Fenofibrate Reduces the Asthma-Related Fibroblast-To-Myofibroblast Transition by TGF-Β/Smad2/3 Signaling Attenuation and Connexin 43-Dependent Phenotype Destabilization

 ,
,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
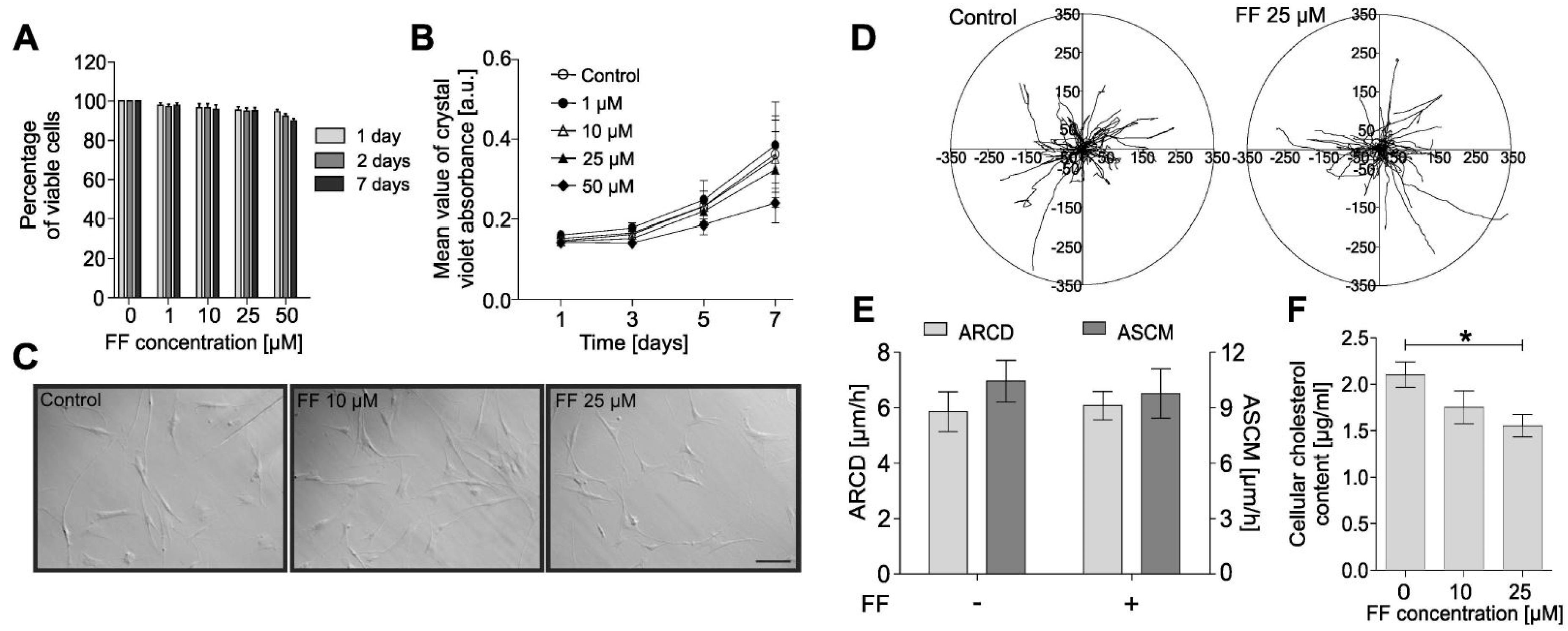
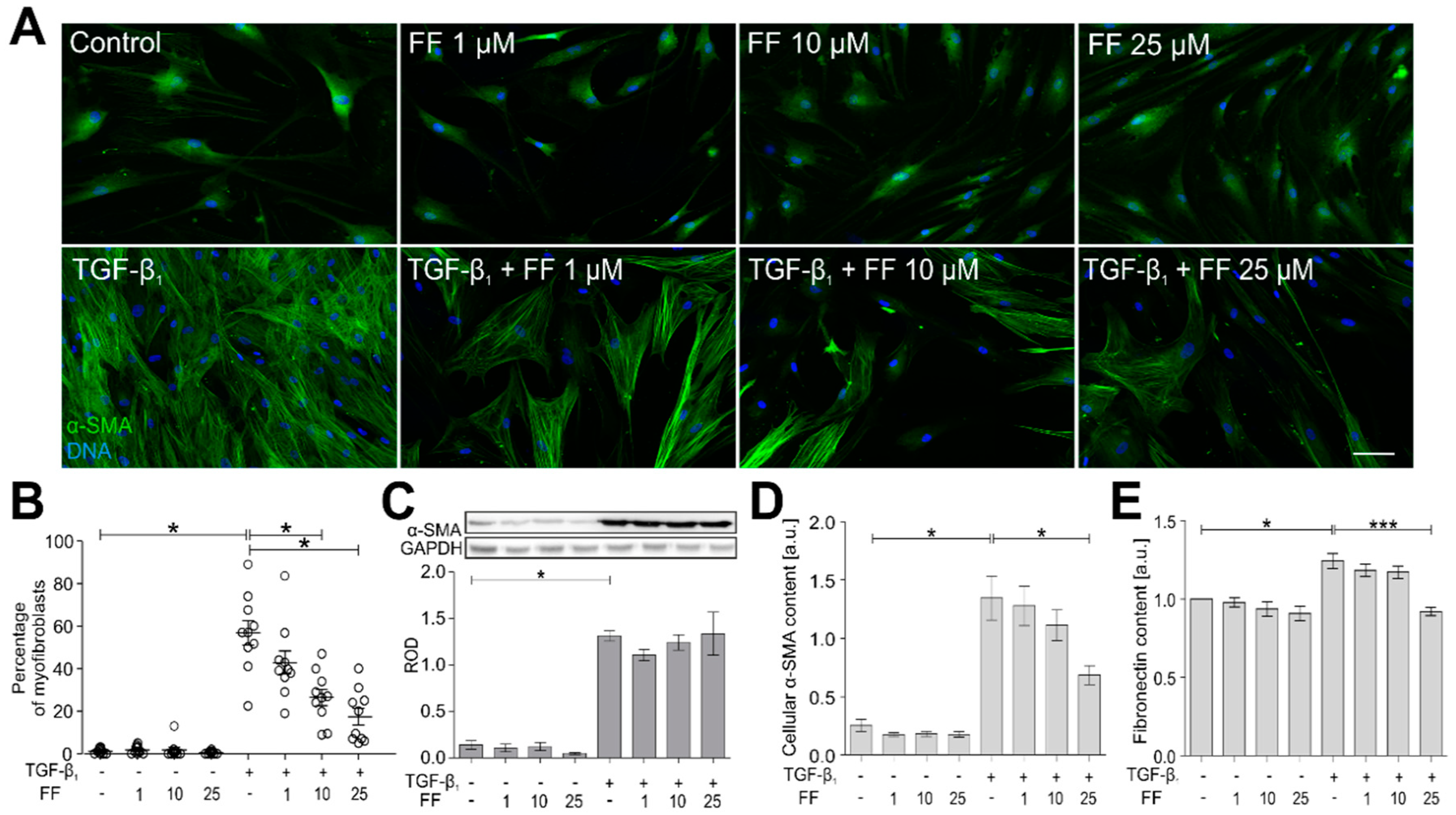
2.1. Fenofibrate Attenuates the TGF-Β1-Induced Differentiation of Human Bronchial Fibroblasts into Myofibroblasts
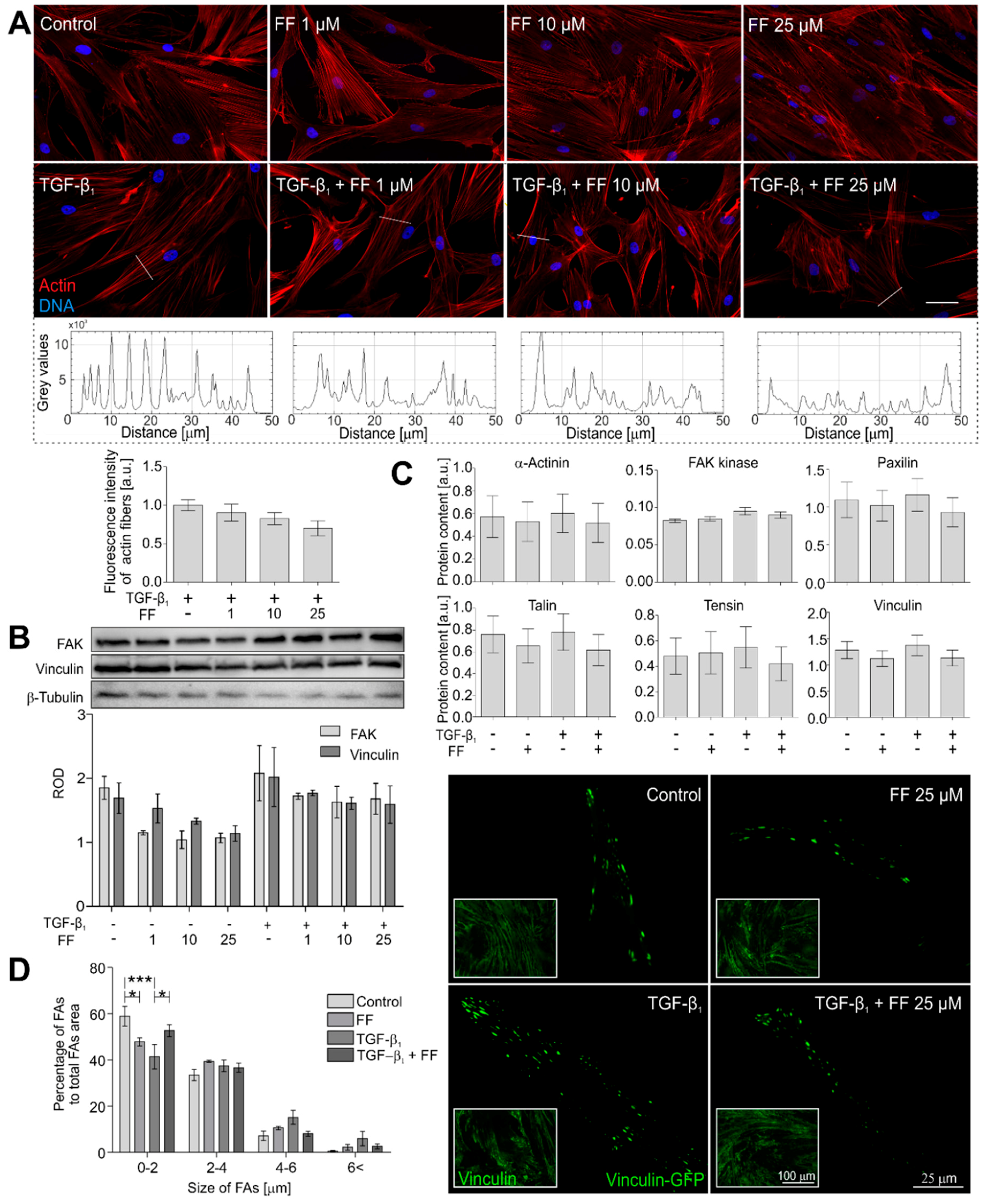
2.2. Actin Cytoskeleton Architecture Is Reorganized by Fenofibrate in TGF-β1-Treated HBFs
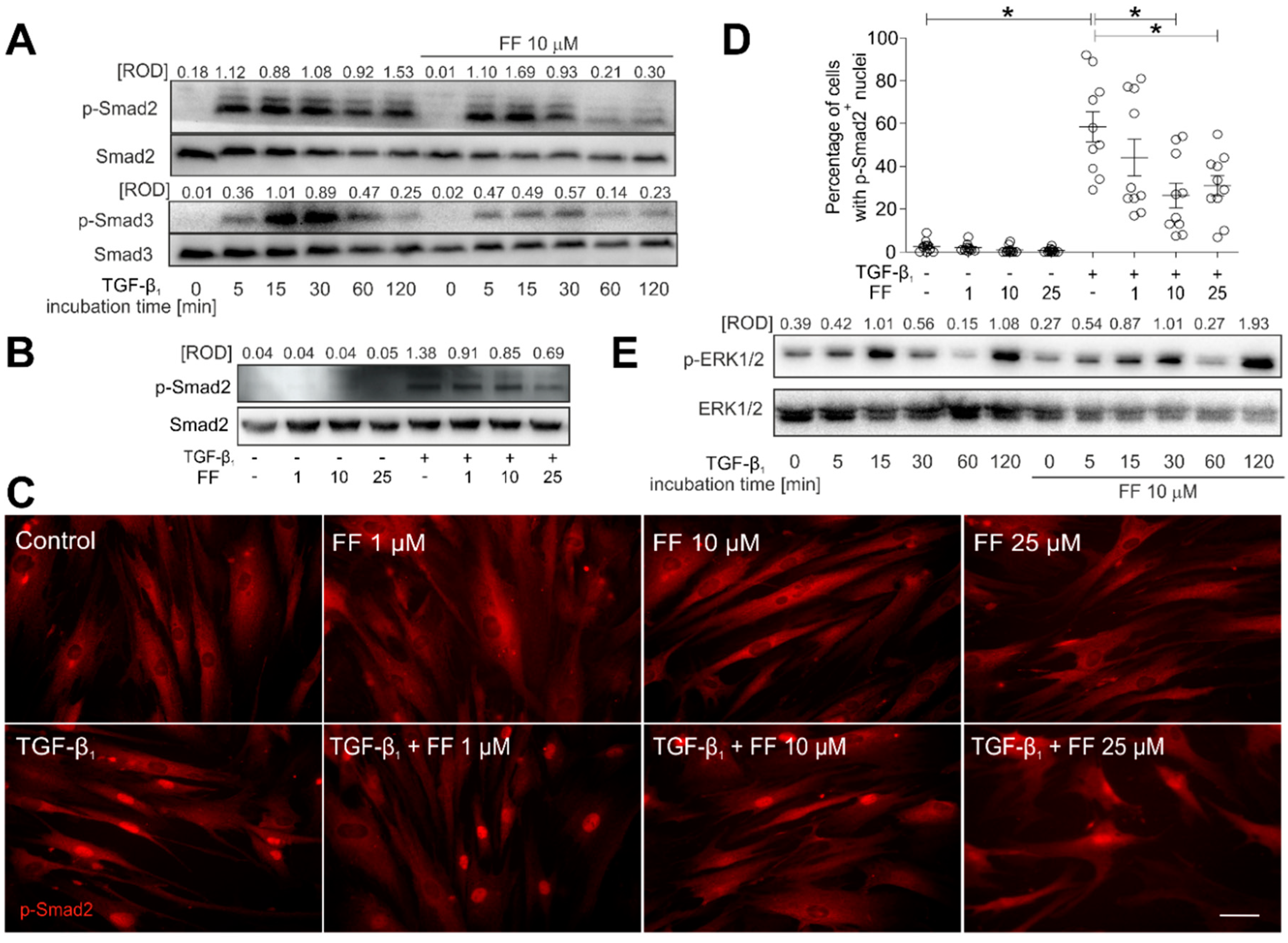
2.3. Fenofibrate Inhibits the TGF-β1/Smad Signaling Pathway in TGF-β1-Treated HBFs
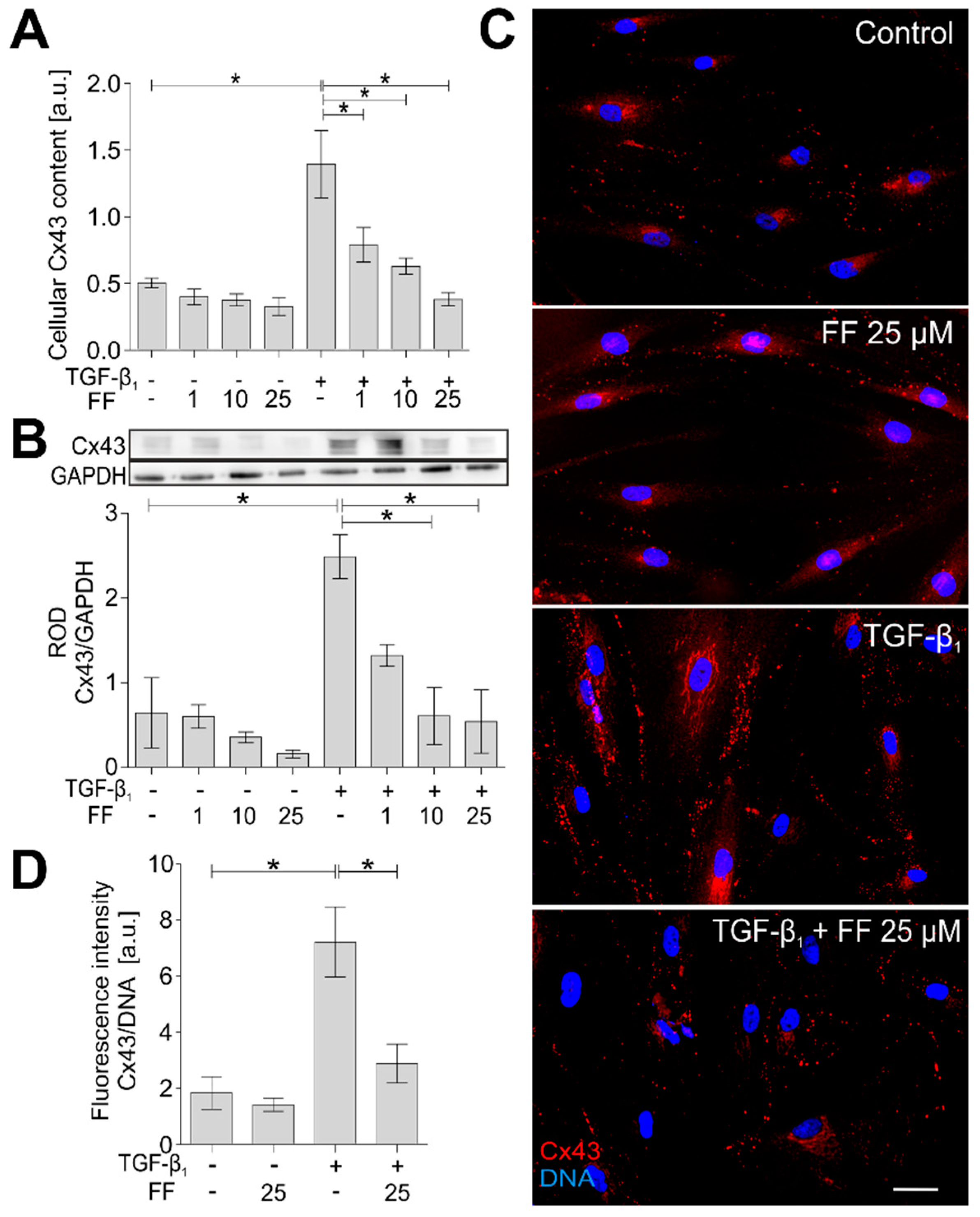
2.4. Connexin 43 Is Involved in Fenofibrate-Induced Attenuation of the FMT in TGF-β1-Treated HBFs
3. Discussion
4. Materials and Methods
4.1. HBF In Vitro Cultures
4.2. Viability and Proliferation Tests
4.3. Movement of Individual Cells—Time-Lapse Monitoring
4.4. Cholesterol Content Assay
4.5. Immunofluorescence Staining
4.6. Focal Adhesion Imaging
4.7. Protein Extraction and Western Blotting
4.8. Measurement of Protein Levels Using In-Cell ELISA
4.9. Statistics
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bai, T.R.; Knight, D.A. Structural changes in the airways in asthma: Observations and consequences. Clin. Sci. 2005, 108, 463–477. [Google Scholar] [CrossRef] [PubMed]
- Anandan, C.; Nurmatov, U.; van Schayck, O.C.P.; Sheikh, A. Is the prevalence of asthma declining? Systematic review of epidemiological studies. Allergy 2010, 65, 152–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Muhsen, S.; Johnson, J.R.; Hamid, Q. Remodeling in asthma. J. Allergy Clin. Immunol. 2011, 128, 451–462. [Google Scholar] [CrossRef] [PubMed]
- Jeffery, P.K. Remodeling in Asthma and Chronic Obstructive Lung Disease. Am. J. Respir. Crit. Care Med. 2001, 164, S28–S38. [Google Scholar] [CrossRef] [PubMed]
- Global Strategy for Asthma Management and Prevention (2016 Update). Gina Report. 2016. Available online: https://ginasthma.org/wp-content/uploads/2016/04/GINA-2016-main-report_tracked.pdf (accessed on 15 August 2018).
- Anderson, W.C.; Szefler, S.J. New and future strategies to improve asthma control in children. J. Allergy Clin. Immunol. 2015, 136, 848–859. [Google Scholar] [CrossRef] [PubMed]
- Papi, A.; Cazzola, M. Combination treatment in asthma: Reviewing old and new options. Pulm. Pharmacol. Ther. 2015, 34, 72–74. [Google Scholar] [CrossRef] [PubMed]
- Mauad, T.; Bel, E.H.; Sterk, P.J. Asthma therapy and airway remodeling. J. Allergy Clin. Immunol. 2007, 120, 997–1009. [Google Scholar] [CrossRef] [PubMed]
- Bourdin, A.; Kleis, S.; Chakra, M.; Vachier, I.; Paganin, F.; Godard, P.; Chanez, P. Limited Short-term Steroid Responsiveness Is Associated With Thickening of Bronchial Basement Membrane in Severe Asthma. Chest 2012, 141, 1504–1511. [Google Scholar] [CrossRef] [PubMed]
- Scaffidi, A.K.; Moodley, Y.P.; Weichselbaum, M.; Thompson, P.J.; Knight, D.A. Regulation of human lung fibroblast phenotype and function by vitronectin and vitronectin integrins. J. Cell Sci. 2001, 114, 3507–3516. [Google Scholar] [PubMed]
- Benayoun, L.; Druilhe, A.; Dombret, M.-C.; Aubier, M.; Pretolani, M. Airway Structural Alterations Selectively Associated with Severe Asthma. Am. J. Respir. Crit. Care Med. 2003, 167, 1360–1368. [Google Scholar] [CrossRef] [PubMed]
- Phan, S.H. Biology of Fibroblasts and Myofibroblasts. Proc. Am. Thorac. Soc. 2008, 5, 334–337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michalik, M.; Wójcik-Pszczoła, K.; Paw, M.; Wnuk, D.; Koczurkiewicz, P.; Sanak, M.; Pękala, E.; Madeja, Z. Fibroblast-to-myofibroblast transition in bronchial asthma. Cell. Mol. Life Sci. 2018. [Google Scholar] [CrossRef] [PubMed]
- Michalik, M.; Pierzchalska, M.; Legutko, A.; Ura, M.; Ostaszewska, A.; Soja, J.; Sanak, M. Asthmatic bronchial fibroblasts demonstrate enhanced potential to differentiate into myofibroblasts in culture. Med. Sci. Monit. 2009, 15, BR194–BR201. [Google Scholar] [PubMed]
- Trunk-Black Juel, C.; Ali, Z.; Nilas, L.; Ulrik, C. Asthma and obesity: Does weight loss improve asthma control? a systematic review. J. Asthma Allergy 2012, 5, 21–26. [Google Scholar] [CrossRef]
- Mohanan, S.; Tapp, H.; McWilliams, A.; Dulin, M. Obesity and asthma: Pathophysiology and implications for diagnosis and management in primary care. Exp. Biol. Med. 2014, 239, 1531–1540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michalik, M.; Soczek, E.; Kosińska, M.; Rak, M.; Wójcik, K.A.; Lasota, S.; Pierzchalska, M.; Czyż, J.; Madeja, Z. Lovastatin-induced decrease of intracellular cholesterol level attenuates fibroblast-to-myofibroblast transition in bronchial fibroblasts derived from asthmatic patients. Eur. J. Pharmacol. 2013, 704, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Grabacka, M.; Reiss, K. Anticancer Properties of PPARα - Effects on Cellular Metabolism and Inflammation. PPAR Res. 2008, 2008, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Elaidy, S.M.; Essawy, S.S.; Hussain, M.A.; El-Kherbetawy, M.K.; Hamed, E.R. Modulation of the IL-23/IL-17 axis by fenofibrate ameliorates the ovalbumin/lipopolysaccharide-induced airway inflammation and bronchial asthma in rats. Naunyn Schmiedebergs Arch. Pharmacol. 2018, 391, 309–321. [Google Scholar] [CrossRef] [PubMed]
- Delayre-Orthez, C.; Becker, J.; Guenon, I.; Lagente, V.; Auwerx, J.; Frossard, N.; Pons, F. PPARα downregulates airway inflammation induced by lipopolysaccharide in the mouse. Respir. Res. 2005, 6, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delayre-Orthez, C.; Becker, J.; Auwerx, J.; Frossard, N.; Pons, F. Suppression of allergen-induced airway inflammation and immune response by the peroxisome proliferator-activated receptor-alpha agonist fenofibrate. Eur. J. Pharmacol. 2008, 581, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Samah, M.; El-Aidy, A.E.-R.; Tawfik, M.K.; Ewais, M.M.S. Evaluation of the antifibrotic effect of fenofibrate and rosiglitazone on bleomycin-induced pulmonary fibrosis in rats. Eur. J. Pharmacol. 2012, 689, 186–193. [Google Scholar] [CrossRef] [PubMed]
- Paw, M.; Borek, I.; Wnuk, D.; Ryszawy, D.; Piwowarczyk, K.; Kmiotek, K.; Wójcik-Pszczoła, K.A.; Pierzchalska, M.; Madeja, Z.; Sanak, M.; et al. Connexin43 Controls the Myofibroblastic Differentiation of Bronchial Fibroblasts from Patients with Asthma. Am. J. Respir. Cell Mol. Biol. 2017, 57, 100–110. [Google Scholar] [CrossRef] [PubMed]
- Michalik, M.; Wójcik, K.A.; Jakieła, B.; Szpak, K.; Pierzchalska, M.; Sanak, M.; Madeja, Z.; Czyż, J. Lithium attenuates TGF-β1-induced fibroblasts to myofibroblasts transition in bronchial fibroblasts derived from asthmatic patients. J. Allergy 2012, 2012, 206109. [Google Scholar] [CrossRef] [PubMed]
- Moustakas, A.; Pardali, K.; Gaal, A.; Heldin, C.H. Mechanisms of TGF-beta signaling in regulation of cell growth and differentiation. Immunol. Lett. 2002, 82, 85–91. [Google Scholar] [CrossRef]
- Evans, R.A.; Tian, Y.C.; Steadman, R.; Phillips, A.O. TGF-β1-mediated fibroblast–myofibroblast terminal differentiation—The role of smad proteins. Exp. Cell Res. 2003, 282, 90–100. [Google Scholar] [CrossRef]
- Rockey, D.C.; Weymouth, N.; Shi, Z. Smooth Muscle α Actin (Acta2) and Myofibroblast Function during Hepatic Wound Healing. PLoS ONE 2013, 8, e77166. [Google Scholar] [CrossRef] [PubMed]
- Omori, K.; Hattori, N.; Senoo, T.; Takayama, Y.; Masuda, T.; Nakashima, T.; Iwamoto, H.; Fujitaka, K.; Hamada, H.; Kohno, N. Inhibition of Plasminogen Activator Inhibitor-1 Attenuates Transforming Growth Factor-β-Dependent Epithelial Mesenchymal Transition and Differentiation of Fibroblasts to Myofibroblasts. PLoS ONE 2016, 11, e0148969. [Google Scholar] [CrossRef] [PubMed]
- Royce, S.G.; Cheng, V.; Samuel, C.S.; Tang, M.L.K. The regulation of fibrosis in airway remodeling in asthma. Mol. Cell. Endocrinol. 2012, 351, 167–175. [Google Scholar] [CrossRef] [PubMed]
- Wójcik-Pszczoła, K.; Hińcza, K.; Wnuk, D.; Kądziołka, D.; Koczurkiewicz, P.; Sanak, M.; Madeja, Z.; Pękala, E.; Michalik, M. Pentoxifylline and its active metabolite lisofylline attenuate transforming growth factor β1-induced asthmatic bronchial fibroblast-to-myofibroblast transition. Acta Biochim. Pol. 2016, 63, 437–442. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.-M.; Tang, P.M.-K.; Li, J.; Lan, H.Y. TGF-β/Smad signaling in renal fibrosis. Front. Physiol. 2015, 6, 82. [Google Scholar] [CrossRef] [PubMed]
- Ogata, T.; Miyauchi, T.; Sakai, S.; Irukayama-Tomobe, Y.; Goto, K.; Yamaguchi, I. Stimulation of peroxisome-proliferator-activated receptor α (PPAR α) attenuates cardiac fibrosis and endothelin-1 production in pressure-overloaded rat hearts. Clin. Sci. 2002, 103, 284S–288S. [Google Scholar] [CrossRef] [PubMed]
- Ogata, T.; Miyauchi, T.; Sakai, S.; Takanashi, M.; Irukayama-Tomobe, Y.; Yamaguchi, I. Myocardial fibrosis and diastolic dysfunction in deoxycorticosterone acetate-salt hypertensive rats is ameliorated by the peroxisome proliferator-activated receptor-alpha activator fenofibrate, partly by suppressing inflammatory responses associated with. J. Am. Coll. Cardiol. 2004, 43, 1481–1488. [Google Scholar] [CrossRef] [PubMed]
- Diep, Q.N.; Benkirane, K.; Amiri, F.; Cohn, J.S.; Endemann, D.; Schiffrin, E.L. PPAR alpha activator fenofibrate inhibits myocardial inflammation and fibrosis in angiotensin II-infused rats. J. Mol. Cell. Cardiol. 2004, 36, 295–304. [Google Scholar] [CrossRef] [PubMed]
- Calkin, A.C.; Giunti, S.; Jandeleit-Dahm, K.A.; Allen, T.J.; Cooper, M.E.; Thomas, M.C. PPAR-α and -γ agonists attenuate diabetic kidney disease in the apolipoprotein E knockout mouse. Nephrol. Dial. Transplant. 2006, 21, 2399–2405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michalik, M.; Pierzchalska, M.; Włodarczyk, A.; Wójcik, K.A.; Czyż, J.; Sanak, M.; Madeja, Z. Transition of asthmatic bronchial fibroblasts to myofibroblasts is inhibited by cell–cell contacts. Respir. Med. 2011, 105, 1467–1475. [Google Scholar] [CrossRef] [PubMed]
- Halwani, R.; Al-Muhsen, S.; Al-Jahdali, H.; Hamid, Q. Role of Transforming Growth Factor–β in Airway Remodeling in Asthma. Am. J. Respir. Cell Mol. Biol. 2011, 44, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Doser, K.; Guserle, R.; Nitsche, V.; Arnold, G. Comparative steady state study with 2 fenofibrate 250 mg slow release capsules. An example of bioequivalence assessment with a highly variable drug. Int. J. Clin. Pharmacol. Ther. 1996, 34, 345–348. [Google Scholar] [PubMed]
- Kajosaari, L.I.; Backman, J.T.; Neuvonen, M.; Laitila, J.; Neuvonen, P.J. Lack of effect of bezafibrate and fenofibrate on the pharmacokinetics and pharmacodynamics of repaglinide. Br. J. Clin. Pharmacol. 2004, 58, 390–396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, L.; Wu, H.; Niu, F.; Yan, C.; Yang, X.; Jia, Y. Design of fenofibrate microemulsion for improved bioavailability. Int. J. Pharm. 2011, 420, 251–255. [Google Scholar] [CrossRef] [PubMed]
- Piwowarczyk, K.; Wybieralska, E.; Baran, J.; Borowczyk, J.; Rybak, P.; Kosińska, M.; Włodarczyk, A.J.; Michalik, M.; Siedlar, M.; Madeja, Z.; et al. Fenofibrate enhances barrier function of endothelial continuum within the metastatic niche of prostate cancer cells. Expert Opin. Ther. Targets 2015, 19, 163–176. [Google Scholar] [CrossRef] [PubMed]
- Varet, J.; Vincent, L.; Mirshahi, P.; Pille, J.V.; Legrand, E.; Opolon, P.; Mishal, Z.; Soria, J.; Li, H.; Soria, C. Fenofibrate inhibits angiogenesis in vitro and in vivo. Cell. Mol. Life Sci. 2003, 60, 810–819. [Google Scholar] [PubMed]
- Li, L.; Emmett, N.; Mann, D.; Zhao, X. Fenofibrate attenuates tubulointerstitial fibrosis and inflammation through suppression of nuclear factor-κB and transforming growth factor-β1/Smad3 in diabetic nephropathy. Exp. Biol. Med. 2010, 235, 383–391. [Google Scholar] [CrossRef] [PubMed]
- Fruchart, J.-C.; Duriez, P. Mode of action of fibrates in the regulation of triglyceride and HDL-cholesterol metabolism. Drugs Today 2006, 42, 39–64. [Google Scholar] [CrossRef] [PubMed]
- Valasek, M.A.; Clarke, S.L.; Repa, J.J. Fenofibrate reduces intestinal cholesterol absorption via PPARα-dependent modulation of NPC1L1 expression in mouse. J. Lipid Res. 2007, 48, 2725–2735. [Google Scholar] [CrossRef] [PubMed]
- Cao, H.; Wen, G.; Li, H. Role of peroxisome proliferator-activated receptor α in atherosclerosis. Mol. Med. Rep. 2014, 9, 1755–1760. [Google Scholar] [CrossRef] [PubMed]
- Brusselmans, K.; Timmermans, L.; Van de Sande, T.; Van Veldhoven, P.P.; Guan, G.; Shechter, I.; Claessens, F.; Verhoeven, G.; Swinnen, J.V. Squalene Synthase, a Determinant of Raft-associated Cholesterol and Modulator of Cancer Cell Proliferation. J. Biol. Chem. 2007, 282, 18777–18785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Czyż, J.; Szpak, K.; Madeja, Z. The role of connexins in prostate cancer promotion and progression. Nat. Rev. Urol. 2012, 9, 274–282. [Google Scholar] [CrossRef] [PubMed]
- Czyż, J.; Piwowarczyk, K.; Paw, M.; Luty, M.; Wróbel, T.; Catapano, J.; Madeja, Z.; Ryszawy, D. Connexin-dependent intercellular stress signaling in tissue homeostasis and tumor development. Acta Biochim. Pol. 2017, 64, 377–389. [Google Scholar] [CrossRef] [PubMed]
- Dbouk, H.A.; Mroue, R.M.; El-Sabban, M.E.; Talhouk, R.S. Connexins: A myriad of functions extending beyond assembly of gap junction channels. Cell Commun. Signal. 2009, 7, 4. [Google Scholar] [CrossRef] [PubMed]
- Asazuma-Nakamura, Y.; Dai, P.; Harada, Y.; Jiang, Y.; Hamaoka, K.; Takamatsu, T. Cx43 contributes to TGF-β signaling to regulate differentiation of cardiac fibroblasts into myofibroblasts. Exp. Cell Res. 2009, 315, 1190–1199. [Google Scholar] [CrossRef] [PubMed]
- Pierzchalska, M.; Szabó, Z.; Sanak, M.; Soja, J.; Szczeklik, A. Deficient prostaglandin E2 production by bronchial fibroblasts of asthmatic patients, with special reference to aspirin-induced asthma. J. Allergy Clin. Immunol. 2003, 111, 1041–1048. [Google Scholar] [CrossRef] [PubMed]
- Sarna, M.; Wojcik, K.A.; Hermanowicz, P.; Wnuk, D.; Burda, K.; Sanak, M.; Czyż, J.; Michalik, M. Undifferentiated Bronchial Fibroblasts Derived from Asthmatic Patients Display Higher Elastic Modulus than Their Non-Asthmatic Counterparts. PLoS ONE 2015, 10, e0116840. [Google Scholar] [CrossRef] [PubMed]
- Galanty, A.; Michalik, M.; Sȩdek, Ł.; Podolak, I. The influence of LTS-4, a saponoside from Lysimachia thyrsiflora L., on human skin fibroblasts and human melanoma cells. Cell. Mol. Biol. Lett. 2008, 13, 585–598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galanty, A.; Koczurkiewicz, P.; Wnuk, D.; Paw, M.; Karnas, E.; Podolak, I.; Węgrzyn, M.; Borusiewicz, M.; Madeja, Z.; Czyż, J.; Michalik, M. Usnic acid and atranorin exert selective cytostatic and anti-invasive effects on human prostate and melanoma cancer cells. Toxicol. Vitr. 2017, 40, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [PubMed]
- Piwowarczyk, K.; Paw, M.; Ryszawy, D.; Rutkowska-Zapała, M.; Madeja, Z.; Siedlar, M.; Czyż, J. Connexin43 high prostate cancer cells induce endothelial connexin43 up-regulation through the activation of intercellular ERK1/2-dependent signaling axis. Eur. J. Cell Biol. 2017, 96, 337–346. [Google Scholar] [CrossRef] [PubMed]
- Horzum, U.; Ozdil, B.; Pesen-Okvur, D. Step-by-step quantitative analysis of focal adhesions. MethodsX 2014, 1, 56–59. [Google Scholar] [CrossRef] [PubMed]





© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paw, M.; Wnuk, D.; Kądziołka, D.; Sęk, A.; Lasota, S.; Czyż, J.; Madeja, Z.; Michalik, M. Fenofibrate Reduces the Asthma-Related Fibroblast-To-Myofibroblast Transition by TGF-Β/Smad2/3 Signaling Attenuation and Connexin 43-Dependent Phenotype Destabilization. Int. J. Mol. Sci. 2018, 19, 2571. https://doi.org/10.3390/ijms19092571
Paw M, Wnuk D, Kądziołka D, Sęk A, Lasota S, Czyż J, Madeja Z, Michalik M. Fenofibrate Reduces the Asthma-Related Fibroblast-To-Myofibroblast Transition by TGF-Β/Smad2/3 Signaling Attenuation and Connexin 43-Dependent Phenotype Destabilization. International Journal of Molecular Sciences. 2018; 19(9):2571. https://doi.org/10.3390/ijms19092571
Chicago/Turabian StylePaw, Milena, Dawid Wnuk, Dominika Kądziołka, Aleksandra Sęk, Sławomir Lasota, Jarosław Czyż, Zbigniew Madeja, and Marta Michalik. 2018. "Fenofibrate Reduces the Asthma-Related Fibroblast-To-Myofibroblast Transition by TGF-Β/Smad2/3 Signaling Attenuation and Connexin 43-Dependent Phenotype Destabilization" International Journal of Molecular Sciences 19, no. 9: 2571. https://doi.org/10.3390/ijms19092571