Epigenetics of Subcellular Structure Functioning in the Origin of Risk or Resilience to Comorbidity of Neuropsychiatric and Cardiometabolic Disorders


Abstract
:
1. Introduction
2. Mitochondria
2.1. Mitochondria/ Nuclear DNA Interactions
2.2. Cross-Talk between Mitochondria and Nucleus and Epigenetic Marks in Nuclear DNA
2.3. Mitochondria Determine Epigenetic Modification Reactions
2.4. Mitochondrial Biogenesis Is Determined by the Epigenetic Program
2.5. Mitochondrial Biogenesis in Neuropsychiatric and Cardiometabolic Disorders
3. Endoplasmic Reticulum
4. Nucleus
4.1. Telomeres
4.2. DNA Reparation
5. Possible Interventions to Promote the Modification of Epigenetic Cues that Alter Subcellular Functioning
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| Acetyl CoA | Acetyl Coenzyme A |
| AMPK | AMP-Activated Protein Kinase |
| ATP | Adenosine Triphosphate |
| BD | Bipolar Disorder |
| BER | Base Excision Repair |
| Ca2+ | Calcium Ion |
| cAMP | Cyclic Adenosine Monophosphate |
| CMD | Cardiometabolic Disorders |
| CpG | Cytosine Phosphate Guanine |
| CREB | cAMP Response Element-Binding Protein |
| DNA | Deoxyribonucleic Acid |
| ER | Endoplasmic Reticulum |
| Fe-S | Iron-Sulfur Centers |
| FOXOs | Forkhead Transcription Factors |
| Hsp | Heat Shock Proteins |
| JNK | c-Jun N-Terminal Kinase |
| MDD | Major Depressive Disorder |
| mtPTP | Mitochondrial Permeability Transition Pore |
| NAD+ | β-Nicotinamide Adenine Dinucleotide |
| NADH | Reduced Nicotinamide Adenine Dinucleotide |
| NER | Nucleotide Excision Repair |
| NF-κB | Nuclear Factor Kappa-Light Chain Enhancer of Activated B Cells |
| NPD | Neuropsychiatric Disorders |
| OGG1 | 8-Oxoguanine Glycosylase-1 |
| PCG1α | Peroxisome Proliferator-Activated Receptor gamma Coactivator 1 Alpha |
| PKA | cAMP-Dependent Protein Kinase |
| PTSD | Post-Traumatic Stress Disorder |
| ROS | Reactive Oxygen Species |
| SIRT1 | Sirtuin 1 |
| UPR | Unfolded Protein Response |
References
- Nakatochi, M.; Ichihara, S.; Yamamoto, K.; Naruse, K.; Yokota, S.H.; Asano, H.; Matsubara, T.; Yokota, M. Epigenome-wide association of myocardial infarction with DNA methylation sites at loci related to cardiovascular disease. Clin. Epigenet. 2017, 9, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Liu, X.; Deng, Y.; Qing, H. DNA methylation, a hand behind neurodegenerative diseases. Front. Aging Neurosci. 2013, 5, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Roth, G.A.; Johnson, C.; Abajobir, A.; Abd-Allah, F.; Abera, S.F.; Abyu, G.; Murray, C. Global, regional, and national burden of cardiovascular diseases for 10 causes, 1990 to 2015. J. Am. Coll. Cardiol. 2017, 70, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Whiteford, H.A.; Ferrari, A.J.; Degenhardt, L.; Feigin, V.; Vos, T. The global burden of mental, neurological and substance use disorders: An analysis from the Global Burden of Disease Study 2010. PLoS ONE 2015, 10, e0116820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duric, V.; Clayton, S.; Leong, M.L.; Yuan, L.L. Comorbidity factors and brain mechanisms linking chronic stress and systemic illness. Neural Plasticity 2016, 5460732. [Google Scholar] [CrossRef] [PubMed]
- Bankier, B.; Januzzi, J.L.; Littman, A.B. The high prevalence of multiple psychiatric disorders in stable outpatients with coronary heart disease. Psychosom. Med. 2004, 66, 645–650. [Google Scholar] [CrossRef] [PubMed]
- Roest, A.M.; Martens, E.J.; de Jonge, P.; Denollet, J. Anxiety and risk of incident coronary heart disease: A meta-analysis. J. Am. Coll. Cardiol. 2010, 56, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Arzate-Mejía, R.G.; Valle-García, D.; Recillas-Targa, F. Signaling epigenetics: Novel insights on cell signaling and epigenetic regulation. IUBMB Life 2011, 63, 881–895. [Google Scholar] [CrossRef] [PubMed]
- Zapata-Martín del Campo, C.M.; Martínez-Rosas, M.; Guarner-Lans, V. Epigenetic Programming of Synthesis, Release, and/or Receptor Expression of Common Mediators Participating in the Risk/Resilience for Comorbid Stress-Related Disorders and Coronary Artery Disease. Int. J. Mol. Sci. 2018, 19, 1224. [Google Scholar] [CrossRef] [PubMed]
- Babenko, O.; Kovalchuk, I.; Metz, G.A. Epigenetic programming of neurodegenerative diseases by an adverse environment. Brain Res. 2012, 1444, 96–111. [Google Scholar] [CrossRef] [PubMed]
- Jia, H.; Morris, C.D.; Williams, R.M.; Loring, J.F.; Thomas, E.A. HDAC inhibition imparts beneficial transgenerational effects in Huntington’s disease mice via altered DNA and histone methylation. Proc. Natl. Acad. Sci. USA 2015, 112, E56–E64. [Google Scholar] [CrossRef] [PubMed]
- El Osta, A.; Brasacchio, D.; Yao, D.; Pocai, A.; Jones, P.L.; Roeder, R.G.; Cooper, M.E.; Brownlee, M. Transient high glucose causes persistent epigenetic changes and altered gene expression during subsequent normoglycemia. J. Exp. Med. 2008, 205, 2409–2417. [Google Scholar] [CrossRef] [PubMed]
- Brasacchio, D.; Okabe, J.; Tikellis, C.; Balcerczyk, A.; George, P.; Baker, E.K.; Calkin, A.C.; Brownlee, M.; Cooper, M.E.; El-Osta, A. Hyperglycemia induces a dynamic cooperativity of histone methylase and demethylase enzymes associated with gene-activating epigenetic marks that coexist on the lysine tail. Diabetes 2009, 58, 1229–1236. [Google Scholar] [CrossRef] [PubMed]
- Pirola, L.; Balcerczyk, A.; Tothill, R.W.; Haviv, I.; Kaspi, A.; Lunke, S.; Ziemann, M.; Karagiannis, T.; Tonna, S.; Kowalczyk, A.; et al. Genome-wide analysis distinguishes hyperglycemia regulated epigenetic signatures of primary vascular cells. Genome Res. 2011, 21, 1601–1615. [Google Scholar] [CrossRef] [PubMed]
- Takizawa, F.; Mizutani, S.; Ogawa, Y.; Sawada, N. Glucose-independent persistence of PAI-1 gene expression and H3K4 tri-methylation in type 1 diabetic mouse endothelium: Implication in metabolic memory. Biochem. Biophys. Res. Commun. 2013, 433, 66–72. [Google Scholar] [CrossRef] [PubMed]
- Vecellio, M.; Spallotta, F.; Nanni, S.; Colussi, C.; Cencioni, C.; Derlet, A.; Bassetti, B.; Tilenni, M.; Carena, M.C.; Farsetti, A.; et al. The histone acetylase activator pentadecylidenemalonate 1b rescues proliferation and differentiation in human cardiac mesenchymal cells of type 2 diabetic patients. Diabetes 2014, 63, 2132–2147. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.C. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: A dawn for evolutionary medicine. Ann. Rev. Genet. 2005, 39, 359–407. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.C.; Fan, W. Energetics, epigenetics, mitochondrial genetics. Mitochondrion 2010, 10, 12–31. [Google Scholar] [CrossRef] [PubMed]
- Shaughnessy, D.T.; McAllister, K.; Worth, L.; Haugen, A.C.; Meyer, J.N.; Domann, F.E.; Van Houten, B.; Mostoslavsky, R.; Bultman, S.J.; Baccarelli, A.A.; et al. Mitochondria, energetics, epigenetics, and cellular responses to stress. Environ. Health Perspect. 2014, 122, 1271–1278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Devall, M.; Mill, J.; Lunnon, K. The mitochondrial epigenome: A role in Alzheimer’s disease? Epigenomics 2014, 6, 665–675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mulder, H.; Ling, C. Mitochondrial dysfunction in pancreatic beta-cells in Type 2 diabetes. Mol. Cell. Endocrinol. 2009, 297, 34–40. [Google Scholar] [CrossRef] [PubMed]
- Rocha, M.; Apostolova, N.; Herance, J.R.; Rovira-Llopis, S.; Hernandez-Mijares, A.; Victor, V.M. Perspectives and potential applications of mitochondria-targeted antioxidants in cardiometabolic diseases and type 2 diabetes. Med. Res. Rev. 2014, 34, 160–189. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Z.; Almeida, F.A. Mitochondrial alteration in type 2 diabetes and obesity. An epigenetic link. Cell Cycle 2014, 13, 890–897. [Google Scholar] [CrossRef] [PubMed]
- Fillmore, N.; Mori, J.; Lopaschuk, G.D. Mitochondrial fatty acid oxidation alterations in heart failure, ischaemic heart disease and diabetic cardiomyopathy. Br. J. Pharmacol. 2014, 171, 2080–2090. [Google Scholar] [CrossRef] [PubMed]
- Marazziti, D.; Baroni, S.; Picchetti, M.; Landi, P.; Silvestri, S.; Vatteroni, E.; Catena Dell’Osso, M. Psychiatric disorders and mitochondrial dysfunctions. Eur. Rev. Med. Pharmacol. Sci. 2012, 16, 270–275. [Google Scholar] [PubMed]
- Li, C.H.; Casanueva, O. Epigenetic inheritance of proteostasis and ageing. Essays Biochem. 2016, 60, 191–202. [Google Scholar] [CrossRef] [PubMed]
- Oakes, S.A.; Papa, F.R. The role of endoplasmic reticulum stress in human pathology. Annu. Rev. Pathol. 2015, 10, 173–194. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.L.; Gao, D.S. Endoplasmic reticulum proteins quality control and the unfolded protein response: The regulative mechanism of organisms against stress injuries. Biofactors 2014, 40, 569–585. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Arrigo, A.P.; Currie, R.W. Heat shock treatment suppresses angiotensin II-induced activation of NF-kappaB pathway and heart inflammation: A role for IKK depletion by heat shock? Am. J. Physiol. Heart Circ. Physiol. 2004, 287, H1104–H1114. [Google Scholar] [CrossRef] [PubMed]
- Mozzini, C.; Cominacini, L.; Garbin, U.; Fratta Pasini, A.M. Endoplasmic Reticulum Stress, NRF2 Signalling and Cardiovascular Diseases in a Nutshell. Curr. Atheroscler. Rep. 2017, 19, 33. [Google Scholar] [CrossRef] [PubMed]
- Minamino, T.; Kitakaze, M. ER stress in cardiovascular disease. J. Mol. Cell. Cardiol. 2010, 48, 1105–1110. [Google Scholar] [CrossRef] [PubMed]
- Kannan, S.; Muthusamy, V.R.; Whitehead, K.J.; Wang, L.; Gomes, A.V.; Litwin, S.E.; Kensler, T.W.; Abel, E.D.; Hoidal, J.R.; Rajasekaran, N.S. Nrf2 deficiency prevents reductive stress-induced hypertrophic cardiomyopathy. Cardiovasc. Res. 2013, 100, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Xie, H.B.; Cammarato, A.; Rajasekaran, N.S.; Zhang, H.; Suggs, J.A.; Lin, H.C.; Bernstein, S.I.; Benjamin, I.J.; Golic, K.G. The NADPH metabolic network regulates human αB-crystallin cardiomyopathy and reductive stress in Drosophila melanogaster. PLoS Genet. 2013, 9, e1003544. [Google Scholar] [CrossRef] [PubMed]
- Doyle, K.M.; Kennedy, D.; Gorman, A.M.; Gupta, S.; Healy, S.J.M.; Samali, A. Unfolded proteins and endoplasmic reticulum stress in neurodegenerative disorders. J. Cell. Mol. Med. 2011, 15, 2025–2039. [Google Scholar] [CrossRef] [PubMed]
- Gapp, K.; Woldemichael, B.T.; Bohacek, J.; Mansuy, I.M. Epigenetic regulation in neurodevelopment and neurodegenerative diseases. Neuroscience 2014, 264, 99–111. [Google Scholar] [CrossRef] [PubMed]
- Blasco, M.A. The epigenetic regulation of mammalian telomeres. Nat. Rev. Genet. 2007, 8, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Wong, L.H. Epigenetic regulation of telomere chromatin integrity in pluripotent embryonic stem cells. Epigenomics 2010, 2, 639–655. [Google Scholar] [CrossRef] [PubMed]
- Dan, J.; Yang, J.; Liu, Y.; Xiao, A.; Liu, L. Roles for Histone Acetylation in Regulation of Telomere Elongation and Two-cell State in Mouse ES Cells. J. Cell Physiol. 2015, 230, 2337–2344. [Google Scholar] [CrossRef] [PubMed]
- Lindqvist, D.; Epe, E.S.; Mellonc, S.H.; Penninx, B.W.; Révészd, D.; Verhoeven, J.E.; Reus, V.I.; Lin, J.; Mahanb, L.; Hough, C.M.; et al. Psychiatric disorders and leukocyte telomere length: Underlying mechanisms linking mental illness with cellular aging. Neurosci. Biobehav. Rev. 2015, 55, 333–364. [Google Scholar] [CrossRef] [PubMed]
- Sibille, K.T.; Witek-Janusek, L.; Mathews, H.L.; Fillingim, R.B. Telomeres and epigenetics: Potential relevance to chronic pain. Pain 2012, 153, 1789–1793. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, P.M.; Tufvesson, H.; Leosdottir, M.; Melander, O. Telomeres and cardiovascular disease risk: An update 2013. Transl. Res. 2013, 162, 371–380. [Google Scholar] [CrossRef] [PubMed]
- Bellacosa, A.; Drohat, A.C. Role of base excision repair in maintaining the genetic and epigenetic integrity of CpG sites. DNA Repair 2015, 32, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Moore, S.P.; Toomire, K.J.; Strauss, P.R. DNA modifications repaired by base excision repair are epigenetic. DNA Repair 2013, 12, 1152–1158. [Google Scholar] [CrossRef] [PubMed]
- Fleming, A.M.; Ding, Y.; Burrows, C.J. Oxidative DNA damage is epigenetic by regulating gene transcription via base excision repair. Proc. Natl. Acad. Sci. USA 2017, 114, 2604–2609. [Google Scholar] [CrossRef] [PubMed]
- Markkanen, E.; Meyer, U.; Dianov, G.L. DNA Damage and Repair in Schizophrenia and Autism: Implications for Cancer Comorbidity and Beyond. Int. J. Mol. Sci. 2016, 17, 856. [Google Scholar] [CrossRef] [PubMed]
- Uryga, A.; Gray, K.; Bennett, M. DNA Damage and Repair in Vascular Disease. Annu. Rev. Physiol. 2016, 78, 45–66. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Z.; Ristow, M. Mitochondria and metabolic homeostasis. Antioxid. Redox Signal 2013, 19, 240–242. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulos, V.; Miller, W.L. Role of mitochondria in steroidogenesis. Best Pract. Res. Clin. Endocrinol. Metab. 2012, 26, 771–790. [Google Scholar] [CrossRef] [PubMed]
- Suen, D.F.; Norris, K.L.; Youle, R.J. Mitochondrial dynamics and apoptosis. Genes Dev. 2008, 22, 1577–1590. [Google Scholar] [CrossRef] [PubMed]
- Tait, S.W.; Green, D.R. Mitochondria and cell signalling. J. Cell Sci. 2012, 125, 807–815. [Google Scholar] [CrossRef] [PubMed]
- Zemirli, N.; Arnoult, D. Mitochondrial anti-viral immunity. Int. J. Biochem. Cell Biol. 2012, 44, 1473–1476. [Google Scholar] [CrossRef] [PubMed]
- Cyr, A.R.; Domann, F.E. The redox basis of epigenetic modifications: From mechanisms to functional consequences. Antioxid. Redox Signal. 2011, 15, 551–589. [Google Scholar] [CrossRef] [PubMed]
- Donohoe, D.R.; Bultman, S.J. Metaboloepigenetics: Interrelationships between energy metabolism and epigenetic control of gene expression. J. Cell Physiol. 2012, 227, 3169–3177. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Pastor, B.; Cosentino, C.; Mostoslavsky, R. A tale of metabolites: The cross-talk between chromatin and energy metabolism. Cancer Discov. 2013, 3, 497–501. [Google Scholar] [CrossRef] [PubMed]
- Lane, N. Oxygen: The Molecule That Made the World; Oxford: Oxford University Press: New York, NY, USA, 2002; pp. 1–384. ISBN 978-0-19-860783-0. [Google Scholar]
- Lane, N. Power, Sex, Suicide: Mitochondria and the Meaning of Life; Oxford University Press: New York, NY, USA, 2005; pp. 1–368. ISBN 0192-80481-2. [Google Scholar]
- Hitchler, M.J.; Domann, F.E. Metabolic defects provide a spark for the epigenetic switch in cancer. Free Radic. Biol. Med. 2009, 47, 115–127. [Google Scholar] [CrossRef] [PubMed]
- Chan, D.C. Fusion and Fission: Interlinked Processes Critical for Mitochondrial Health. Annu. Rev. Genet. 2012, 46, 265–287. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Chomyn, A.; Chan, D.C. Disruption of fusion results in mitochondrial heterogeneity and dysfunction. J. Biol. Chem. 2005, 280, 26185–26192. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Detmer, S.A.; Ewald, A.J.; Griffin, E.E.; Fraser, S.E.; Chan, D.C. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J. Cell Biol. 2003, 160, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Cipolat, S.; Martins de Brito, O.; Dal Zilio, B.; Scorrano, L. OPA1 requires mitofusin 1 to promote mitochondrial fusion. Proc. Natl. Acad. Sci. USA 2004, 101, 15927–15932. [Google Scholar] [CrossRef] [PubMed]
- Gandre-Babbe, S.; van der Bliek, A.M. The novel tail-anchored membrane protein Mff controls mitochondrial and peroxisomal fission in mammalian cells. Mol. Biol. Cell 2008, 19, 2402–2412. [Google Scholar] [CrossRef] [PubMed]
- James, D.I.; Parone, P.A.; Mattenberger, Y.; Martinou, J.C. hFis1, a novel component of the mammalian mitochondrial fission machinery. J. Biol. Chem. 2003, 278, 36373–36379. [Google Scholar] [CrossRef] [PubMed]
- Smirnova, E.; Griparic, L.; Shurland, D.L.; van der Bliek, A.M. Dynamin-related protein Drp1 is required for mitochondrial division in mammalian cells. Mol. Biol. Cell 2001, 12, 2245–2256. [Google Scholar] [CrossRef] [PubMed]
- Yoon, Y.; Krueger, E.W.; Oswald, B.J.; McNiven, M.A. The mitochondrial protein hFis1 regulates mitochondrial fission in mammalian cells through an interaction with the dynamin-like protein DLP1. Mol. Cell. Biochem. 2003, 23, 5409–5420. [Google Scholar] [CrossRef]
- Fariss, M.W.; Chan, C.B.; Patel, M.; Van Houten, B.; Orrenius, S. Role of mitochondria in toxic oxidative stress. Mol. Interv. 2005, 5, 94–111. [Google Scholar] [CrossRef] [PubMed]
- Van Houten, B.; Woshner, V.; Santos, J.H. Role of mitochondrial DNA in toxic responses to oxidative stress. DNA Repair 2006, 5, 145–152. [Google Scholar] [CrossRef] [PubMed]
- Wallace, S.S. Base excision repair: A critical player in many games. DNA Repair 2014, 19, 14–26. [Google Scholar] [CrossRef] [PubMed]
- Toker, L.; Agam, G. Mitochondrial dysfunction in psychiatric morbidity: Current evidence and therapeutic prospects. Neuropsychiatr. Dis. Treat. 2015, 11, 2441–2447. [Google Scholar] [PubMed]
- Streck, E.; Gonçalves, C.L.; Furlanetto, C.B.; Scaini, G.; Dal-Pizzol, F.; Quevedo, J. Mitochondria and the central nervous system: Searching for a pathophysiological basis of psychiatric disorders. Rev. Bras. Psiquiatr. 2014, 36, 156–167. [Google Scholar] [CrossRef] [PubMed]
- Andreazza, A.C. Combining redox-proteomics and epigenomics to explain the involvement of oxidative stress in psychiatric disorders. Mol. Biosyst. 2012, 8, 2503–2512. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhou, R.; Li, X.; Ursano, R.J.; Li, H. Stress-induced change of mitochondria membrane potential regulated by genomic and non-genomic GR signaling: A possible mechanism for hippocampus atrophy in PTSD. Med. Hypotheses 2006, 66, 1205–1208. [Google Scholar] [CrossRef] [PubMed]
- Picard, M.; Juster, R.P.; McEwen, B.S. Mitochondrial allostatic load puts the ‘gluc’ back in glucocorticoids. Nat. Rev. Endocrinol. 2014, 10, 303–310. [Google Scholar] [CrossRef] [PubMed]
- Atlante, A.; Calissano, P.; Bobba, A.; Giannattasio, S.; Marra, E.; Passarella, S. Glutamate neurotoxicity, oxidative stress and mitochondria. FEBS Lett. 2001, 497, 1–5. [Google Scholar] [CrossRef]
- Toya, S.; Takatsuru, Y.; Kokubo, M.; Amano, I.; Shimokawa, N.; Koibuchi, N. Early-life-stress affects the homeostasis of glutamatergic synapses. Eur. J. Neurosci. 2014, 40, 3627–3634. [Google Scholar] [CrossRef] [PubMed]
- Marín-García, J. Mitochondrial DNA repair: A novel therapeutic target for heart failure. Heart Fail. Rev. 2016, 21, 475–487. [Google Scholar] [CrossRef] [PubMed]
- Shen, G.X. Mitochondrial dysfunction, oxidative stress and diabetic cardiovascular disorders. Cardiovasc. Hematol. Disord. Drug Targets 2012, 12, 106–112. [Google Scholar] [CrossRef] [PubMed]
- Vilne, B.; Skogsberg, J.; Foroughi, A.H.; Talukdar, H.A.; Kessler, T.; Björkegren, J.L.M.; Schunkert, H. Network analysis reveals a causal role of mitochondrial gene activity in atherosclerotic lesion formation. Atherosclerosis 2017, 267, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Madamanchi, N.R.; Runge, M.S. Mitochondrial dysfunction in atherosclerosis. Circ. Res. 2007, 100, 460–473. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C.; Chevet, E.; Oakes, S.A. Proteostasis control by the unfolded protein response. Nat. Cell Biol. 2015, 17, 829–838. [Google Scholar] [CrossRef] [PubMed]
- Görlach, A.; Bertram, K.; Hudecova, S.; Krizanova, O. Calcium and ROS: A mutual interplay. Redox Biol. 2015, 6, 260–271. [Google Scholar] [CrossRef] [PubMed]
- Chaudhari, N.; Talwar, P.; Parimisetty, A.; Lefebvre d’Hellencourt, C.; Ravanan, P. A molecular web: Endoplasmic reticulum stress, inflammation, and oxidative stress. Front. Cell. Neurosci. 2014, 8, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Song, Y.; Loscalzo, J. Regulation of the protein disulfide proteome by mitochondria in mammalian cells. Proc. Natl. Acad. Sci. USA 2007, 104, 10813–10817. [Google Scholar] [CrossRef] [PubMed]
- Walter, P.; Ron, D. The unfolded protein response: From stress pathway to homeostatic regulation. Science 2011, 334, 1081–1086. [Google Scholar] [CrossRef] [PubMed]
- Guarner, V.; Rubio-Ruiz, M.E. Low-grade systemic inflammation connects aging, metabolic syndrome and cardiovascular disease. Interdiscip. Top. Gerontol. 2015, 40, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Basseri, S.; Austin, R.C. Endoplasmic reticulum stress and lipid metabolism: Mechanisms and therapeutic potential. Biochem. Res. Int. 2012, 2012, 841362. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.Z.; Zhao, D.; Khan, S.H.; Yang, L. Unfolded protein response pathways in neurodegenerative diseases. J. Mol. Neurosci. 2015, 57, 529–537. [Google Scholar] [CrossRef] [PubMed]
- Penney, J.; Mendell, A.; Zeng, M.; Tran, K.; Lymer, J.; Turner, P.V.; Choleris, E.; MacLusky, N.; Lu, R. LUMAN/CREB3 is a key regulator of glucocorticoid-mediated stress responses. Mol. Cell. Endocrinol. 2017, 439, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Von Zglinicki, T. Role of oxidative stress in telomere length regulation and replicative senescence. Ann. N. Y. Acad. Sci. 2000, 908, 99–110. [Google Scholar] [CrossRef] [PubMed]
- Starnino, L.; Busque, L.; Tardif, J.C.; D’Antono, B. Psychological Profiles in the Prediction of Leukocyte Telomere Length in Healthy Individuals. PLoS ONE 2016, 11, e0165482. [Google Scholar] [CrossRef] [PubMed]
- Verhoeven, J.E.; van Oppen, P.; Révész, D.; Wolkowitz, O.M.; Penninx, B.W.J.H. Depressive and Anxiety Disorders Showing Robust, but Non-Dynamic, 6-Year Longitudinal Association with Short Leukocyte Telomere Length. Am. J. Psychiatry 2016, 173, 617–624. [Google Scholar] [CrossRef] [PubMed]
- Zou1, Y.; Leong, W.; Yao, M.; Hu, X.; Lu, S.; Zhu, X.; Chen, L.; Tong, J.; Shi, J.; Gilson, E.; et al. Test anxiety and telomere length: Academic stress in adolescents may not cause rapid telomere erosion. Oncotarget 2017, 8, 10836–10844. [Google Scholar] [CrossRef] [PubMed]
- Shalev, I. Early life stress and telomere length: Investigating the connection and possible mechanisms: A critical survey of the evidence base, research methodology and basic biology. Bioessays 2012, 34, 943–952. [Google Scholar] [CrossRef] [PubMed]
- Lopizzo, N.; Tosato, S.; Begni, V.; Tomassi, S.; Cattane, N.; Barcella, M.; Turco, G.; Ruggeri, M.; Riva, M.A.; Pariante, C.M.; et al. Transcriptomic analyses and leukocyte telomere length measurement in subjects exposed to severe recent stressful life events. Transl. Psychiatry 2017, 7, e1042. [Google Scholar] [CrossRef] [PubMed]
- Monaghan, P. Organismal stress, telomeres and life histories Organismal stress, telomeres and life histories. J. Exp. Biol. 2014, 217, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Robles, T.F.; Carroll, J.E.; Bai, S.; Bridget, M.; Reynolds, B.M.; Esquivel, S.; Repetti, R.L. Emotions and Family Interactions in Childhood: Associations with Leukocyte Telomere Length. Psychoneuroendocrinology 2016, 63, 343–350. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, J.; Mihara, K.; Bhattacharjee, D.; Mukherjee, M. Telomere length as a potential biomarker of coronary artery disease. Indian J. Med. Res. 2017, 145, 730–737. [Google Scholar] [CrossRef] [PubMed]
- Fyhrquist, F.; Saijonmaa, O. Telomere length and cardiovascular aging. Ann. Med. 2012, 44 (Suppl. S1), S138–S142. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, J.; Spyridopoulos, I. Telomere length in cardiovascular disease: New challenges in measuring this marker of cardiovascular aging. Future Cardiol. 2011, 7, 789–803. [Google Scholar] [CrossRef] [PubMed]
- Dexheimer, T. DNA Repair Pathways and Mechanisms. In DNA Repair of Cancer Stem Cells; Mathews, L., Cabarcas, S., Hurt, E., Eds.; Springer: Dordrecht, The Netherlands, 2013; pp. 19–32. ISBN 978-9400745896. [Google Scholar]
- Shaposhnikov, M.; Proshkina, E.; Shilova, L.; Zhavoronkov, A.; Moskalev, A. Lifespan and Stress Resistance in Drosophila with Overexpressed DNA Repair Genes. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef] [PubMed]
- Bohra, V.A.; Stevnsnerb, T.; Souza-Pintoa, N.C.de. Mitochondrial DNA repair of oxidative damage in mammalian cells. Gene 2002, 286, 127–134. [Google Scholar] [CrossRef]
- Fleming, A.M.; Burrows, C.J. 8-Oxo-7,8-dihydroguanine, friend and foe: Epigenetic-like regulator versus initiator of mutagenesis. DNA Repair 2017, 56, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Fishel, M.L.; Vasko, M.R.; Kelley, M.R. DNA repair in neurons: So if they don’t divide what’s to repair? Mutat. Res. 2007, 614, 24–36. [Google Scholar] [CrossRef] [PubMed]
- Raza, M.U.; Tufan, T.; Wang, Y.; Hill, C.; Zhu, M.Y. DNA Damage in Major Psychiatric Diseases. Neurotox. Res. 2016, 30, 251–267. [Google Scholar] [CrossRef] [PubMed]
- Czarny, P.; Kwiatkowski, D.; Toma, M.; Kubiak, J.; Sliwinska, A.; Talarowska, M.; Szemraj, J.; Maes, M.; Galecki, P.; Sliwinski, T. Impact of Single Nucleotide Polymorphisms of Base Excision Repair Genes on DNA Damage and Efficiency of DNA Repair in Recurrent Depression Disorder. Mol. Neurobiol. 2017, 54, 4150–4159. [Google Scholar] [CrossRef] [PubMed]
- Cengiz, M.; Bayoglu, B.; Alansal, N.O.; Cengiz, S.; Dirican, A.; Kocabasoglu, N. Pro198Leu polymorphism in the oxidative stress gene, glutathione peroxidase-1, is associated with a gender-specific risk for panic disorder. Int. J. Psychiatry Clin. Pract. 2015, 19, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Sarga, L.; Hart, N.; Koch, L.; Britton, S.; Hajas, G.; Boldogh, I.; Ba, X.; Radak, Z. Aerobic endurance capacity affects spatial memory and SIRT1 is a potent modulator of 8-oxoguanine repair. Neuroscience 2013, 252, 326–336. [Google Scholar] [CrossRef] [PubMed]
- Gavin, D.P.; Chase, K.A.; Sharma, R.P. Active DNA demethylation in post-mitotic neurons: A reason for optimism. Neuropharmacology 2013, 75, 233–245. [Google Scholar] [CrossRef] [PubMed]
- Mantha, A.K.; Sarkar, B.; Tell, G. A short review on the implications of base excision repair pathway for neurons: Relevance to neurodegenerative diseases. Mitochondrion 2014, 16, 38–49. [Google Scholar] [CrossRef] [PubMed]
- Mahmoudi, M.; Mercer, J.; Bennett, M. DNA damage and repair in atherosclerosis. Cardiovasc. Res. 2006, 71, 259–268. [Google Scholar] [CrossRef] [PubMed]
- Vakonaki, E.; Tsarouhas, K.; Spandidos, D.A.; Tsatsakis, A.M. Complex interplay of DNA damage, DNA repair genes, and oxidative stress in coronary artery disease. Anatol. J. Cardiol. 2016, 16, 939. [Google Scholar] [CrossRef] [PubMed]
- Gray, K.; Kumar, S.; Figg, N.; Harrison, J.; Baker, L.; Mercer, J.; Littlewood, T.; Bennett, M. Effects of DNA damage in smooth muscle cells in atherosclerosis. Circ. Res. 2015, 116, 816–826. [Google Scholar] [CrossRef] [PubMed]
- Gorenne, I.; Kumar, S.; Gray, K.; Figg, N.; Yu, H.; Mercer, J.; Bennett, M. Vascular smooth muscle cell sirtuin 1 protects against DNA damage and inhibits atherosclerosis. Circulation 2013, 127, 386–396. [Google Scholar] [CrossRef] [PubMed]
- Vinci, M.C.; Polvani, G.; Pesce, M. Epigenetic programming and risk: The birthplace of cardiovascular disease? Stem Cell Rev. 2013, 9, 241–253. [Google Scholar] [CrossRef] [PubMed]
- Takigawa-Imamura, H.; Sekine, T.; Murata, M.; Takayama, K.; Nakazawa, K.; Nakagawa, J. Stimulation of glucose uptake in muscle cells by prolonged treatment with scriptide, a histone deacetylase inhibitor. Biosci. Biotechnol. Biochem. 2003, 67, 1499–1506. [Google Scholar] [CrossRef] [PubMed]
- Galmozzi, A.; Mitro, N.; Ferrari, A.; Gers, E.; Gilardi, F.; Godio, C.; Cermenati, G.; Gualerzi, A.; Donetti, E.; Rotili, D.; et al. Inhibition of class I histone deacetylases unveils a mitochondrial signature and enhances oxidative metabolism in skeletal muscle and adipose tissue. Diabetes 2013, 62, 732–742. [Google Scholar] [CrossRef] [PubMed]
- Crosson, C.E.; Mani, S.K.; Husain, S.; Alsarraf, O.; Menick, D.R. Inhibition of histone deacetylase protects the retina from ischemic injury. Investig. Ophthalmol. Vis. Sci. 2010, 51, 3639–3645. [Google Scholar] [CrossRef] [PubMed]
- Lenoir, O.; Flosseau, K.; Ma, F.X.; Blondeau, B.; Mai, A.; Bassel-Duby, R.; Ravassard, P.; Olson, E.N.; Haumaitre, C.; Scharfmann, R. Specific control of pancreatic endocrine beta- and delta-cell mass by class IIa histone deacetylases HDAC4, HDAC5, and HDAC9. Diabetes 2011, 60, 2861–2871. [Google Scholar] [CrossRef] [PubMed]
- Cencioni, C.; Spallotta, F.; Martelli, F.; Valente, S.; Mai, A.; Zeiher, A.M.; Gaetano, C. Oxidative stress and epigenetic regulation in ageing and age-related diseases. Int. J. Mol. Sci. 2013, 14, 17643–17663. [Google Scholar] [CrossRef] [PubMed]
- Lewis, E.C.; Blaabjerg, L.; Storling, J.; Ronn, S.G.; Mascagni, P.; Dinarello, C.A.; Mandrup-Poulsen, T. The oral histone deacetylase inhibitor ITF2357 reduces cytokines and protects islet beta cells in vivo and in vitro. Mol. Med. 2011, 17, 369–377. [Google Scholar] [CrossRef] [PubMed]
- Tedong, L.; Madiraju, P.; Martineau, L.C.; Vallerand, D.; Arnason, J.T.; Desire, D.D.; Lavoie, L.; Kamtchouing, P.; Haddad, P.S. Hydro-ethanolic extract of cashew tree (Anacardium occidentale) nut and its principal compound, anacardic acid, stimulate glucose uptake in C2C12 muscle cells. Mol. Nutr. Food Res. 2010, 54, 1753–1762. [Google Scholar] [CrossRef] [PubMed]
- Halili, M.A.; Andrews, M.R.; Labzin, L.I.; Schroder, K.; Matthias, G.; Cao, C.; Lovelace, E.; Reid, R.C.; Le, G.T.; Hume, D.A.; et al. Differential effects of selective HDAC inhibitors on macrophage inflammatory responses to the Toll-like receptor 4 agonist LPS. J. Leukoc. Biol. 2010, 87, 1103–1114. [Google Scholar] [CrossRef] [PubMed]
- Luna Ortiz, P.; Guarner, V.; Farías, J.M.; Hernández-Pacheco, G.; Martínez, M. Importance of metabolic memory in the development of vascular complications in diabetic patients. J. Cardiothorac. Vasc. Anesth. 2016, 30, 1369–1378. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Lan, F.; Matson, C.; Mulligan, P.; Whetstine, J.R.; Cole, P.A.; Casero, R.A. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 2004, 119, 941–953. [Google Scholar] [CrossRef] [PubMed]
- Tsukada, Y.; Fang, J.; Erdjument-Bromage, H.; Warren, M.E.; Borchers, C.H.; Tempst, P.; Zhang, Y. Histone demethylation by a family of JmjC domain-containing proteins. Nature 2006, 439, 811–816. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
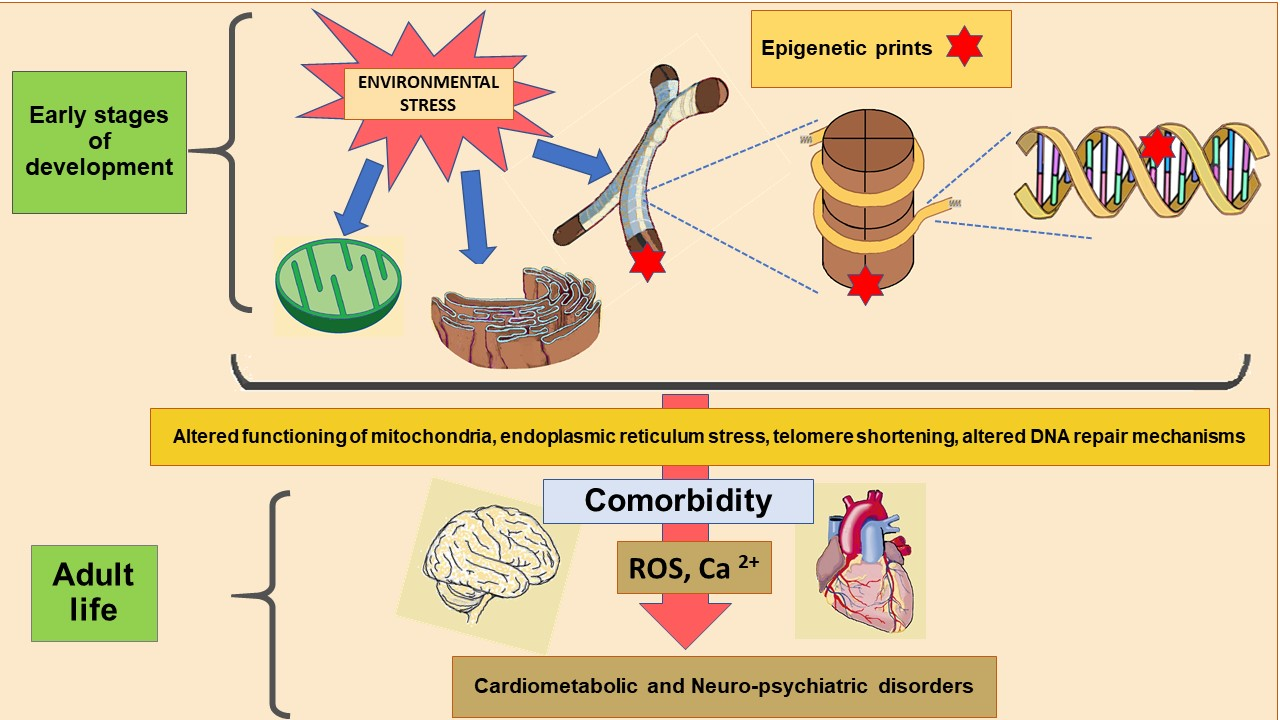
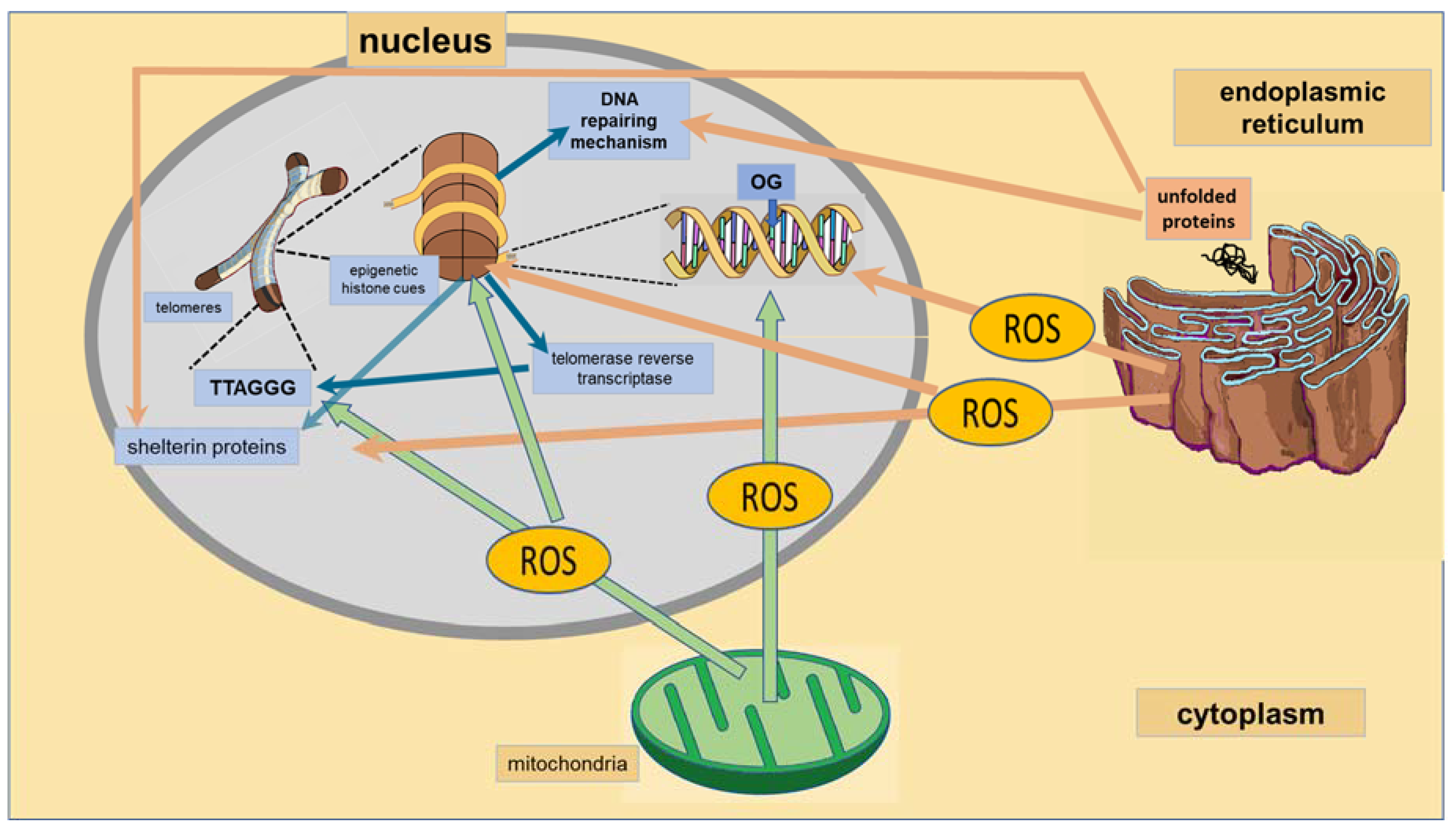
| Subcellular Structure | Epigenetics and the Structure | Related Disorders | |
|---|---|---|---|
| Mitochondria | Crosstalk with nucleus: -Leaves epigenetic markers through the generation of metabolites by the tricarboxylic acid cycle [17,18,19]. -Regulates mitochondrial biogenesis through acetylation of FOXOs and PCG-1α [17,18,19]. | -neurodegenerative diseases: Alzheimer’s disease [20]. | |
| -metabolic diseases: type 2 diabetes mellitus [21,22]. | |||
| -cardiovascular diseases: myocardial ischemia, cardiomyopathy and heart failure [23,24]. | |||
| -psychiatric disorders: bipolar disorder, schizophrenia, autism, attention deficit-hyperactivity disorder [25]. | |||
| Endoplasmic Reticulum | Expression of chaperones and heat shock proteins that prevent unfolding or misfolding of proteins is a target of epigenetic markers [26]. | -metabolic diseases: metabolic syndrome, obesity and diabetes [27,28]. | |
| -cardiovascular diseases: aggregation cardiomyopathies [29,30,31,32,33]. | |||
| -neurodegenerative diseases: Parkinson’s, Alzheimer’s and Huntington’s disease, amyotrophic lateral sclerosis and Machado–Joseph disease [34,35]. | |||
| Nucleus | Telomeres | Telomeres are rich in epigenetic markers that determine their shortening [36,37,38]. | -psychiatric disorders: chronic stress, pain, MDD, BD, PTSD, schizophrenia, anxiety disorders [39,40]. |
| -cardiovascular diseases: coronary heart disease, left ventricular hypertrophy [41]. | |||
| DNA Reparation | The BER pathway is important for maintaining both the genetic stability and the methylation status [42,43,44]. | Psychiatric disorders; schizophrenia, autism spectrum disorders [45]. | |
| The BER substrate, 8-oxoguanine, is an epigenetic marker modulating transcription factor recognition/binding [44]. | Cardiovascular diseases: atherosclerosis, vascular smooth muscle cell dysfunction [46]. | ||
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zapata-Martín del Campo, C.M.; Martínez-Rosas, M.; Guarner-Lans, V. Epigenetics of Subcellular Structure Functioning in the Origin of Risk or Resilience to Comorbidity of Neuropsychiatric and Cardiometabolic Disorders. Int. J. Mol. Sci. 2018, 19, 1456. https://doi.org/10.3390/ijms19051456
Zapata-Martín del Campo CM, Martínez-Rosas M, Guarner-Lans V. Epigenetics of Subcellular Structure Functioning in the Origin of Risk or Resilience to Comorbidity of Neuropsychiatric and Cardiometabolic Disorders. International Journal of Molecular Sciences. 2018; 19(5):1456. https://doi.org/10.3390/ijms19051456
Chicago/Turabian StyleZapata-Martín del Campo, Carlos Manuel, Martín Martínez-Rosas, and Verónica Guarner-Lans. 2018. "Epigenetics of Subcellular Structure Functioning in the Origin of Risk or Resilience to Comorbidity of Neuropsychiatric and Cardiometabolic Disorders" International Journal of Molecular Sciences 19, no. 5: 1456. https://doi.org/10.3390/ijms19051456