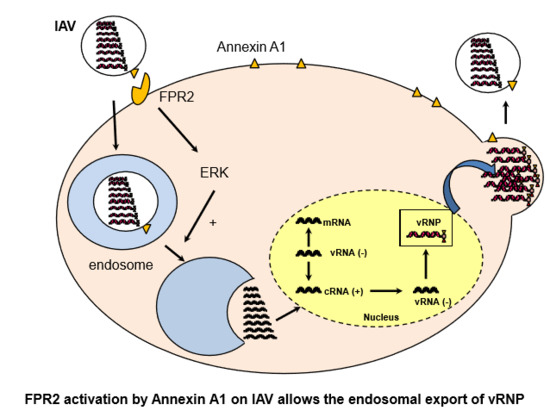
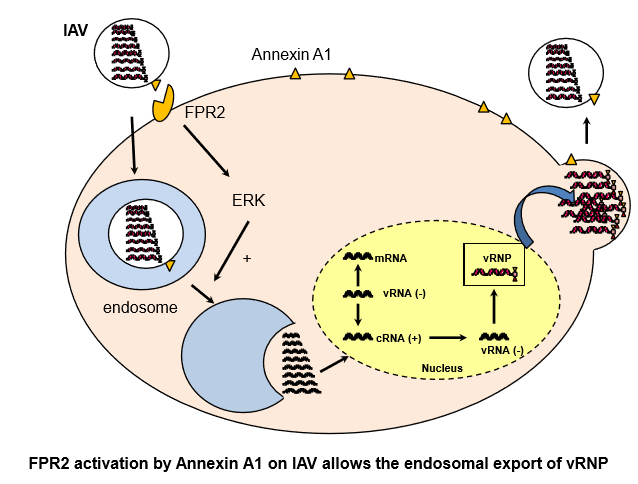
The Annexin A1 Receptor FPR2 Regulates the Endosomal Export of Influenza Virus

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
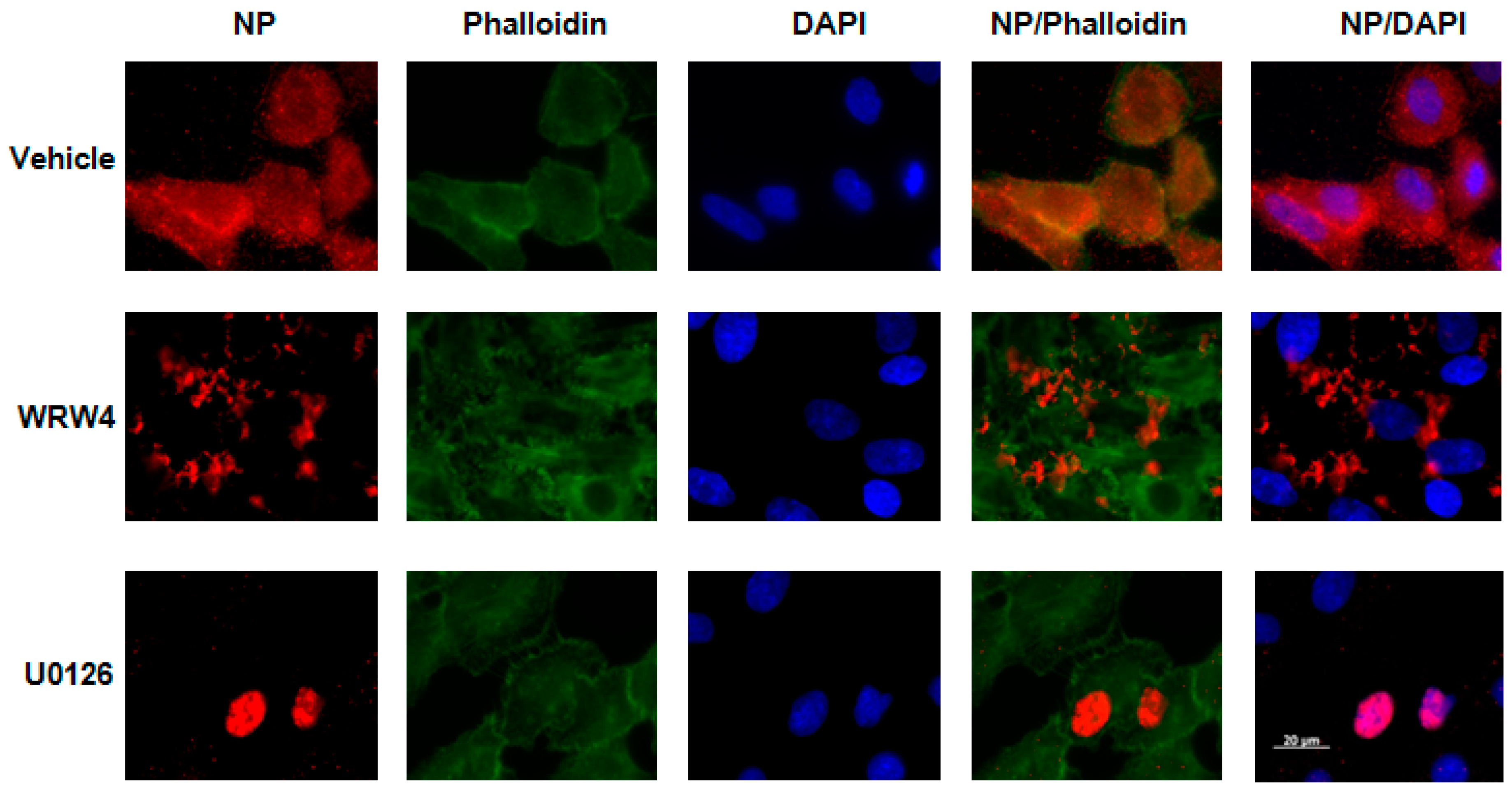
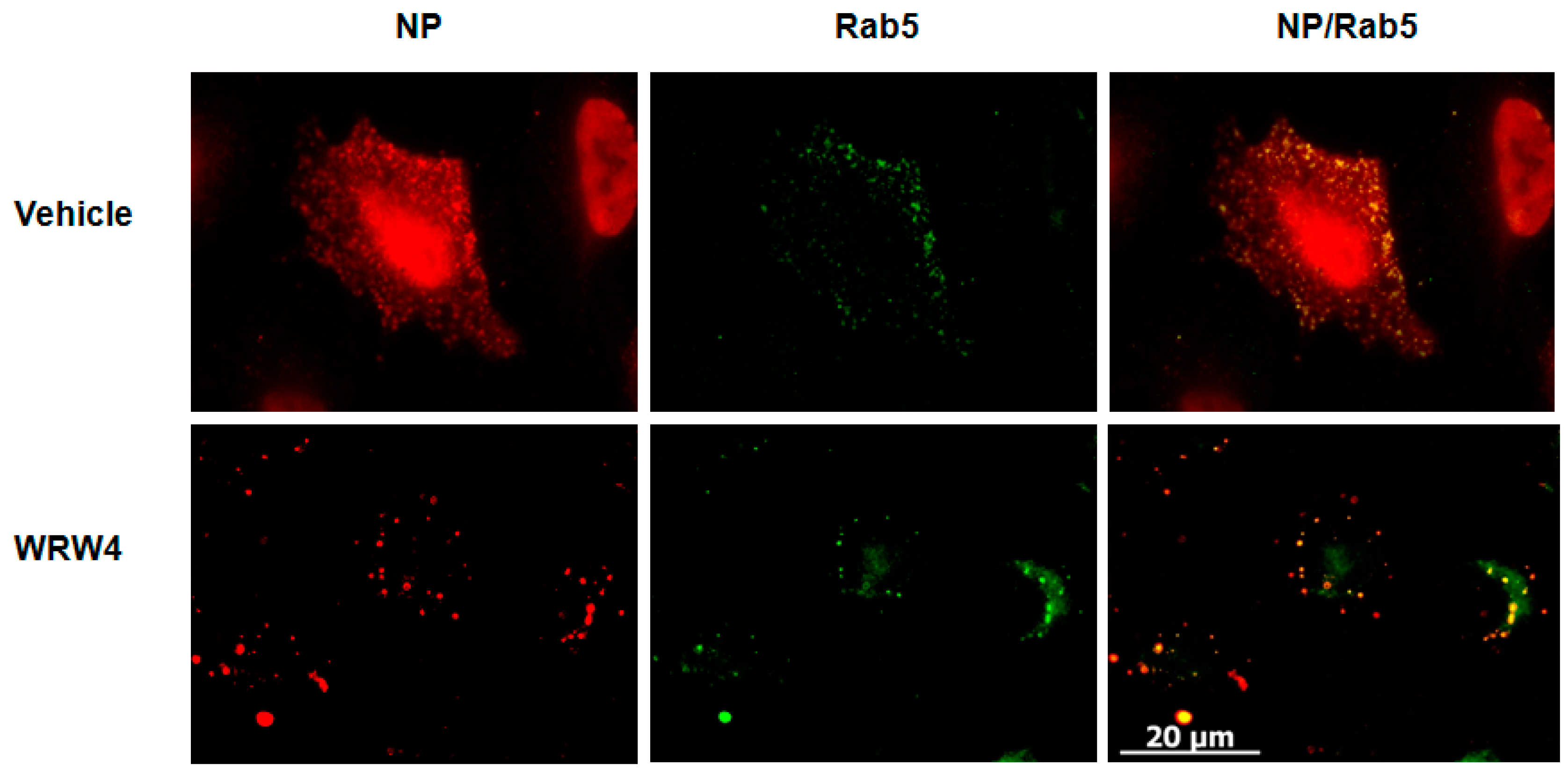
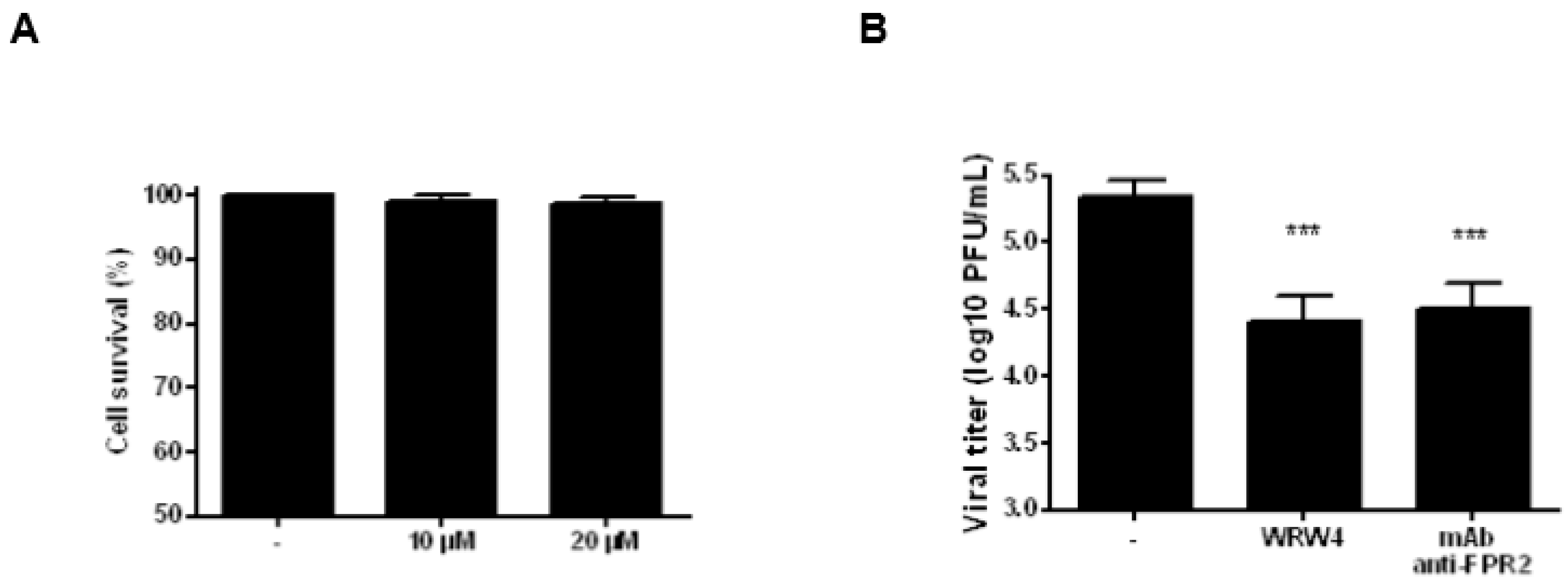
2.1. Treatment of A549 Cells with WRW4 Blocks IAVTrafficking
2.2. Treatment of A549 Cells with an Anti-FPR2 Antibody Blocks Virus Replication
2.3. Treatment of A549 Cells with the Anti-FPR2 mAb, FN-1D6-AI Affects Virus Trafficking
3. Discussion
4. Materials and Methods
4.1. Reagents Cells and Viruses
4.2. Infection Experiments and Cell Viability
4.3. Titration Experiment
4.4. Fluorescence Microscopy Experiments
5. Statistical Analysis
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Kuiken, T.; Riteau, B.; Fouchier, R.A.; Rimmelzwaan, G.F. Pathogenesis of influenza virus infections: The good, the bad and the ugly. Curr. Opin. Virol. 2012, 2, 276–286. [Google Scholar] [CrossRef] [PubMed]
- Lipatov, A.S.; Govorkova, E.A.; Webby, R.J.; Ozaki, H.; Peiris, M.; Guan, Y.; Poon, L.; Webster, R.G. Influenza: Emergence and control. J. Virol. 2004, 78, 8951–8959. [Google Scholar] [CrossRef] [PubMed]
- Berri, F.; Le, V.B.; Jandrot-Perrus, M.; Lina, B.; Riteau, B. Switch from protective to adverse inflammation during influenza: Viral determinants and hemostasis are caught as culprits. Cell. Mol. Life Sci. CMLS 2014, 71, 885–898. [Google Scholar] [CrossRef] [PubMed]
- Xiong, X.; McCauley, J.W.; Steinhauer, D.A. Receptor binding properties of the influenza virus hemagglutinin as a determinant of host range. Curr. Top. Microb. Immunol. 2014, 385, 63–91. [Google Scholar]
- Wilks, S.; de Graaf, M.; Smith, D.J.; Burke, D.F. A review of influenza haemagglutinin receptor binding as it relates to pandemic properties. Vaccine 2012, 30, 4369–4376. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Whittaker, G.R. Entry of influenza virus. Adv. Exp. Med. Biol. 2013, 790, 72–82. [Google Scholar] [PubMed]
- Hutchinson, E.C.; Fodor, E. Transport of the influenza virus genome from nucleus to nucleus. Viruses 2013, 5, 2424–2446. [Google Scholar] [CrossRef] [PubMed]
- Samji, T. Influenza A: Understanding the viral life cycle. Yale J. Biol. Med. 2009, 82, 153–159. [Google Scholar] [PubMed]
- Nayak, D.P.; Balogun, R.A.; Yamada, H.; Zhou, Z.H.; Barman, S. Influenza virus morphogenesis and budding. Virus Res. 2009, 143, 147–161. [Google Scholar] [CrossRef] [PubMed]
- LeBouder, F.; Morello, E.; Rimmelzwaan, G.F.; Bosse, F.; Pechoux, C.; Delmas, B.; Riteau, B. Annexin II incorporated into influenza virus particles supports virus replication by converting plasminogen into plasmin. J. Virol. 2008, 82, 6820–6828. [Google Scholar] [CrossRef] [PubMed]
- Berri, F.; Haffar, G.; Le, V.B.; Sadewasser, A.; Paki, K.; Lina, B.; Wolff, T.; Riteau, B. Annexin V incorporated into influenza virus particles inhibits gamma interferon signaling and promotes viral replication. J. Virol. 2014, 88, 11215–11228. [Google Scholar] [CrossRef] [PubMed]
- Tcherniuk, S.; Cenac, N.; Comte, M.; Frouard, J.; Errazuriz-Cerda, E.; Galabov, A.; Morange, P.E.; Vergnolle, N.; Si-Tahar, M.; Alessi, M.C.; et al. Formyl Peptide Receptor 2 Plays a Deleterious Role During Influenza A Virus Infections. J. Infect. Dis. 2016, 214, 237–247. [Google Scholar] [CrossRef] [PubMed]
- Shaw, M.L.; Stone, K.L.; Colangelo, C.M.; Gulcicek, E.E.; Palese, P. Cellular proteins in influenza virus particles. PLoS Pathog. 2008, 4, e1000085. [Google Scholar] [CrossRef] [PubMed]
- Courtin, N.; Fotso, A.F.; Fautrad, P.; Mas, F.; Alessi, M.C.; Riteau, B. Antiviral activity of formyl peptide receptor 2 antagonists against influenza viruses. Antivir. Res. 2017, 143, 252–261. [Google Scholar] [CrossRef] [PubMed]
- Marjuki, H.; Gornitzky, A.; Marathe, B.M.; Ilyushina, N.A.; Aldridge, J.R.; Desai, G.; Webby, R.J.; Webster, R.G. Influenza A virus-induced early activation of ERK and PI3K mediates V-ATPase-dependent intracellular pH change required for fusion. Cell. Microbiol. 2011, 13, 587–601. [Google Scholar] [CrossRef] [PubMed]
- Pleschka, S.; Wolff, T.; Ehrhardt, C.; Hobom, G.; Planz, O.; Rapp, U.R.; Ludwig, S. Influenza virus propagation is impaired by inhibition of the Raf/MEK/ERK signalling cascade. Nat. Cell Boil. 2001, 3, 301–305. [Google Scholar] [CrossRef] [PubMed]
- Bae, Y.S.; Lee, H.Y.; Jo, E.J.; Kim, J.I.; Kang, H.K.; Ye, R.D.; Kwak, J.Y.; Ryu, S.H. Identification of peptides that antagonize formyl peptide receptor-like 1-mediated signaling. J. Immunol. 2004, 173, 607–614. [Google Scholar] [CrossRef] [PubMed]
- Portela, A.; Digard, P. The influenza virus nucleoprotein: A multifunctional RNA-binding protein pivotal to virus replication. J. Gen. Virol. 2002, 83 Pt 4, 723–734. [Google Scholar] [CrossRef] [PubMed]
- Casadevall, A. Antibody-based therapies for emerging infectious diseases. Emerg. Infect. Dis. 1996, 2, 200–208. [Google Scholar] [CrossRef] [PubMed]
- Norling, L.V.; Dalli, J.; Flower, R.J.; Serhan, C.N.; Perretti, M. Resolvin D1 limits polymorphonuclear leukocyte recruitment to inflammatory loci: Receptor-dependent actions. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 1970–1978. [Google Scholar] [CrossRef] [PubMed]
- De Santo, C.; Arscott, R.; Booth, S.; Karydis, I.; Jones, M.; Asher, R.; Salio, M.; Middleton, M.; Cerundolo, V. Invariant NKT cells modulate the suppressive activity of IL-10-secreting neutrophils differentiated with serum amyloid A. Nat. Immunol. 2010, 11, 1039–1046. [Google Scholar] [CrossRef] [PubMed]
- Alessi, M.C.; Cenac, N.; Si-Tahar, M.; Riteau, B. FPR2: A Novel Promising Target for the Treatment of Influenza. Front. Microb. 2017, 8, 1719. [Google Scholar] [CrossRef] [PubMed]
- Arora, S.; Lim, W.; Bist, P.; Perumalsamy, R.; Lukman, H.M.; Li, F.; Welker, L.B.; Yan, B.; Sethi, G.; Tambyah, P.A.; et al. Influenza A virus enhances its propagation through the modulation of Annexin-A1 dependent endosomal trafficking and apoptosis. Cell Death Differ. 2016, 23, 1243–1256. [Google Scholar] [CrossRef] [PubMed]
- Rescher, U.; Gerke, V. Annexins—Unique membrane binding proteins with diverse functions. J. Cell Sci. 2004, 117 Pt 13, 2631–2639. [Google Scholar] [CrossRef] [PubMed]
- Hoque, M.; Rentero, C.; Cairns, R.; Tebar, F.; Enrich, C.; Grewal, T. Annexins-scaffolds modulating PKC localization and signaling. Cell. Signal. 2014, 26, 1213–1225. [Google Scholar] [CrossRef] [PubMed]
- Rescher, U.; Grewal, T. Highlight: Annexins in health and disease. Biol. Chem. 2016, 397, 947–948. [Google Scholar] [CrossRef] [PubMed]
- LeBouder, F.; Lina, B.; Rimmelzwaan, G.F.; Riteau, B. Plasminogen promotes influenza A virus replication through an annexin 2-dependent pathway in the absence of neuraminidase. J. Gen. Virol. 2010, 91 Pt 11, 2753–2761. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Sun, J.; Gu, L.; Bao, H.; Zhao, Y.; Shi, L.; Yao, W.; Tian, G.; Wang, X.; Chen, H. Annexin A2 (ANXA2) interacts with nonstructural protein 1 and promotes the replication of highly pathogenic H5N1 avian influenza virus. BMC Microbiol. 2017, 17, 191. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.T.; Lichtenberg, B.; Rick, O. Involvement of annexin V in the entry of influenza viruses and role of phospholipids in infection. FEBS Lett. 1996, 392, 59–62. [Google Scholar] [CrossRef]
- Musiol, A.; Gran, S.; Ehrhardt, C.; Ludwig, S.; Grewal, T.; Gerke, V.; Rescher, U. Annexin A6-balanced late endosomal cholesterol controls influenza A replication and propagation. MBio 2013, 4, e00608-13. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Kien, F.; Maniere, M.; Zhang, Y.; Lagarde, N.; Tse, K.S.; Poon, L.L.; Nal, B. Human annexin A6 interacts with influenza a virus protein M2 and negatively modulates infection. J. Virol. 2012, 86, 1789–1801. [Google Scholar] [CrossRef] [PubMed]
- Kuehnl, A.; Musiol, A.; Raabe, C.A.; Rescher, U. Emerging functions as host cell factors—An encyclopedia of annexin-pathogen interactions. Biol. Chem. 2016, 397, 949–959. [Google Scholar] [CrossRef] [PubMed]
- Backes, P.; Quinkert, D.; Reiss, S.; Binder, M.; Zayas, M.; Rescher, U.; Gerke, V.; Bartenschlager, R.; Lohmann, V. Role of annexin A2 in the production of infectious hepatitis C virus particles. J. Virol. 2010, 84, 5775–5789. [Google Scholar] [CrossRef] [PubMed]
- Derry, M.C.; Sutherland, M.R.; Restall, C.M.; Waisman, D.M.; Pryzdial, E.L. Annexin 2-mediated enhancement of cytomegalovirus infection opposes inhibition by annexin 1 or annexin 5. J. Gen. Virol. 2007, 88 Pt 1, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Nobusawa, E.; Sato, K. Comparison of the mutation rates of human influenza A and B viruses. J. Virol. 2006, 80, 3675–3678. [Google Scholar] [CrossRef] [PubMed]
- Poland, G.A.; Jacobson, R.M.; Ovsyannikova, I.G. Influenza virus resistance to antiviral agents: A plea for rational use. Clin. Infect. Dis. 2009, 48, 1254–1256. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, S. Disruption of virus-host cell interactions and cell signaling pathways as an anti-viral approach against influenza virus infections. Biol. Chem. 2011, 392, 837–847. [Google Scholar] [CrossRef] [PubMed]
- Planz, O. Development of cellular signaling pathway inhibitors as new antivirals against influenza. Antivir. Res. 2013, 98, 457–468. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, S. Targeting cell signalling pathways to fight the flu: Towards a paradigm change in anti-influenza therapy. J. Antimicrob. Chemother. 2009, 64, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Scheuch, G.; Canisius, S.; Nocker, K.; Hofmann, T.; Naumann, R.; Pleschka, S.; Ludwig, S.; Welte, T.; Planz, O. Targeting intracellular signaling as an antiviral strategy: Aerosolized LASAG for the treatment of influenza in hospitalized patients. Emerg. Microbes Infect. 2018, 7, 21. [Google Scholar] [CrossRef] [PubMed]
- Haasbach, E.; Muller, C.; Ehrhardt, C.; Schreiber, A.; Pleschka, S.; Ludwig, S.; Planz, O. The MEK-inhibitor CI-1040 displays a broad anti-influenza virus activity in vitro and provides a prolonged treatment window compared to standard of care in vivo. Antivir. Res. 2017, 142, 178–184. [Google Scholar] [CrossRef] [PubMed]
- Carter, P.J. Potent antibody therapeutics by design. Nat. Rev. Immunol. 2006, 6, 343–357. [Google Scholar] [CrossRef] [PubMed]








© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rahman, F.; Chebbo, M.; Courtin, N.; Fotso Fotso, A.; Alessi, M.-C.; Riteau, B. The Annexin A1 Receptor FPR2 Regulates the Endosomal Export of Influenza Virus. Int. J. Mol. Sci. 2018, 19, 1400. https://doi.org/10.3390/ijms19051400
Rahman F, Chebbo M, Courtin N, Fotso Fotso A, Alessi M-C, Riteau B. The Annexin A1 Receptor FPR2 Regulates the Endosomal Export of Influenza Virus. International Journal of Molecular Sciences. 2018; 19(5):1400. https://doi.org/10.3390/ijms19051400
Chicago/Turabian StyleRahman, Fryad, Mohammad Chebbo, Noémie Courtin, Aurelien Fotso Fotso, Marie-Christine Alessi, and Béatrice Riteau. 2018. "The Annexin A1 Receptor FPR2 Regulates the Endosomal Export of Influenza Virus" International Journal of Molecular Sciences 19, no. 5: 1400. https://doi.org/10.3390/ijms19051400
APA StyleRahman, F., Chebbo, M., Courtin, N., Fotso Fotso, A., Alessi, M.-C., & Riteau, B. (2018). The Annexin A1 Receptor FPR2 Regulates the Endosomal Export of Influenza Virus. International Journal of Molecular Sciences, 19(5), 1400. https://doi.org/10.3390/ijms19051400