induces hepatocyte epithelial mesenchymal transition and alterations in survival signaling pathways. Hepatology 2008, 48, 909–919. [Google Scholar] [CrossRef] [PubMed]
- Caja, L.; Ortiz, C.; Bertran, E.; Murillo, M.M.; Miro-Obradors, M.J.; Palacios, E.; Fabregat, I. Differential intracellular signalling induced by TGF-β in rat adult hepatocytes and hepatoma cells: Implications in liver carcinogenesis. Cell. Signal. 2007, 19, 683–694. [Google Scholar] [CrossRef] [PubMed]
- Zeisberg, E.M.; Potenta, S.E.; Sugimoto, H.; Zeisberg, M.; Kalluri, R. Fibroblasts in kidney fibrosis emerge via endothelial-to-mesenchymal transition. J. Am. Soc. Nephrol. JASN 2008, 19, 2282–2287. [Google Scholar] [CrossRef] [PubMed]
- Iwano, M.; Plieth, D.; Danoff, T.M.; Xue, C.; Okada, H.; Neilson, E.G. Evidence that fibroblasts derive from epithelium during tissue fibrosis. J. Clin. Investig. 2002, 110, 341–350. [Google Scholar] [CrossRef] [PubMed]
- Humphreys, B.D.; Lin, S.L.; Kobayashi, A.; Hudson, T.E.; Nowlin, B.T.; Bonventre, J.V.; Valerius, M.T.; McMahon, A.P.; Duffield, J.S. Fate tracing reveals the pericyte and not epithelial origin of myofibroblasts in kidney fibrosis. Am. J. Pathol. 2010, 176, 85–97. [Google Scholar] [CrossRef] [PubMed]
- Grande, M.T.; Sanchez-Laorden, B.; Lopez-Blau, C.; De Frutos, C.A.; Boutet, A.; Arevalo, M.; Rowe, R.G.; Weiss, S.J.; Lopez-Novoa, J.M.; Nieto, M.A. Snail1-induced partial epithelial-to-mesenchymal transition drives renal fibrosis in mice and can be targeted to reverse established disease. Nat. Med. 2015, 21, 989–997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lovisa, S.; LeBleu, V.S.; Tampe, B.; Sugimoto, H.; Vadnagara, K.; Carstens, J.L.; Wu, C.C.; Hagos, Y.; Burckhardt, B.C.; Pentcheva-Hoang, T.; et al. Epithelial-to-mesenchymal transition induces cell cycle arrest and parenchymal damage in renal fibrosis. Nat. Med. 2015, 21, 998–1009. [Google Scholar] [CrossRef] [PubMed]
- Tanjore, H.; Xu, X.C.; Polosukhin, V.V.; Degryse, A.L.; Li, B.; Han, W.; Sherrill, T.P.; Plieth, D.; Neilson, E.G.; Blackwell, T.S.; et al. Contribution of epithelial-derived fibroblasts to bleomycin-induced lung fibrosis. Am. J. Respir. Crit. Care Med. 2009, 180, 657–665. [Google Scholar] [CrossRef] [PubMed]
- DeMaio, L.; Buckley, S.T.; Krishnaveni, M.S.; Flodby, P.; Dubourd, M.; Banfalvi, A.; Xing, Y.; Ehrhardt, C.; Minoo, P.; Zhou, B.; et al. Ligand-independent transforming growth factor-β type I receptor signalling mediates type I collagen-induced epithelial-mesenchymal transition. J. Pathol. 2012, 226, 633–644. [Google Scholar] [CrossRef] [PubMed]
- Rock, J.R.; Barkauskas, C.E.; Cronce, M.J.; Xue, Y.; Harris, J.R.; Liang, J.; Noble, P.W.; Hogan, B.L. Multiple stromal populations contribute to pulmonary fibrosis without evidence for epithelial to mesenchymal transition. Proc. Natl. Acad. Sci. USA 2011, 108, E1475–E1483. [Google Scholar] [CrossRef] [PubMed]
- Zeisberg, M.; Yang, C.; Martino, M.; Duncan, M.B.; Rieder, F.; Tanjore, H.; Kalluri, R. Fibroblasts derive from hepatocytes in liver fibrosis via epithelial to mesenchymal transition. J. Biol. Chem. 2007, 282, 23337–23347. [Google Scholar] [CrossRef] [PubMed]
- Taura, K.; Miura, K.; Iwaisako, K.; Osterreicher, C.H.; Kodama, Y.; Penz-Osterreicher, M.; Brenner, D.A. Hepatocytes do not undergo epithelial-mesenchymal transition in liver fibrosis in mice. Hepatology 2009, 51, 1027–1036. [Google Scholar] [CrossRef] [PubMed]
- Chu, A.S.; Diaz, R.; Hui, J.J.; Yanger, K.; Zong, Y.; Alpini, G.; Stanger, B.Z.; Wells, R.G. Lineage tracing demonstrates no evidence of cholangiocyte epithelial-to-mesenchymal transition in murine models of hepatic fibrosis. Hepatology 2011, 53, 1685–1695. [Google Scholar] [CrossRef] [PubMed]
- Rowe, R.G.; Lin, Y.; Shimizu-Hirota, R.; Hanada, S.; Neilson, E.G.; Greenson, J.K.; Weiss, S.J. Hepatocyte-derived Snail1 propagates liver fibrosis progression. Mol. Cell. Biol. 2011, 31, 2392–2403. [Google Scholar] [CrossRef] [PubMed]
- Flier, S.N.; Tanjore, H.; Kokkotou, E.G.; Sugimoto, H.; Zeisberg, M.; Kalluri, R. Identification of epithelial to mesenchymal transition as a novel source of fibroblasts in intestinal fibrosis. J. Biol. Chem. 2010, 285, 20202–20212. [Google Scholar] [CrossRef] [PubMed]
- Baulida, J. Epithelial-to-mesenchymal transition transcription factors in cancer-associated fibroblasts. Mol. Oncol. 2017, 11, 847–859. [Google Scholar] [CrossRef] [PubMed]
- Shiga, K.; Hara, M.; Nagasaki, T.; Sato, T.; Takahashi, H.; Takeyama, H. Cancer-associated fibroblasts: Their characteristics and their roles in tumor growth. Cancers 2015, 7, 2443–2458. [Google Scholar] [CrossRef] [PubMed]
- Petersen, O.W.; Nielsen, H.L.; Gudjonsson, T.; Villadsen, R.; Rank, F.; Niebuhr, E.; Bissell, M.J.; Ronnov-Jessen, L. Epithelial to mesenchymal transition in human breast cancer can provide a nonmalignant stroma. Am. J. Pathol. 2003, 162, 391–402. [Google Scholar] [CrossRef]
- Mink, S.R.; Vashistha, S.; Zhang, W.; Hodge, A.; Agus, D.B.; Jain, A. Cancer-associated fibroblasts derived from EGFR-TKI-resistant tumors reverse EGFR pathway inhibition by EGFR-TKIs. Mol. Cancer Res. MCR 2010, 8, 809–820. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Wu, C.P.; Pan, J.Y.; Zheng, W.W.; Cao, X.J.; Fan, G.K. Cancer-associated fibroblasts in a human HEp-2 established laryngeal xenografted tumor are not derived from cancer cells through epithelial-mesenchymal transition, phenotypically activated but karyotypically normal. PLoS ONE 2015, 10, e0117405. [Google Scholar]
- Stanisavljevic, J.; Loubat-Casanovas, J.; Herrera, M.; Luque, T.; Pena, R.; Lluch, A.; Albanell, J.; Bonilla, F.; Rovira, A.; Pena, C.; et al. Snail1-expressing fibroblasts in the tumor microenvironment display mechanical properties that support metastasis. Cancer Res. 2015, 75, 284–295. [Google Scholar] [CrossRef] [PubMed]
- Herrera, A.; Herrera, M.; Alba-Castellon, L.; Silva, J.; Garcia, V.; Loubat-Casanovas, J.; Alvarez-Cienfuegos, A.; Miguel Garcia, J.; Rodriguez, R.; Gil, B.; et al. Protumorigenic effects of Snail-expression fibroblasts on colon cancer cells. Int. J. Cancer 2014, 134, 2984–2990. [Google Scholar] [CrossRef] [PubMed]
- Sung, C.O.; Lee, K.W.; Han, S.; Kim, S.H. Twist1 is up-regulated in gastric cancer-associated fibroblasts with poor clinical outcomes. Am. J. Pathol. 2011, 179, 1827–1838. [Google Scholar] [CrossRef] [PubMed]
- Galvan, J.A.; Helbling, M.; Koelzer, V.H.; Tschan, M.P.; Berger, M.D.; Hadrich, M.; Schnuriger, B.; Karamitopoulou, E.; Dawson, H.; Inderbitzin, D.; et al. TWIST1 and TWIST2 promoter methylation and protein expression in tumor stroma influence the epithelial-mesenchymal transition-like tumor budding phenotype in colorectal cancer. Oncotarget 2015, 6, 874–885. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Palmero, I.; Torres, S.; Bartolome, R.A.; Pelaez-Garcia, A.; Larriba, M.J.; Lopez-Lucendo, M.; Pena, C.; Escudero-Paniagua, B.; Munoz, A.; Casal, J.I. Twist1-induced activation of human fibroblasts promotes matrix stiffness by upregulating palladin and collagen α1VI. Oncogene 2016, 35, 5224–5236. [Google Scholar] [CrossRef] [PubMed]
- Bronsert, P.; Kohler, I.; Timme, S.; Kiefer, S.; Werner, M.; Schilling, O.; Vashist, Y.; Makowiec, F.; Brabletz, T.; Hopt, U.T.; et al. Prognostic significance of Zinc finger E-box binding homeobox 1 (ZEB1) expression in cancer cells and cancer-associated fibroblasts in pancreatic head cancer. Surgery 2014, 156, 97–108. [Google Scholar] [CrossRef] [PubMed]
- Galvan, J.A.; Zlobec, I.; Wartenberg, M.; Lugli, A.; Gloor, B.; Perren, A.; Karamitopoulou, E. Expression of E-cadherin repressors SNAIL, ZEB1 and ZEB2 by tumour and stromal cells influences tumour-budding phenotype and suggests heterogeneity of stromal cells in pancreatic cancer. Br. J. Cancer 2015, 112, 1944–1950. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Xiao, C.H.; Tan, L.D.; Wang, Q.S.; Li, X.Q.; Feng, Y.M. Cancer-associated fibroblasts induce epithelial-mesenchymal transition of breast cancer cells through paracrine TGF-β signalling. Br. J. Cancer 2014, 110, 724–732. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, J.; Lu, Q.; Shen, B.; Huang, X.; Shen, L.; Zheng, X.; Huang, R.; Yan, J.; Guo, H. TGFb1 secreted by cancer-associated fibroblasts induces epithelial-mesenchymal transition of bladder cancer cells through lncRNA-ZEB2NAT. Sci. Rep. 2015, 5, 11924. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.; Jia, H.H.; Xu, Y.Q.; Zhou, X.; Zhao, X.H.; Wang, Y.F.; Song, X.; Zhu, Z.Y.; Sun, T.; Dou, Y.; et al. Paracrine and epigenetic control of CAF-induced metastasis: The role of HOTAIR stimulated by TGF-ss1 secretion. Mol. Cancer 2018, 17, 5. [Google Scholar] [CrossRef] [PubMed]
- Yeung, T.L.; Leung, C.S.; Wong, K.K.; Samimi, G.; Thompson, M.S.; Liu, J.; Zaid, T.M.; Ghosh, S.; Birrer, M.J.; Mok, S.C. TGF-β modulates ovarian cancer invasion by upregulating CAF-derived versican in the tumor microenvironment. Cancer Res. 2013, 73, 5016–5028. [Google Scholar] [CrossRef] [PubMed]
- Calon, A.; Espinet, E.; Palomo-Ponce, S.; Tauriello, D.V.; Iglesias, M.; Cespedes, M.V.; Sevillano, M.; Nadal, C.; Jung, P.; Zhang, X.H.; et al. Dependency of colorectal cancer on a TGF-b-driven program in stromal cells for metastasis initiation. Cancer Cell 2012, 22, 571–584. [Google Scholar] [CrossRef] [PubMed]
- Razani, B.; Zhang, X.L.; Bitzer, M.; von Gersdorff, G.; Bottinger, E.P.; Lisanti, M.P. Caveolin-1 regulates transforming growth factor (TGF)-β/SMAD signaling through an interaction with the TGF-β type I receptor. J. Biol. Chem. 2001, 276, 6727–6738. [Google Scholar] [CrossRef] [PubMed]
- Guido, C.; Whitaker-Menezes, D.; Capparelli, C.; Balliet, R.; Lin, Z.; Pestell, R.G.; Howell, A.; Aquila, S.; Ando, S.; Martinez-Outschoorn, U.; et al. Metabolic reprogramming of cancer-associated fibroblasts by TGF-b drives tumor growth connecting TGF-b signaling with “Warburg-like” cancer metabolism and L-lactate production. Cell Cycle 2012, 11, 3019–3035. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Wang, Y.; Shi, Z.; Liu, J.; Sun, P.; Hou, X.; Zhang, J.; Zhao, S.; Zhou, B.P.; Mi, J. Metabolic reprogramming of cancer-associated fibroblasts by IDH3α downregulation. Cell Rep. 2015, 10, 1335–1348. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.G.; Kang, H.R.; Homer, R.J.; Chupp, G.; Elias, J.A. Transgenic modeling of transforming growth factor-β(1): Role of apoptosis in fibrosis and alveolar remodeling. Proc. Am. Thorac. Soc. 2006, 3, 418–423. [Google Scholar] [CrossRef] [PubMed]
- Barbas-Filho, J.V.; Ferreira, M.A.; Sesso, A.; Kairalla, R.A.; Carvalho, C.R.; Capelozzi, V.L. Evidence of type II pneumocyte apoptosis in the pathogenesis of idiopathic pulmonary fibrosis (IFP)/usual interstitial pneumonia (UIP). J. Clin. Pathol. 2001, 54, 132–138. [Google Scholar] [CrossRef] [PubMed]
- Wang, K. Molecular mechanisms of hepatic apoptosis. Cell Death Dis. 2014, 5, e996. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Duffhues, G.; Garcia de Vinuesa, A.; Ten Dijke, P. Endothelial-to-mesenchymal transition in cardiovascular diseases: Developmental signaling pathways gone awry. Dev. Dyn. 2017, 247, 492–508. [Google Scholar] [CrossRef] [PubMed]
- Zeisberg, E.M.; Tarnavski, O.; Zeisberg, M.; Dorfman, A.L.; McMullen, J.R.; Gustafsson, E.; Chandraker, A.; Yuan, X.; Pu, W.T.; Roberts, A.B.; et al. Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat. Med. 2007, 13, 952–961. [Google Scholar] [CrossRef] [PubMed]
- Xavier, S.; Vasko, R.; Matsumoto, K.; Zullo, J.A.; Chen, R.; Maizel, J.; Chander, P.N.; Goligorsky, M.S. Curtailing endothelial TGF-β signaling is sufficient to reduce endothelial-mesenchymal transition and fibrosis in CKD. J. Am. Soc. Nephrol. JASN 2015, 26, 817–829. [Google Scholar] [CrossRef] [PubMed]
- Kokudo, T.; Suzuki, Y.; Yoshimatsu, Y.; Yamazaki, T.; Watabe, T.; Miyazono, K. Snail is required for TGFβ-induced endothelial-mesenchymal transition of embryonic stem cell-derived endothelial cells. J. Cell Sci. 2008, 121 Pt 20, 3317–3324. [Google Scholar] [CrossRef] [PubMed]
- Medici, D.; Potenta, S.; Kalluri, R. Transforming growth factor-β2 promotes Snail-mediated endothelial-mesenchymal transition through convergence of Smad-dependent and Smad-independent signalling. Biochem. J. 2011, 437, 515–520. [Google Scholar] [CrossRef] [PubMed]
- Welch-Reardon, K.M.; Ehsan, S.M.; Wang, K.; Wu, N.; Newman, A.C.; Romero-Lopez, M.; Fong, A.H.; George, S.C.; Edwards, R.A.; Hughes, C.C. Angiogenic sprouting is regulated by endothelial cell expression of Slug. J. Cell Sci. 2014, 127 Pt 9, 2017–2028. [Google Scholar] [CrossRef] [PubMed]
- Braga, T.T.; Agudelo, J.S.; Camara, N.O. Macrophages during the fibrotic process: M2 as friend and foe. Front. Immunol. 2015, 6, 602. [Google Scholar] [CrossRef] [PubMed]
- Murray, L.A.; Chen, Q.; Kramer, M.S.; Hesson, D.P.; Argentieri, R.L.; Peng, X.; Gulati, M.; Homer, R.J.; Russell, T.; van Rooijen, N.; et al. TGF-β driven lung fibrosis is macrophage dependent and blocked by Serum amyloid P. Int. J. Biochem. Cell Biol. 2011, 43, 154–162. [Google Scholar] [CrossRef] [PubMed]
- Lech, M.; Anders, H.J. Macrophages and fibrosis: How resident and infiltrating mononuclear phagocytes orchestrate all phases of tissue injury and repair. Biochim. Biophys. Acta 2013, 1832, 989–997. [Google Scholar] [CrossRef] [PubMed]
- Fan, Q.M.; Jing, Y.Y.; Yu, G.F.; Kou, X.R.; Ye, F.; Gao, L.; Li, R.; Zhao, Q.D.; Yang, Y.; Lu, Z.H.; et al. Tumor-associated macrophages promote cancer stem cell-like properties via transforming growth factor-b1-induced epithelial-mesenchymal transition in hepatocellular carcinoma. Cancer Lett. 2014, 352, 160–168. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Che, D.; Yang, F.; Chi, C.; Meng, H.; Shen, J.; Qi, L.; Liu, F.; Lv, L.; Li, Y.; et al. Tumor-associated macrophages promote tumor metastasis via the TGF-β/SOX9 axis in non-small cell lung cancer. Oncotarget 2017, 8, 99801–99815. [Google Scholar] [CrossRef] [PubMed]
- Standiford, T.J.; Kuick, R.; Bhan, U.; Chen, J.; Newstead, M.; Keshamouni, V.G. TGF-β-induced IRAK-M expression in tumor-associated macrophages regulates lung tumor growth. Oncogene 2011, 30, 2475–2484. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Wang, H.; Wang, X.; Jiang, G.; Liu, H.; Zhang, G.; Wang, H.; Fang, R.; Bu, X.; Cai, S.; et al. TGF-β induces M2-like macrophage polarization via SNAIL-mediated suppression of a pro-inflammatory phenotype. Oncotarget 2016, 7, 52294–52306. [Google Scholar] [CrossRef] [PubMed]
- Byrne, S.N.; Knox, M.C.; Halliday, G.M. TGFβ is responsible for skin tumour infiltration by macrophages enabling the tumours to escape immune destruction. Immunol. Cell Biol. 2008, 86, 92–97. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, H.; Shinto, O.; Yashiro, M.; Yamazoe, S.; Iwauchi, T.; Muguruma, K.; Kubo, N.; Ohira, M.; Hirakawa, K. Transforming growth factor β signaling inhibitor, SB-431542, induces maturation of dendritic cells and enhances anti-tumor activity. Oncol. Rep. 2010, 24, 1637–1643. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, A.; Oberle, N.; Krammer, P.H. Molecular mechanisms of treg-mediated T cell suppression. Front. Immunol. 2012, 3, 51. [Google Scholar] [CrossRef] [PubMed]
- Thomas, D.A.; Massague, J. TGF-β directly targets cytotoxic T cell functions during tumor evasion of immune surveillance. Cancer Cell 2005, 8, 369–380. [Google Scholar] [CrossRef] [PubMed]
- Gorelik, L.; Flavell, R.A. Immune-mediated eradication of tumors through the blockade of transforming growth factor-b signaling in T cells. Nat. Med. 2001, 7, 1118–1122. [Google Scholar] [CrossRef] [PubMed]
- Stephen, T.L.; Rutkowski, M.R.; Allegrezza, M.J.; Perales-Puchalt, A.; Tesone, A.J.; Svoronos, N.; Nguyen, J.M.; Sarmin, F.; Borowsky, M.E.; Tchou, J.; et al. Transforming growth factor B-mediated suppression of antitumor T cells requires FOXP1 transcription factor expression. Immunity 2014, 41, 427–439. [Google Scholar] [CrossRef] [PubMed]
- Das, L.; Levine, A.D. TGF-β inhibits IL-2 production and promotes cell cycle arrest in TCR-activated effector/memory T cells in the presence of sustained TCR signal transduction. J. Immunol. 2008, 180, 1490–1498. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.H.; Seguin-Devaux, C.; Burke, N.A.; Oriss, T.B.; Watkins, S.C.; Clipstone, N.; Ray, A. Transforming growth factor β blocks Tec kinase phosphorylation, Ca2+ influx, and NFATc translocation causing inhibition of T cell differentiation. J. Exp. Med. 2003, 197, 1689–1699. [Google Scholar] [CrossRef] [PubMed]
- Flavell, R.A.; Sanjabi, S.; Wrzesinski, S.H.; Licona-Limon, P. The polarization of immune cells in the tumour environment by TGF b. Nat. Rev. Immunol. 2010, 10, 554–567. [Google Scholar] [CrossRef] [PubMed]
- Ohkura, N.; Kitagawa, Y.; Sakaguchi, S. Development and maintenance of regulatory T cells. Immunity 2013, 38, 414–423. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Jin, W.; Hardegen, N.; Lei, K.J.; Li, L.; Marinos, N.; McGrady, G.; Wahl, S.M. Conversion of peripheral CD4+CD25− naive T cells to CD4+CD25+ regulatory T cells by TGF-β induction of transcription factor Foxp3. J. Exp. Med. 2003, 198, 1875–1886. [Google Scholar] [CrossRef] [PubMed]
- Somasundaram, R.; Jacob, L.; Swoboda, R.; Caputo, L.; Song, H.; Basak, S.; Monos, D.; Peritt, D.; Marincola, F.; Cai, D.; et al. Inhibition of cytolytic T lymphocyte proliferation by autologous CD4+/CD25+ regulatory T cells in a colorectal carcinoma patient is mediated by transforming growth factor-β. Cancer Res. 2002, 62, 5267–5272. [Google Scholar] [PubMed]
- Murakami, M.; Sakamoto, A.; Bender, J.; Kappler, J.; Marrack, P. CD25+CD4+ T cells contribute to the control of memory CD8+ T cells. Proc. Natl. Acad. Sci. USA 2002, 99, 8832–8837. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.L.; Pittet, M.J.; Gorelik, L.; Flavell, R.A.; Weissleder, R.; von Boehmer, H.; Khazaie, K. Regulatory T cells suppress tumor-specific CD8 T cell cytotoxicity through TGF-β signals in vivo. Proc. Natl. Acad. Sci. USA 2005, 102, 419–424. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.G.; Li, C.; Qiao, W.; Mamura, M.; Kasprzak, B.; Anver, M.; Wolfraim, L.; Hong, S.; Mushinski, E.; Potter, M.; et al. Smad4 signalling in T cells is required for suppression of gastrointestinal cancer. Nature 2006, 441, 1015–1019. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Lu, Y.; Lin, Y.Y.; Zheng, Z.Y.; Fang, J.H.; He, S.; Zhuang, S.M. Vascular mimicry formation is promoted by paracrine TGF-β and SDF1 of cancer-associated fibroblasts and inhibited by miR-101 in hepatocellular carcinoma. Cancer Lett. 2016, 383, 18–27. [Google Scholar] [CrossRef] [PubMed]
- Esebanmen, G.E.; Langridge, W.H.R. The role of TGF-β signaling in dendritic cell tolerance. Immunol. Res. 2017, 65, 987–994. [Google Scholar] [CrossRef] [PubMed]
- Fattovich, G.; Stroffolini, T.; Zagni, I.; Donato, F. Hepatocellular carcinoma in cirrhosis: Incidence and risk factors. Gastroenterology 2004, 127, S35–S50. [Google Scholar] [CrossRef] [PubMed]
- Khemlina, G.; Ikeda, S.; Kurzrock, R. The biology of Hepatocellular carcinoma: Implications for genomic and immune therapies. Mol. Cancer 2017, 16, 149. [Google Scholar] [CrossRef] [PubMed]
- Saffioti, F.; Pinzani, M. Development and Regression of Cirrhosis. Dig. Dis. 2016, 34, 374–381. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, R.; Devhare, P.; Ray, R.B.; Ray, R. Hepatitis C virus-induced tumor-initiating cancer stem-like cells activate stromal fibroblasts in a xenograft tumor model. Hepatology 2017, 66, 1766–1778. [Google Scholar] [CrossRef] [PubMed]
- Budhu, S.; Schaer, D.A.; Li, Y.; Toledo-Crow, R.; Panageas, K.; Yang, X.; Zhong, H.; Houghton, A.N.; Silverstein, S.C.; Merghoub, T.; et al. Blockade of surface-bound TGF-β on regulatory T cells abrogates suppression of effector T cell function in the tumor microenvironment. Sci. Signal. 2017, 10, eaak9702. [Google Scholar] [CrossRef] [PubMed]
- Seki, E.; de Minicis, S.; Osterreicher, C.H.; Kluwe, J.; Osawa, Y.; Brenner, D.A.; Schwabe, R.F. TLR4 enhances TGF-β signaling and hepatic fibrosis. Nat. Med. 2007, 13, 1324–1332. [Google Scholar] [CrossRef] [PubMed]
- Omori, K.; Hattori, N.; Senoo, T.; Takayama, Y.; Masuda, T.; Nakashima, T.; Iwamoto, H.; Fujitaka, K.; Hamada, H.; Kohno, N. Inhibition of plasminogen activator inhibitor-1 attenuates transforming growth factor-β-dependent epithelial mesenchymal transition and differentiation of fibroblasts to myofibroblasts. PLoS ONE 2016, 11, e0148969. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.M.; Wang, S.; Huang, X.R.; Yang, C.; Xiao, J.; Zhang, Y.; To, K.F.; Nikolic-Paterson, D.J.; Lan, H.Y. Inflammatory macrophages can transdifferentiate into myofibroblasts during renal fibrosis. Cell Death Dis. 2016, 7, e2495. [Google Scholar] [CrossRef] [PubMed]
- Mack, M.; Yanagita, M. Origin of myofibroblasts and cellular events triggering fibrosis. Kidney Int. 2015, 87, 297–307. [Google Scholar] [CrossRef] [PubMed]
- Roulis, M.; Flavell, R.A. Fibroblasts and myofibroblasts of the intestinal lamina propria in physiology and disease. Differ. Res. Biol. Divers. 2016, 92, 116–131. [Google Scholar] [CrossRef] [PubMed]
- Mifflin, R.C.; Pinchuk, I.V.; Saada, J.I.; Powell, D.W. Intestinal myofibroblasts: Targets for stem cell therapy. Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 300, G684–G696. [Google Scholar] [CrossRef] [PubMed]
- Hinz, B. The role of myofibroblasts in wound healing. Curr. Res. Transl. Med. 2016, 64, 171–177. [Google Scholar] [CrossRef] [PubMed]
- Jeong, W.I.; Park, O.; Radaeva, S.; Gao, B. STAT1 inhibits liver fibrosis in mice by inhibiting stellate cell proliferation and stimulating NK cell cytotoxicity. Hepatology 2006, 44, 1441–1451. [Google Scholar] [CrossRef] [PubMed]
- Rastogi, V.; Sharma, R.; Misra, S.R.; Yadav, L.; Sharma, V. Emperipolesis—A review. J. Clin. Diagn. Res. JCDR 2014, 8, ZM01-2. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Zhao, J.; Zhang, X.; Cheng, Y.; Hu, J.; Li, Y.; Zhao, X.; Shang, Q.; Sun, Y.; Tu, B.; et al. Activated hepatic stellate cells impair NK cell anti-fibrosis capacity through a TGF-β-dependent emperipolesis in HBV cirrhotic patients. Sci. Rep. 2017, 7, 44544. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, P.; Pellicoro, A.; Vernon, M.A.; Boulter, L.; Aucott, R.L.; Ali, A.; Hartland, S.N.; Snowdon, V.K.; Cappon, A.; Gordon-Walker, T.T.; et al. Differential Ly-6C expression identifies the recruited macrophage phenotype, which orchestrates the regression of murine liver fibrosis. Proc. Natl. Acad. Sci. USA 2012, 109, E3186–E3195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- An, S.Y.; Jang, Y.J.; Lim, H.J.; Han, J.; Lee, J.; Lee, G.; Park, J.Y.; Park, S.Y.; Kim, J.H.; Do, B.R.; et al. Milk fat globule-EGF factor 8, secreted by mesenchymal stem cells, protects against liver fibrosis in mice. Gastroenterology 2017, 152, 1174–1186. [Google Scholar] [CrossRef] [PubMed]
- Malfettone, A.; Soukupova, J.; Bertran, E.; Crosas-Molist, E.; Lastra, R.; Fernando, J.; Koudelkova, P.; Rani, B.; Fabra, A.; Serrano, T.; et al. Transforming growth factor-β-induced plasticity causes a migratory stemness phenotype in hepatocellular carcinoma. Cancer Lett. 2017, 392, 39–50. [Google Scholar] [CrossRef] [PubMed]
- Fransvea, E.; Angelotti, U.; Antonaci, S.; Giannelli, G. Blocking transforming growth factor-β up-regulates E-cadherin and reduces migration and invasion of hepatocellular carcinoma cells. Hepatology 2008, 47, 1557–1566. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Caceres, J.; Mainez, J.; Mayoral, R.; Martin-Sanz, P.; Egea, G.; Fabregat, I. Caveolin-1-dependent activation of the metalloprotease TACE/ADAM17 by TGF-β in hepatocytes requires activation of Src and the NADPH oxidase NOX1. FEBS J. 2016, 283, 1300–1310. [Google Scholar] [CrossRef] [PubMed]
- Mazzocca, A.; Fransvea, E.; Dituri, F.; Lupo, L.; Antonaci, S.; Giannelli, G. Down-regulation of connective tissue growth factor by inhibition of transforming growth factor β blocks the tumor-stroma cross-talk and tumor progression in hepatocellular carcinoma. Hepatology 2010, 51, 523–534. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Chen, S.; Wang, W.; Ning, B.F.; Chen, F.; Shen, W.; Ding, J.; Chen, W.; Xie, W.F.; Zhang, X. Cancer-associated fibroblasts promote hepatocellular carcinoma metastasis through chemokine-activated hedgehog and TGF-β pathways. Cancer Lett. 2016, 379, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Bruix, J.; Qin, S.; Merle, P.; Granito, A.; Huang, Y.H.; Bodoky, G.; Pracht, M.; Yokosuka, O.; Rosmorduc, O.; Breder, V.; et al. Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017, 389, 56–66. [Google Scholar] [CrossRef]
- Azzariti, A.; Mancarella, S.; Porcelli, L.; Quatrale, A.E.; Caligiuri, A.; Lupo, L.; Dituri, F.; Giannelli, G. Hepatic stellate cells induce hepatocellular carcinoma cell resistance to sorafenib through the laminin-332/α3 integrin axis recovery of focal adhesion kinase ubiquitination. Hepatology 2016, 64, 2103–2117. [Google Scholar] [CrossRef] [PubMed]
- Dituri, F.; Serio, G.; Filannino, D.; Mascolo, A.; Sacco, R.; Villa, E.; Giannelli, G. Circulating TGF-β1-related biomarkers in patients with hepatocellular carcinoma and their association with HCC staging scores. Cancer Lett. 2014, 353, 264–271. [Google Scholar] [CrossRef] [PubMed]
- Giannelli, G.; Rani, B.; Dituri, F.; Cao, Y.; Palasciano, G. Moving towards personalised therapy in patients with hepatocellular carcinoma: The role of the microenvironment. Gut 2014, 63, 1668–1676. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Agarwal, R.; Dituri, F.; Lupo, L.; Trerotoli, P.; Mancarella, S.; Winter, P.; Giannelli, G. NGS-based transcriptome profiling reveals biomarkers for companion diagnostics of the TGF-β receptor blocker galunisertib in HCC. Cell Death Dis. 2017, 8, e2634. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, R.; Cao, Y.; Hoffmeier, K.; Krezdorn, N.; Jost, L.; Meisel, A.R.; Jungling, R.; Dituri, F.; Mancarella, S.; Rotter, B.; et al. Precision medicine for hepatocelluar carcinoma using molecular pattern diagnostics: Results from a preclinical pilot study. Cell Death Dis. 2017, 8, e2867. [Google Scholar] [CrossRef] [PubMed]
- Critelli, R.; Milosa, F.; Faillaci, F.; Condello, R.; Turola, E.; Marzi, L.; Lei, B.; Dituri, F.; Andreani, S.; Sighinolfi, P.; et al. Microenvironment inflammatory infiltrate drives growth speed and outcome of hepatocellular carcinoma: A prospective clinical study. Cell Death Dis. 2017, 8, e3017. [Google Scholar] [CrossRef] [PubMed]
- Nault, J.C.; De Reynies, A.; Villanueva, A.; Calderaro, J.; Rebouissou, S.; Couchy, G.; Decaens, T.; Franco, D.; Imbeaud, S.; Rousseau, F.; et al. A hepatocellular carcinoma 5-gene score associated with survival of patients after liver resection. Gastroenterology 2013, 145, 176–187. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Bru, C.; Bruix, J. Prognosis of hepatocellular carcinoma: The BCLC staging classification. Semin. Liver Dis. 1999, 19, 329–338. [Google Scholar] [CrossRef] [PubMed]
- Hoshida, Y.; Nijman, S.M.; Kobayashi, M.; Chan, J.A.; Brunet, J.P.; Chiang, D.Y.; Villanueva, A.; Newell, P.; Ikeda, K.; Hashimoto, M.; et al. Integrative transcriptome analysis reveals common molecular subclasses of human hepatocellular carcinoma. Cancer Res. 2009, 69, 7385–7392. [Google Scholar] [CrossRef] [PubMed]
- Villa, E.; Critelli, R.; Lei, B.; Marzocchi, G.; Camma, C.; Giannelli, G.; Pontisso, P.; Cabibbo, G.; Enea, M.; Colopi, S.; et al. Neoangiogenesis-related genes are hallmarks of fast-growing hepatocellular carcinomas and worst survival. Results from a prospective study. Gut 2016, 65, 861–869. [Google Scholar] [CrossRef] [PubMed]




© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Caja, L.; Dituri, F.; Mancarella, S.; Caballero-Diaz, D.; Moustakas, A.; Giannelli, G.; Fabregat, I. TGF-β and the Tissue Microenvironment: Relevance in Fibrosis and Cancer. Int. J. Mol. Sci. 2018, 19, 1294. https://doi.org/10.3390/ijms19051294
Caja L, Dituri F, Mancarella S, Caballero-Diaz D, Moustakas A, Giannelli G, Fabregat I. TGF-β and the Tissue Microenvironment: Relevance in Fibrosis and Cancer. International Journal of Molecular Sciences. 2018; 19(5):1294. https://doi.org/10.3390/ijms19051294
Chicago/Turabian StyleCaja, Laia, Francesco Dituri, Serena Mancarella, Daniel Caballero-Diaz, Aristidis Moustakas, Gianluigi Giannelli, and Isabel Fabregat. 2018. "TGF-β and the Tissue Microenvironment: Relevance in Fibrosis and Cancer" International Journal of Molecular Sciences 19, no. 5: 1294. https://doi.org/10.3390/ijms19051294