Oxidative Stress in Preeclampsia and Placental Diseases


 and
and 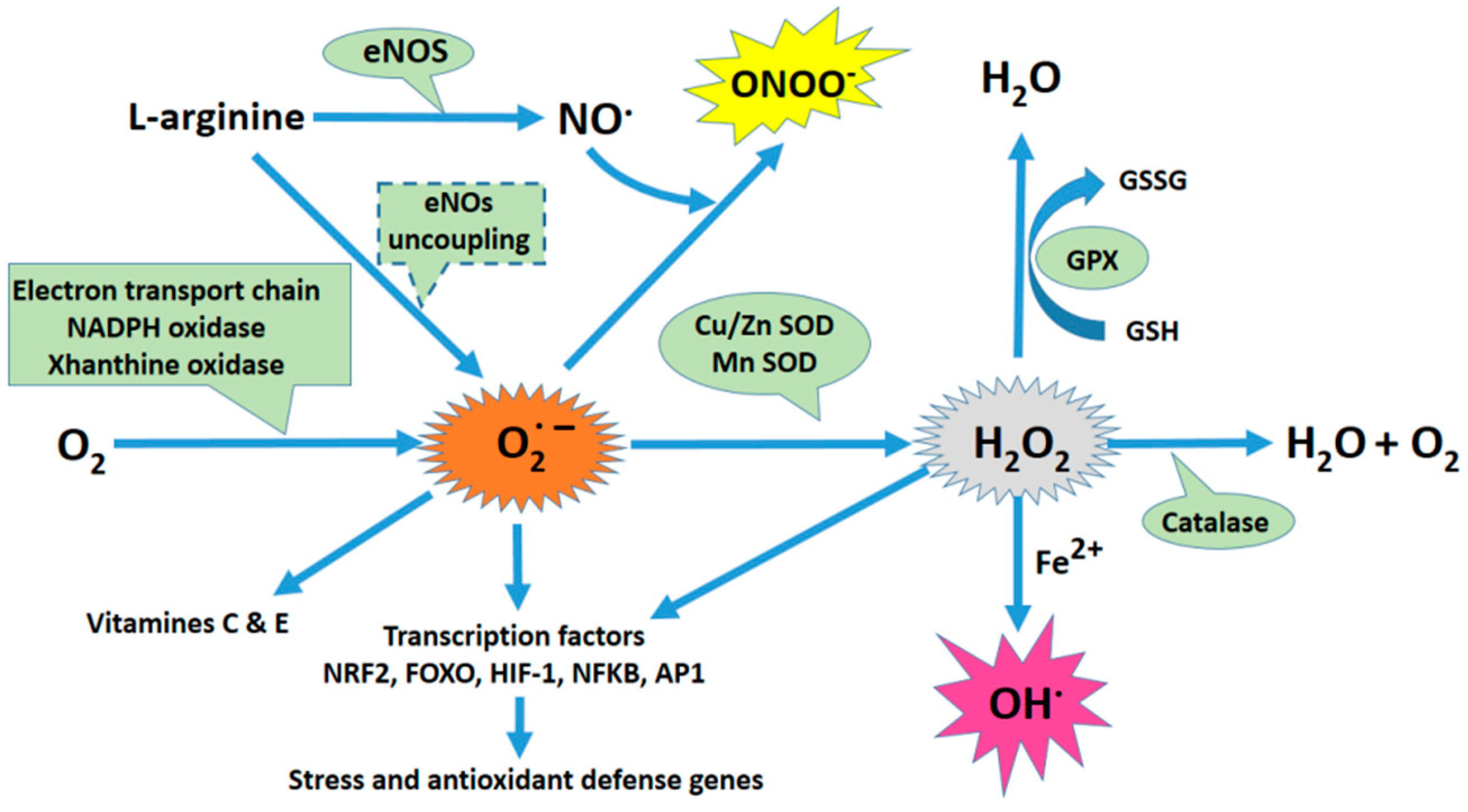{kind=link}
{kind=link}
{kind=link}
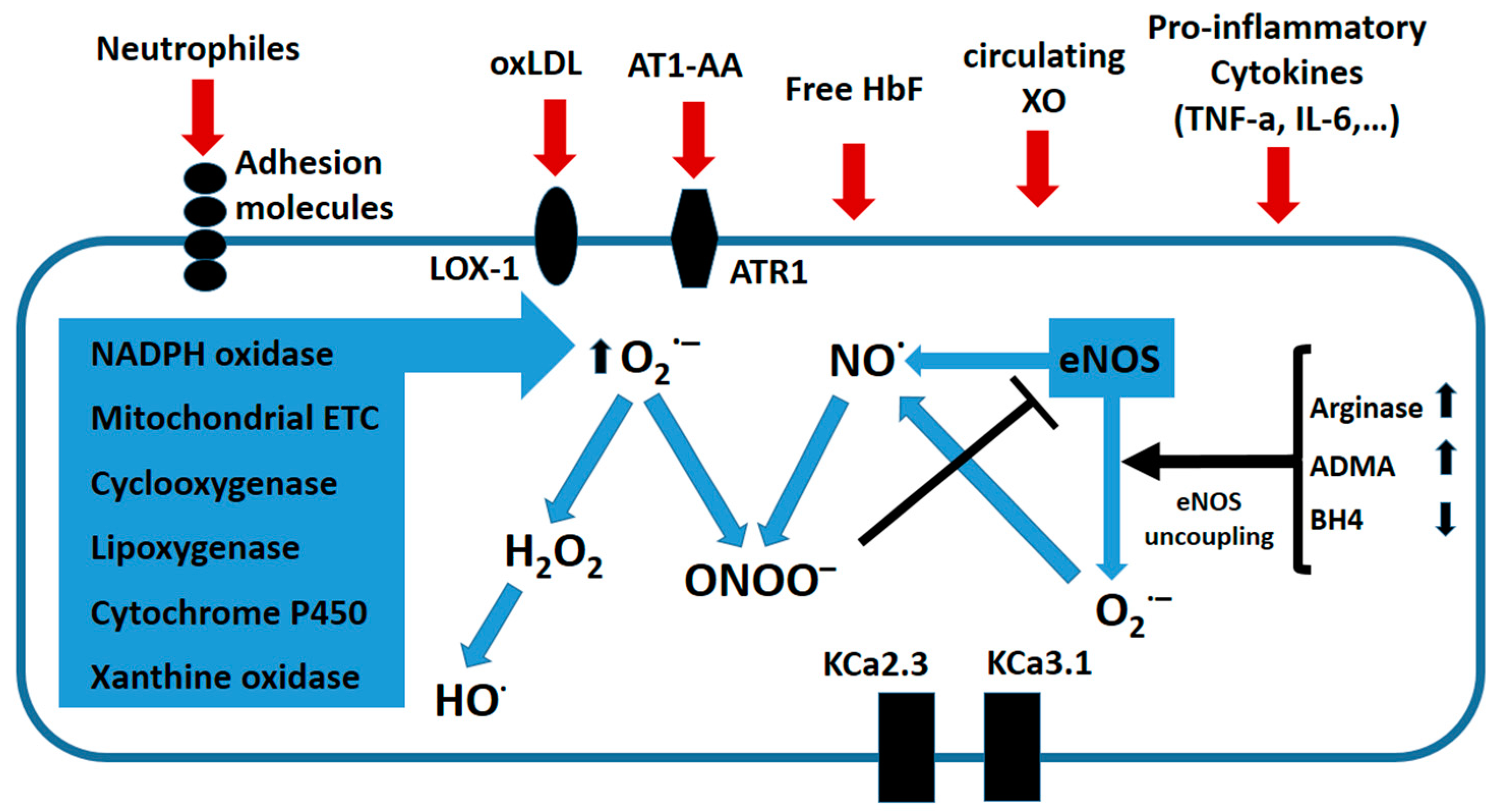{kind=link}
Abstract
:1. Introduction
1.1. Preeclampsia
1.2. Oxidative Stress
2. Major Enzymatic and Cellular Systems Involved in the Generation of Free Radicals
2.1. Nitric Oxide (NO) Synthases
2.2. Role of the NADPH Oxidase (NOX) as a Source of ROS
2.3. Mitochondrial Reactive Oxygen Species (ROS) Production
2.4. Xanthine Oxidase and ROS
3. Cellular Sources of Oxidative Stress in the Human Placenta under Normal and Pathological Conditions
3.1. Cell Types of the Placenta and the Origins of Oxidative Stress
3.2. Oxidative Stress from the Trophoblasts
3.3. Oxidative Stress Origin and Regulation in Hofbauer Cells
3.4. Consequences of Oxidative Stress on the Physiology of Placental Cells
3.4.1. Cell Models
3.4.2. Animal Models
3.5. Consequences of Oxidative Stress on Protein Post-Translational Modifications, Lipid Alterations, and DNA Damage in the Placenta
3.6. Oxidative Stress in Placental Pathologies Other than Preeclampsia
3.6.1. Spontaneous Pregnancy Loss
3.6.2. Intra-Uterine Growth Restriction (IUGR)
3.6.3. Gestational Diabetes Mellitus
4. Role of Oxidative Stress on the Maternal Endothelial Dysfunction in Preeclampsia
4.1. Preeclampsia: An Oxidative Stress-Mediated Inflammatory Disease of the Maternal Endothelium
4.2. Antioxidant Therapeutic Approaches to Treat Endothelial Dysfunction in PE
5. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
Abbreviations
| A1M | Alpha 1-microglobulin |
| ADMA | asymmetric dimethyl arginine |
| AEGs | advanced end glycation products |
| AngII | angiotensin II |
| AOPPS | advanced oxidation proteins products |
| AT1-AA | Agonist autoimmune antibodies against the angiotensin II receptor type I |
| ATLs | Aspirin-triggered lipoxins |
| BH4 | tetrahydrobiopterin |
| CAT | catalase |
| CBS | cystathionine-β-synthase |
| COX1 | Cyclooxygenase 1 = PTGS1 (Prostaglandin synthase 1) |
| COX2 | Cyclooxygenase 2 = PTGS2 (Prostaglandin synthase 2) |
| CSE | cystathionine-γ-lyase |
| ECs | endothelial cells |
| EDR | endothelium-dependent relaxation |
| ENG | Endoglin |
| eNOS | endothelial nitric oxide synthase |
| ETC | electron-transport chain |
| EVT | extravillous trophoblasts |
| EVT | extravillous trophoblasts |
| GDM | Gestational diabetes mellitus |
| GPx | glutathione peroxidase |
| GSH | Glutathione |
| H2O2 | hydrogen peroxide hydroxyl radical |
| HbF | free fetal hemoglobin |
| HCG | Chorionic Gonadotropin |
| HMOX1 | Heme Oxygenase 1 |
| HUVEC | Human Umbilical Vascular Endothelial Cell |
| HO-1 | hemoxygenase |
| •HO | hydroxyl radical |
| ICAM-1 | intercellular adhesion molecule-1 |
| iNOs | inducible nitric oxide synthase |
| IUGR | Intra-uterine Growth Restriction |
| KcaS | calcium-activated potassium channels |
| LOX-1 | LDL receptor-1 |
| MDA | Malondialdehyde |
| MG | Methylglyoxal |
| MMPs | Matrix metalloproteinases |
| NADH | Nicotinamide adenine dinucleotide |
| NF-κB | nuclear factor-kappa B |
| nNOS | neuronal nitric oxide synthase |
| •NO | nitric oxide |
| NOX | NADPH oxidase |
| NSAID | Non-Steroidal Anti-Inflammatory Drug |
| O2•− | superoxide |
| ONOO− | peroxynitrite |
| OS | Oxidative Stress |
| oxLDL | oxidized LDL |
| PC2 | polycystin-2 |
| PDCD1LG2 | Programmed Cell Death 1 Ligand 2 |
| PDGFD | Platelet Derived Growth Factor D |
| PIF | Preimplantation factor |
| PLC | phospholipase C |
| PPIs | Proton Pump Inhibitors |
| PRKG1B | Protein Kinase, CGMP-Dependent, Type I |
| RAS | renin-angiotensin system |
| RNS | reactive nitrogen species |
| ROS | reactive oxygen species |
| ROS | reactive oxygen species |
| SCT | syncytiotrophoblasts |
| SELE | E-selectin |
| SELP | P-selectin |
| sENG | soluble endoglin |
| SERCA | calcium-ATPase of the endoplasmic reticulum |
| sFlt1 | soluble fms-like tyrosine kinase-1 |
| SOD | superoxide dismutase |
| SSAO | semicarbazide-sensitive monoamine oxidase |
| TBARS | Thiobarbituric acid reactive substances |
| TGFBR2 | Transforming Growth Factor Beta Receptor 2 |
| TRX | Thioredoxin |
| VCAM-1 | vascular cell adhesion molecule-1 |
| VCT | villous cytrophoblasts |
| VLDLR | Very Low Density Lipoprotein Receptor |
| XO | Xanthine Oxydase |
References
- Steegers, E.A.; von Dadelszen, P.; Duvekot, J.J.; Pijnenborg, R. Pre-eclampsia. Lancet 2010, 376, 631–644. [Google Scholar] [CrossRef]
- Zabul, P.; Wozniak, M.; Slominski, A.T.; Preis, K.; Gorska, M.; Korozan, M.; Wieruszewski, J.; Zmijewski, M.A.; Zabul, E.; Tuckey, R.; et al. A proposed molecular mechanism of high-dose vitamin d3 supplementation in prevention and treatment of preeclampsia. Int. J. Mol. Sci. 2015, 16, 13043–13064. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, G.; Grewal, J.; Mannisto, T.; Mendola, P.; Chen, Z.; Xie, Y.; Laughon, S.K. Racial/ethnic differences in pregnancy-related hypertensive disease in nulliparous women. Ethn. Dis. 2014, 24, 283–289. [Google Scholar] [PubMed]
- Hod, T.; Cerdeira, A.S.; Karumanchi, S.A. Molecular mechanisms of preeclampsia. Cold Spring Harb. Perspect. Med. 2015, 5, a023473. [Google Scholar] [CrossRef] [PubMed]
- Goulopoulou, S.; Davidge, S.T. Molecular mechanisms of maternal vascular dysfunction in preeclampsia. Trends Mol. Med. 2015, 21, 88–97. [Google Scholar] [CrossRef] [PubMed]
- Sies, H.; Jones, D. Oxidative Stress, 2nd ed.; Elsevier: Amsterdam, The Netherlands, 2007; Volume 3. [Google Scholar]
- Pacher, P.; Beckman, J.S.; Liaudet, L. Nitric oxide and peroxynitrite in health and disease. Physiol. Rev. 2007, 87, 315–424. [Google Scholar] [CrossRef] [PubMed]
- Espinosa-Diez, C.; Miguel, V.; Mennerich, D.; Kietzmann, T.; Sanchez-Perez, P.; Cadenas, S.; Lamas, S. Antioxidant responses and cellular adjustments to oxidative stress. Redox Biol. 2015, 6, 183–197. [Google Scholar] [CrossRef] [PubMed]
- Craige, S.M.; Kant, S.; Keaney, J.F., Jr. Reactive oxygen species in endothelial function—From disease to adaptation. Circ. J. 2015, 79, 1145–1155. [Google Scholar] [CrossRef] [PubMed]
- Mittal, M.; Siddiqui, M.R.; Tran, K.; Reddy, S.P.; Malik, A.B. Reactive oxygen species in inflammation and tissue injury. Antioxid. Redox Signal. 2014, 20, 1126–1167. [Google Scholar] [CrossRef] [PubMed]
- Knowles, R.G.; Moncada, S. Nitric oxide synthases in mammals. Biochem. J. 1994, 298, 249–258. [Google Scholar] [CrossRef] [PubMed]
- Forstermann, U.; Sessa, W.C. Nitric oxide synthases: Regulation and function. Eur. Heart J. 2012, 33, 829–837. [Google Scholar] [CrossRef] [PubMed]
- Laursen, J.B.; Somers, M.; Kurz, S.; McCann, L.; Warnholtz, A.; Freeman, B.A.; Tarpey, M.; Fukai, T.; Harrison, D.G. Endothelial regulation of vasomotion in apoe-deficient mice: Implications for interactions between peroxynitrite and tetrahydrobiopterin. Circulation 2001, 103, 1282–1288. [Google Scholar] [CrossRef] [PubMed]
- Forstermann, U.; Munzel, T. Endothelial nitric oxide synthase in vascular disease: From marvel to menace. Circulation 2006, 113, 1708–1714. [Google Scholar] [CrossRef] [PubMed]
- Heiss, E.H.; Dirsch, V.M. Regulation of enos enzyme activity by posttranslational modification. Curr. Pharm. Des. 2014, 20, 3503–3513. [Google Scholar] [CrossRef] [PubMed]
- Bedard, K.; Krause, K.H. The NOX family of ros-generating nadph oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef] [PubMed]
- Touyz, R.M.; Montezano, A.C. Vascular NOX4: A multifarious nadph oxidase. Circ. Res. 2012, 110, 1159–1161. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Luo, Y.X.; Chen, H.Z.; Liu, D.P. Mitochondria, endothelial cell function, and vascular diseases. Front. Physiol. 2014, 5, 175. [Google Scholar] [CrossRef] [PubMed]
- Widlansky, M.E.; Gutterman, D.D. Regulation of endothelial function by mitochondrial reactive oxygen species. Antioxid. Redox Signal. 2011, 15, 1517–1530. [Google Scholar] [CrossRef] [PubMed]
- Pacher, P.; Nivorozhkin, A.; Szabo, C. Therapeutic effects of xanthine oxidase inhibitors: Renaissance half a century after the discovery of allopurinol. Pharmacol. Rev. 2006, 58, 87–114. [Google Scholar] [CrossRef] [PubMed]
- Tsang, J.C.H.; Vong, J.S.L.; Ji, L.; Poon, L.C.Y.; Jiang, P.; Lui, K.O.; Ni, Y.B.; To, K.F.; Cheng, Y.K.Y.; Chiu, R.W.K.; et al. Integrative single-cell and cell-free plasma rna transcriptomics elucidates placental cellular dynamics. Proc. Natl. Acad. Sci. USA 2017, 114, E7786–E7795. [Google Scholar] [CrossRef] [PubMed]
- Webster, K.A. Hypoxia: Life on the edge. Antioxid. Redox Signal. 2007, 9, 1303–1307. [Google Scholar] [CrossRef] [PubMed]
- Danilov, C.A.; Fiskum, G. Hyperoxia promotes astrocyte cell death after oxygen and glucose deprivation. Glia 2008, 56, 801–808. [Google Scholar] [CrossRef] [PubMed]
- Jauniaux, E.; Watson, A.; Ozturk, O.; Quick, D.; Burton, G. In-vivo measurement of intrauterine gases and acid-base values early in human pregnancy. Hum. Reprod. 1999, 14, 2901–2904. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.; Pillay, P.; Naicker, T.; Moodley, J.; Mackraj, I. Placental hypoxia inducible factor -1alpha & chop immuno-histochemical expression relative to maternal circulatory syncytiotrophoblast micro-vesicles in preeclamptic and normotensive pregnancies. Eur. J. Obstet. Gynecol. Reprod. Biol. 2018, 220, 18–24. [Google Scholar] [PubMed]
- Abou-Kheir, W.; Barrak, J.; Hadadeh, O.; Daoud, G. HTR-8/SVneo cell line contains a mixed population of cells. Placenta 2017, 50, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Deng, Q.; Yin, N.; Chen, Y.; Shan, N.; Liu, X.; Qi, H. Downregulated N-acetylglucosaminyltransferase iii is involved in attenuating trophoblast migration and invasion under hypoxia-reoxygenation condition. J. Matern. Fetal Neonatal Med. 2018, 21, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Rao, H.; Bai, Y.; Zhang, F.; Li, Q.; Zhuang, B.; Luo, X.; Qi, H. The role of SATB1 in HTR8/SVneo cells and pathological mechanism of preeclampsia. J. Matern. Fetal Neonatal Med. 2018, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Kwon, H.S.; Park, J.H.; Hwang, H.S.; Sohn, I.S.; Kim, Y.H.; Cho, S. Effect of DJ-1 downregulation on the functions of the first trimester extravillous trophoblasts. Reprod. Sci. 2017. [Google Scholar] [CrossRef] [PubMed]
- Park, H.R.; Kamau, P.W.; Loch-Caruso, R. Involvement of reactive oxygen species in brominated diphenyl ether-47-induced inflammatory cytokine release from human extravillous trophoblasts in vitro. Toxicol. Appl. Pharmacol. 2014, 274, 283–292. [Google Scholar] [CrossRef] [PubMed]
- Hassan, I.; Kumar, A.M.; Park, H.R.; Lash, L.H.; Loch-Caruso, R. Reactive oxygen stimulation of interleukin-6 release in the human trophoblast cell line HTR-8/SVneo by the trichlorethylene metabolite s-(1,2-dichloro)-l-cysteine. Biol. Reprod. 2016, 95, 66. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.; Zuo, Q.; Huang, S.; Yu, X.; Jiang, Z.; Zou, S.; Fan, M.; Sun, L. Resveratrol inhibits trophoblast apoptosis through oxidative stress in preeclampsia-model rats. Molecules 2014, 19, 20570–20579. [Google Scholar] [CrossRef] [PubMed]
- Bolnick, J.M.; Kilburn, B.A.; Bolnick, A.D.; Diamond, M.P.; Singh, M.; Hertz, M.; Dai, J.; Armant, D.R. Sildenafil stimulates human trophoblast invasion through nitric oxide and guanosine 3’,5’-cyclic monophosphate signaling. Fertil. Steril. 2015, 103, 1587–1595. [Google Scholar] [CrossRef] [PubMed]
- Bolnick, A.D.; Bolnick, J.M.; Kohan-Ghadr, H.R.; Kilburn, B.A.; Pasalodos, O.J.; Singhal, P.K.; Dai, J.; Diamond, M.P.; Armant, D.R.; Drewlo, S. Enhancement of trophoblast differentiation and survival by low molecular weight heparin requires heparin-binding EGF-like growth factor. Hum. Reprod. 2017, 32, 1218–1229. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Y.; Zhang, X.; Li, Q.; Xu, J.; Zhou, X.; Wang, T.; Xing, Q.; Liu, Y.; Wang, L.; He, L.; et al. Promoter hypomethylation of TIMP3 is associated with pre-eclampsia in a chinese population. Mol. Hum. Reprod. 2013, 19, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Barnea, E.R.; Vialard, F.; Moindjie, H.; Ornaghi, S.; Dieudonne, M.N.; Paidas, M.J. Preimplantation factor (PIF*) endogenously prevents preeclampsia: Promotes trophoblast invasion and reduces oxidative stress. J. Reprod. Immunol. 2016, 114, 58–64. [Google Scholar] [CrossRef] [PubMed]
- Watson, A.L.; Skepper, J.N.; Jauniaux, E.; Burton, G.J. Changes in concentration, localization and activity of catalase within the human placenta during early gestation. Placenta 1998, 19, 27–34. [Google Scholar] [CrossRef]
- Sisino, G.; Bouckenooghe, T.; Aurientis, S.; Fontaine, P.; Storme, L.; Vambergue, A. Diabetes during pregnancy influences hofbauer cells, a subtype of placental macrophages, to acquire a pro-inflammatory phenotype. Biochim. Biophys. Acta 2013, 1832, 1959–1968. [Google Scholar] [CrossRef] [PubMed]
- Holwerda, K.M.; Bos, E.M.; Rajakumar, A.; Ris-Stalpers, C.; van Pampus, M.G.; Timmer, A.; Erwich, J.J.; Faas, M.M.; van Goor, H.; Lely, A.T. Hydrogen sulfide producing enzymes in pregnancy and preeclampsia. Placenta 2012, 33, 518–521. [Google Scholar] [CrossRef] [PubMed]
- Holwerda, K.M.; Weedon-Fekjaer, M.S.; Staff, A.C.; Nolte, I.M.; van Goor, H.; Lely, A.T.; Faas, M.M. The association of single nucleotide polymorphisms of the maternal cystathionine-β-synthase gene with early-onset preeclampsia. Pregnancy Hypertens. 2016, 6, 60–65. [Google Scholar] [CrossRef] [PubMed]
- Holwerda, K.M.; Karumanchi, S.A.; Lely, A.T. Hydrogen sulfide: Role in vascular physiology and pathology. Curr. Opin. Nephrol. Hypertens. 2015, 24, 170–176. [Google Scholar] [CrossRef] [PubMed]
- Perucci, L.O.; Correa, M.D.; Dusse, L.M.; Gomes, K.B.; Sousa, L.P. Resolution of inflammation pathways in preeclampsia-a narrative review. Immunol. Res. 2017, 65, 774–789. [Google Scholar] [CrossRef] [PubMed]
- Hu, T.X.; Wang, G.; Guo, X.J.; Sun, Q.Q.; He, P.; Gu, H.; Huang, Y.; Gao, L.; Ni, X. Mir 20a,-20b and -200c are involved in hydrogen sulfide stimulation of vegf production in human placental trophoblasts. Placenta 2016, 39, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Hu, T.X.; Guo, X.; Wang, G.; Gao, L.; He, P.; Xia, Y.; Gu, H.; Ni, X. miR133b is involved in endogenous hydrogen sulfide suppression of sflt-1 production in human placenta. Placenta 2017, 52, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Calicchio, R.; Buffat, C.; Mathieu, J.R.; Ben Salem, N.; Mehats, C.; Jacques, S.; Hertig, A.; Berkane, N.; Grevoul-Fresquet, J.; Simeoni, U.; et al. Preeclamptic plasma induces transcription modifications involving the AP-1 transcriptional regulator JDP2 in endothelial cells. Am. J. Pathol. 2013, 183, 1993–2006. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Ahmad, S.; Cai, M.; Rennie, J.; Fujisawa, T.; Crispi, F.; Baily, J.; Miller, M.R.; Cudmore, M.; Hadoke, P.W.; et al. Dysregulation of hydrogen sulfide producing enzyme cystathionine gamma-lyase contributes to maternal hypertension and placental abnormalities in preeclampsia. Circulation 2013, 127, 2514–2522. [Google Scholar] [CrossRef] [PubMed]
- Murata, M.; Fukushima, K.; Takao, T.; Seki, H.; Takeda, S.; Wake, N. Oxidative stress produced by xanthine oxidase induces apoptosis in human extravillous trophoblast cells. J. Reprod. Dev. 2013, 59, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Kudo, Y.; Boyd, C.A.; Kimura, H.; Cook, P.R.; Redman, C.W.; Sargent, I.L. Quantifying the syncytialisation of human placental trophoblast bewo cells grown in vitro. Biochim. Biophys. Acta 2003, 1640, 25–31. [Google Scholar] [CrossRef]
- McAleer, M.F.; Tuan, R.S. Metallothionein protects against severe oxidative stress-induced apoptosis of human trophoblastic cells. In Vitro Mol. Toxicol. 2001, 14, 219–231. [Google Scholar] [CrossRef] [PubMed]
- Tang, C.; Liang, J.; Qian, J.; Jin, L.; Du, M.; Li, M.; Li, D. Opposing role of JNK-p38 kinase and ERK1/2 in hydrogen peroxide-induced oxidative damage of human trophoblast-like JEG-3 cells. Int. J. Clin. Exp. Pathol. 2014, 7, 959–968. [Google Scholar] [PubMed]
- Hung, T.H.; Chen, S.F.; Liou, J.D.; Hsu, J.J.; Li, M.J.; Yeh, Y.L.; Hsieh, T.T. Bax, bak and mitochondrial oxidants are involved in hypoxia-reoxygenation-induced apoptosis in human placenta. Placenta 2008, 29, 565–583. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, B.; Luo, X.; Rao, H.; Li, Q.; Shan, N.; Liu, X.; Qi, H. Oxidative stress-induced c/EBPβ inhibits β-catenin signaling molecule involving in the pathology of preeclampsia. Placenta 2015, 36, 839–846. [Google Scholar] [CrossRef] [PubMed]
- Montalbetti, N.; Cantero, M.R.; Dalghi, M.G.; Cantiello, H.F. Reactive oxygen species inhibit polycystin-2 (TRPP2) cation channel activity in term human syncytiotrophoblast. Placenta 2008, 29, 510–518. [Google Scholar] [CrossRef] [PubMed]
- Bainbridge, S.A.; von Versen-Hoynck, F.; Roberts, J.M. Uric acid inhibits placental system a amino acid uptake. Placenta 2009, 30, 195–200. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Zou, Y.; Ge, Z.; Zuo, Q.; Huang, S.Y.; Sun, L. A role of sFlt-1 in oxidative stress and apoptosis in human and mouse pre-eclamptic trophoblasts. Biol. Reprod. 2015, 93, 73. [Google Scholar] [CrossRef] [PubMed]
- Doridot, L.; Passet, B.; Mehats, C.; Rigourd, V.; Barbaux, S.; Ducat, A.; Mondon, F.; Vilotte, M.; Castille, J.; Breuiller-Fouche, M.; et al. Preeclampsia-like symptoms induced in mice by fetoplacental expression of STOX1 are reversed by aspirin treatment. Hypertension 2013, 61, 662–668. [Google Scholar] [CrossRef] [PubMed]
- Doridot, L.; Chatre, L.; Ducat, A.; Vilotte, J.L.; Lombes, A.; Mehats, C.; Barbaux, S.; Calicchio, R.; Ricchetti, M.; Vaiman, D. Nitroso-redox balance and mitochondrial homeostasis are regulated by STOX1, a pre-eclampsia-associated gene. Antioxid. Redox Signal. 2014, 21, 819–834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beausejour, A.; Bibeau, K.; Lavoie, J.C.; St-Louis, J.; Brochu, M. Placental oxidative stress in a rat model of preeclampsia. Placenta 2007, 28, 52–58. [Google Scholar] [CrossRef] [PubMed]
- Baczyk, D.; Audette, M.C.; Coyaud, E.; Raught, B.; Kingdom, J.C. Spatiotemporal distribution of sumos during human placental development and in response to oxidative and inflammatory stress. J. Physiol. 2018, 596, 1587–1600. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, T.K.; Fujiwara, T.; Kawai, A.; Shimizu, F.; Takami, S.; Hirano, H.; Okuno, S.; Ozaki, K.; Takeda, S.; Shimada, Y.; et al. Cloning, expression, and mapping of UBE2I, a novel gene encoding a human homologue of yeast ubiquitin-conjugating enzymes which are critical for regulating the cell cycle. Cytogenet. Cell Genet. 1996, 72, 86–89. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Xue, Y.; Lu, H.; Chen, G.; Yao, X. A genome-wide analysis of sumoylation-related biological processes and functions in human nucleus. FEBS Lett. 2005, 579, 3369–3375. [Google Scholar] [CrossRef] [PubMed]
- Turko, I.V.; Murad, F. Protein nitration in cardiovascular diseases. Pharmacol. Rev. 2002, 54, 619–634. [Google Scholar] [CrossRef] [PubMed]
- Myatt, L. Reactive oxygen and nitrogen species and functional adaptation of the placenta. Placenta 2010, 31, S66–S69. [Google Scholar] [CrossRef] [PubMed]
- Matsubara, K.; Higaki, T.; Matsubara, Y.; Nawa, A. Nitric oxide and reactive oxygen species in the pathogenesis of preeclampsia. Int. J. Mol. Sci. 2015, 16, 4600–4614. [Google Scholar] [CrossRef] [PubMed]
- Webster, R.P.; Brockman, D.; Myatt, L. Nitration of p38 MAPK in the placenta: Association of nitration with reduced catalytic activity of p38 MAPK in pre-eclampsia. Mol. Hum. Reprod. 2006, 12, 677–685. [Google Scholar] [CrossRef] [PubMed]
- Capobianco, E.; White, V.; Sosa, M.; Di Marco, I.; Basualdo, M.N.; Faingold, M.C.; Jawerbaum, A. Regulation of matrix metalloproteinases 2 and 9 activities by peroxynitrites in term placentas from type 2 diabetic patients. Reprod. Sci. 2012, 19, 814–822. [Google Scholar] [CrossRef] [PubMed]
- Myatt, L.; Kossenjans, W.; Sahay, R.; Eis, A.; Brockman, D. Oxidative stress causes vascular dysfunction in the placenta. J. Matern. Fetal Med. 2000, 9, 79–82. [Google Scholar] [PubMed]
- Vanderlelie, J.; Venardos, K.; Clifton, V.L.; Gude, N.M.; Clarke, F.M.; Perkins, A.V. Increased biological oxidation and reduced anti-oxidant enzyme activity in pre-eclamptic placentae. Placenta 2005, 26, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Zusterzeel, P.L.; Rutten, H.; Roelofs, H.M.; Peters, W.H.; Steegers, E.A. Protein carbonyls in decidua and placenta of pre-eclamptic women as markers for oxidative stress. Placenta 2001, 22, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Shaker, O.G.; Sadik, N.A. Pathogenesis of preeclampsia: Implications of apoptotic markers and oxidative stress. Hum. Exp. Toxicol. 2013, 32, 1170–1178. [Google Scholar] [CrossRef] [PubMed]
- Can, M.; Guven, B.; Bektas, S.; Arikan, I. Oxidative stress and apoptosis in preeclampsia. Tissue Cell 2014, 46, 477–481. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.T.; Wang, S.S.; Zhang, M.; Huang, L.P.; Tian, J.W.; Yu, Y.H.; Wang, Z.J.; Zhong, M. Advanced oxidation protein products enhances soluble fms-like tyrosine kinase 1 expression in trophoblasts: A possible link between oxidative stress and preeclampsia. Placenta 2013, 34, 949–952. [Google Scholar] [CrossRef] [PubMed]
- Tadesse, S.; Kidane, D.; Guller, S.; Luo, T.; Norwitz, N.G.; Arcuri, F.; Toti, P.; Norwitz, E.R. In vivo and in vitro evidence for placental DNA damage in preeclampsia. PLoS ONE 2014, 9, e86791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiktor, H.; Kankofer, M.; Schmerold, I.; Dadak, A.; Lopucki, M.; Niedermuller, H. Oxidative DNA damage in placentas from normal and pre-eclamptic pregnancies. Virchows Arch. 2004, 445, 74–78. [Google Scholar] [CrossRef] [PubMed]
- Fujimaki, A.; Watanabe, K.; Mori, T.; Kimura, C.; Shinohara, K.; Wakatsuki, A. Placental oxidative DNA damage and its repair in preeclamptic women with fetal growth restriction. Placenta 2011, 32, 367–372. [Google Scholar] [CrossRef] [PubMed]
- Ford, H.B.; Schust, D.J. Recurrent pregnancy loss: Etiology, diagnosis, and therapy. Rev. Obstet. Gynecol. 2009, 2, 76–83. [Google Scholar] [PubMed]
- Jauniaux, E.; Watson, A.L.; Hempstock, J.; Bao, Y.P.; Skepper, J.N.; Burton, G.J. Onset of maternal arterial blood flow and placental oxidative stress. A possible factor in human early pregnancy failure. Am. J. Pathol. 2000, 157, 2111–2122. [Google Scholar] [CrossRef]
- Jauniaux, E.; Hempstock, J.; Greenwold, N.; Burton, G.J. Trophoblastic oxidative stress in relation to temporal and regional differences in maternal placental blood flow in normal and abnormal early pregnancies. Am. J. Pathol. 2003, 162, 115–125. [Google Scholar] [CrossRef]
- Hempstock, J.; Jauniaux, E.; Greenwold, N.; Burton, G.J. The contribution of placental oxidative stress to early pregnancy failure. Hum. Pathol. 2003, 34, 1265–1275. [Google Scholar] [CrossRef] [PubMed]
- Ghneim, H.K.; Al-Sheikh, Y.A.; Alshebly, M.M.; Aboul-Soud, M.A.M. Superoxide dismutase activity and gene expression levels in saudi women with recurrent miscarriage. Mol. Med. Rep. 2016, 13, 2606–2612. [Google Scholar] [CrossRef] [PubMed]
- Biri, A.; Bozkurt, N.; Turp, A.; Kavutcu, M.; Himmetoglu, O.; Durak, I. Role of oxidative stress in intrauterine growth restriction. Gynecol. Obstet. Investig. 2007, 64, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Lyu, S.W.; Song, H.; Yoon, J.A.; Chin, M.-U.; Sung, S.R.; Kim, Y.S.; Lee, W.S.; Yoon, T.K.; Cha, D.H.; Shim, S.H. Transcriptional profiling with a pathway-oriented analysis in the placental villi of unexplained miscarriage. Placenta 2013, 34, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Liu, A.-X.; He, W.-H.; Yin, L.-J.; Lv, P.-P.; Zhang, Y.; Sheng, J.-Z.; Leung, P.C.K.; Huang, H.-F. Sustained endoplasmic reticulum stress as a cofactor of oxidative stress in decidual cells from patients with early pregnancy loss. J. Clin. Endocrinol. Metab. 2011, 96, E493–E497. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.-J.; Zhu, Y.-M.; He, W.-H.; Liu, A.-X.; Dong, M.-Y.; Jin, M.; Sheng, J.-Z.; Huang, H.-F. Endoplasmic reticulum stress induced by oxidative stress in decidual cells: A possible mechanism of early pregnancy loss. Mol. Biol. Rep. 2012, 39, 9179–9186. [Google Scholar] [CrossRef] [PubMed]
- Al-Gubory, K.H.; Krawiec, A.; Grange, S.; Faure, P.; Garrel, C. Abortion-prone mating influences placental antioxidant status and adversely affects placental and foetal development. Free Radic. Res. 2014, 48, 1505–1513. [Google Scholar] [CrossRef] [PubMed]
- Ishii, T.; Miyazawa, M.; Takanashi, Y.; Tanigawa, M.; Yasuda, K.; Onouchi, H.; Kawabe, N.; Mitsushita, J.; Hartman, P.S.; Ishii, N. Genetically induced oxidative stress in mice causes thrombocytosis, splenomegaly and placental angiodysplasia that leads to recurrent abortion. Redox Biol. 2014, 2, 679–685. [Google Scholar] [CrossRef] [PubMed]
- Black, R.E. Global prevalence of small for gestational age births. Nestle Nutr. Inst. Worksh. Ser. 2015, 81, 1–7. [Google Scholar]
- Takagi, Y.; Nikaido, T.; Toki, T.; Kita, N.; Kanai, M.; Ashida, T.; Ohira, S.; Konishi, I. Levels of oxidative stress and redox-related molecules in the placenta in preeclampsia and fetal growth restriction. Virchows Arch. 2004, 444, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Madeleneau, D.; Buffat, C.; Mondon, F.; Grimault, H.; Rigourd, V.; Tsatsaris, V.; Letourneur, F.; Vaiman, D.; Barbaux, S.; Gascoin, G. Transcriptomic analysis of human placenta in intrauterine growth restriction. Pediatr. Res. 2015, 77, 799–807. [Google Scholar] [CrossRef] [PubMed]
- Nagai, R.; Watanabe, K.; Wakatsuki, A.; Hamada, F.; Shinohara, K.; Hayashi, Y.; Imamura, R.; Fukaya, T. Melatonin preserves fetal growth in rats by protecting against ischemia/reperfusion-induced oxidative/nitrosative mitochondrial damage in the placenta. J. Pineal Res. 2008, 45, 271–276. [Google Scholar] [CrossRef] [PubMed]
- Jones, M.L.; Mark, P.J.; Lewis, J.L.; Mori, T.A.; Keelan, J.A.; Waddell, B.J. Antioxidant defenses in the rat placenta in late gestation: Increased labyrinthine expression of superoxide dismutases, glutathione peroxidase 3, and uncoupling protein 2. Biol. Reprod. 2010, 83, 254–260. [Google Scholar] [CrossRef] [PubMed]
- Alfaidy, N.; Chauvet, S.; Andrei, S.; Salomon, A.; Saoudi, Y.; Richaud, P.; Aude-Garcia, C.; Hoffmann, P.; Andrieux, A.; Moulis, J.-M.; et al. Prion protein expression and functional importance in developmental angiogenesis: Role in oxidative stress and copper homeostasis. Antioxid. Redox Signal. 2013, 18, 400–411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, D.R.; Qin, K.; Herms, J.W.; Madlung, A.; Manson, J.; Strome, R.; Fraser, P.E.; Kruck, T.; von Bohlen, A.; Schulz-Schaeffer, W.; et al. The cellular prion protein binds copper in vivo. Nature 1997, 390, 684–687. [Google Scholar] [CrossRef] [PubMed]
- Hillman, S.L.; Finer, S.; Smart, M.C.; Mathews, C.; Lowe, R.; Rakyan, V.K.; Hitman, G.A.; Williams, D.J. Novel DNA methylation profiles associated with key gene regulation and transcription pathways in blood and placenta of growth-restricted neonates. Epigenetics 2015, 10, 50–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; McMillan-Ward, E.; Kong, J.; Israels, S.J.; Gibson, S.B. Oxidative stress induces autophagic cell death independent of apoptosis in transformed and cancer cells. Cell Death Differ. 2008, 15, 171–182. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Giordano, S.; Zhang, J. Autophagy, mitochondria and oxidative stress: Cross-talk and redox signalling. Biochem. J. 2012, 441, 523–540. [Google Scholar] [CrossRef] [PubMed]
- Curtis, S.; Jones, C.J.P.; Garrod, A.; Hulme, C.H.; Heazell, A.E.P. Identification of autophagic vacuoles and regulators of autophagy in villous trophoblast from normal term pregnancies and in fetal growth restriction. J. Matern. Fetal Neonatal Med. 2013, 26, 339–346. [Google Scholar] [CrossRef] [PubMed]
- Saad, M.I.; Abdelkhalek, T.M.; Saleh, M.M.; Haiba, M.M.; Tawfik, S.H.; Kamel, M.A. Maternal diabetes impairs oxidative and inflammatory response in murine placenta. SpringerPlus 2016, 5, 532. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Gu, Y.; Zhang, Y.; Lucas, M.J.; Wang, Y. High glucose levels down-regulate glucose transporter expression that correlates with increased oxidative stress in placental trophoblast cells in vitro. J. Soc. Gynecol. Investig. 2004, 11, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Santos, A.; Martinez-Hernandez, M.G.; Contreras-Ramos, A.; Ortega-Camarillo, C.; Baiza-Gutman, L.A. Hyperglycemia-induced mouse trophoblast spreading is mediated by reactive oxygen species. Mol. Reprod. Dev. 2018, 85, 303–315. [Google Scholar] [CrossRef] [PubMed]
- Hahn, T.; Hahn, D.; Blaschitz, A.; Korgun, E.T.; Desoye, G.; Dohr, G. Hyperglycaemia-induced subcellular redistribution of glut1 glucose transporters in cultured human term placental trophoblast cells. Diabetologia 2000, 43, 173–180. [Google Scholar] [CrossRef] [PubMed]
- Araújo, J.R.; Pereira, A.C.; Correia-Branco, A.; Keating, E.; Martel, F. Oxidative stress induced by tert-butylhydroperoxide interferes with the placental transport of glucose: In vitro studies with bewo cells. Eur. J. Pharmacol. 2013, 720, 218–226. [Google Scholar] [CrossRef] [PubMed]
- Lappas, M.; Mitton, A.; Mittion, A.; Permezel, M. In response to oxidative stress, the expression of inflammatory cytokines and antioxidant enzymes are impaired in placenta, but not adipose tissue, of women with gestational diabetes. J. Endocrinol. 2010, 204, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Cahill, P.A.; Redmond, E.M. Vascular endothelium—Gatekeeper of vessel health. Atherosclerosis 2016, 248, 97–109. [Google Scholar] [CrossRef] [PubMed]
- Taylor, R.N.; Roberts, J.M.; Cunningham, F.C.; Lindheimer, M.D. Endothelial cell dysfunction. In Chesley’s Hypertensive Disorders in Pregnancy, 4th ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2015; pp. 181–207. [Google Scholar]
- Chen, X.; Andresen, B.T.; Hill, M.; Zhang, J.; Booth, F.; Zhang, C. Role of reactive oxygen species in tumor necrosis factor-alpha induced endothelial dysfunction. Curr. Hypertens. Rev. 2008, 4, 245–255. [Google Scholar] [CrossRef] [PubMed]
- Gertzberg, N.; Neumann, P.; Rizzo, V.; Johnson, A. NAD(P)H oxidase mediates the endothelial barrier dysfunction induced by TNF-α. Am. J. Physiol. Lung Cell. Mol. Physiol. 2004, 286, L37–L48. [Google Scholar] [CrossRef] [PubMed]
- Takano, M.; Meneshian, A.; Sheikh, E.; Yamakawa, Y.; Wilkins, K.B.; Hopkins, E.A.; Bulkley, G.B. Rapid upregulation of endothelial p-selectin expression via reactive oxygen species generation. Am. J. Physiol. Heart Circ. Physiol. 2002, 283, H2054–H2061. [Google Scholar] [CrossRef] [PubMed]
- Tiruppathi, C.; Naqvi, T.; Sandoval, R.; Mehta, D.; Malik, A.B. Synergistic effects of tumor necrosis factor-alpha and thrombin in increasing endothelial permeability. Am. J. Physiol. Lung Cell. Mol. Physiol. 2001, 281, L958–L968. [Google Scholar] [CrossRef] [PubMed]
- Claesson-Welsh, L. Vascular permeability—The essentials. Ups. J. Med. Sci. 2015, 120, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Gorlach, A.; Bertram, K.; Hudecova, S.; Krizanova, O. Calcium and ros: A mutual interplay. Redox Biol. 2015, 6, 260–271. [Google Scholar] [CrossRef] [PubMed]
- Brennan, L.J.; Morton, J.S.; Davidge, S.T. Vascular dysfunction in preeclampsia. Microcirculation 2014, 21, 4–14. [Google Scholar] [CrossRef] [PubMed]
- Yoshizumi, M.; Perrella, M.A.; Burnett, J.C., Jr.; Lee, M.E. Tumor necrosis factor downregulates an endothelial nitric oxide synthase mrna by shortening its half-life. Circ. Res. 1993, 73, 205–209. [Google Scholar] [CrossRef] [PubMed]
- Schulze-Osthoff, K.; Bakker, A.C.; Vanhaesebroeck, B.; Beyaert, R.; Jacob, W.A.; Fiers, W. Cytotoxic activity of tumor necrosis factor is mediated by early damage of mitochondrial functions. Evidence for the involvement of mitochondrial radical generation. J. Biol. Chem. 1992, 267, 5317–5323. [Google Scholar] [PubMed]
- Hofnagel, O.; Luechtenborg, B.; Stolle, K.; Lorkowski, S.; Eschert, H.; Plenz, G.; Robenek, H. Proinflammatory cytokines regulate LOX-1 expression in vascular smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 1789–1795. [Google Scholar] [CrossRef] [PubMed]
- Cominacini, L.; Rigoni, A.; Pasini, A.F.; Garbin, U.; Davoli, A.; Campagnola, M.; Pastorino, A.M.; Lo Cascio, V.; Sawamura, T. The binding of oxidized low density lipoprotein (OX-LDL) to OX-LDL receptor-1 reduces the intracellular concentration of nitric oxide in endothelial cells through an increased production of superoxide. J. Biol. Chem. 2001, 276, 13750–13755. [Google Scholar] [CrossRef] [PubMed]
- Matsubara, K.; Matsubara, Y.; Hyodo, S.; Katayama, T.; Ito, M. Role of nitric oxide and reactive oxygen species in the pathogenesis of preeclampsia. J. Obstet. Gynaecol. Res. 2010, 36, 239–247. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.; Kim, J.A.; Na, H.Y.; Kim, J.E.; Park, S.; Han, K.H.; Kim, Y.J.; Suh, S.H. Nadph oxidase 2-derived superoxide downregulates endothelial KCA3.1 in preeclampsia. Free Radic. Biol. Med. 2013, 57, 10–21. [Google Scholar] [CrossRef] [PubMed]
- Wallukat, G.; Homuth, V.; Fischer, T.; Lindschau, C.; Horstkamp, B.; Jupner, A.; Baur, E.; Nissen, E.; Vetter, K.; Neichel, D.; et al. Patients with preeclampsia develop agonistic autoantibodies against the angiotensin at1 receptor. J. Clin. Invest. 1999, 103, 945–952. [Google Scholar] [CrossRef] [PubMed]
- Dechend, R.; LaMarca, B.B.; Taylor, R.N. The renin-angiotensin system. Its autoantibodies, and body fluid volume in preeclampsia. In Chesley’s Hypertensive Disorders in Pregnancy, 4th ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2015; pp. 315–334. [Google Scholar]
- Choi, S.; Kim, J.A.; Li, H.Y.; Lee, S.J.; Seok, Y.S.; Kim, T.H.; Han, K.H.; Park, M.H.; Cho, G.J.; Suh, S.H. Altered redox state modulates endothelial KCa2.3 and KCa3.1 levels in normal pregnancy and preeclampsia. Antioxid. Redox Signal. 2018. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Gao, Q.; Jiang, L.; Feng, X.; Zhu, X.; Fan, X.; Mao, C.; Xu, Z. The NOX2-derived reactive oxygen species damaged endothelial nitric oxide system via suppressed BKCa/SKCa in preeclampsia. Hypertens. Res. 2017, 40, 457–464. [Google Scholar] [CrossRef] [PubMed]
- Vasquez-Vivar, J.; Kalyanaraman, B.; Martasek, P.; Hogg, N.; Masters, B.S.; Karoui, H.; Tordo, P.; Pritchard, K.A., Jr. Superoxide generation by endothelial nitric oxide synthase: The influence of cofactors. Proc. Natl. Acad. Sci. USA 1998, 95, 9220–9225. [Google Scholar] [CrossRef] [PubMed]
- Kuzkaya, N.; Weissmann, N.; Harrison, D.G.; Dikalov, S. Interactions of peroxynitrite, tetrahydrobiopterin, ascorbic acid, and thiols: Implications for uncoupling endothelial nitric-oxide synthase. J. Biol. Chem. 2003, 278, 22546–22554. [Google Scholar] [CrossRef] [PubMed]
- Sankaralingam, S.; Xu, H.; Davidge, S.T. Arginase contributes to endothelial cell oxidative stress in response to plasma from women with preeclampsia. Cardiovasc. Res. 2010, 85, 194–203. [Google Scholar] [CrossRef] [PubMed]
- Radi, R. Nitric oxide, oxidants, and protein tyrosine nitration. Proc. Natl. Acad. Sci. USA 2004, 101, 4003–4008. [Google Scholar] [CrossRef] [PubMed]
- Cooke, C.L.; Davidge, S.T. Peroxynitrite increases inos through NF-κB and decreases prostacyclin synthase in endothelial cells. Am. J. Physiol. Cell Physiol. 2002, 282, C395–C402. [Google Scholar] [CrossRef] [PubMed]
- Sankaralingam, S.; Xu, Y.; Sawamura, T.; Davidge, S.T. Increased lectin-like oxidized low-density lipoprotein receptor-1 expression in the maternal vasculature of women with preeclampsia: Role for peroxynitrite. Hypertension 2009, 53, 270–277. [Google Scholar] [CrossRef] [PubMed]
- Sankaralingam, S.; Xu, H.; Jiang, Y.; Sawamura, T.; Davidge, S.T. Evidence for increased methylglyoxal in the vasculature of women with preeclampsia: Role in upregulation of LOX-1 and arginase. Hypertension 2009, 54, 897–904. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Ma, G.; Chen, X. Lipopolysaccharide induced LOX-1 expression via TLR4/MyD88/ROS activated p38MAPK-NF-κb pathway. Vascul. Pharmacol. 2014, 63, 162–172. [Google Scholar] [CrossRef] [PubMed]
- Tsukimori, K.; Fukushima, K.; Tsushima, A.; Nakano, H. Generation of reactive oxygen species by neutrophils and endothelial cell injury in normal and preeclamptic pregnancies. Hypertension 2005, 46, 696–700. [Google Scholar] [CrossRef] [PubMed]
- Tsukimori, K.; Tsushima, A.; Fukushima, K.; Nakano, H.; Wake, N. Neutrophil-derived reactive oxygen species can modulate neutrophil adhesion to endothelial cells in preeclampsia. Am. J. Hypertens. 2008, 21, 587–591. [Google Scholar] [CrossRef] [PubMed]
- Cindrova-Davies, T. The therapeutic potential of antioxidants, er chaperones, NO and H2S donors, and statins for treatment of preeclampsia. Front. Pharmacol. 2014, 5, 119. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Aranguren, L.C.; Prada, C.E.; Riano-Medina, C.E.; Lopez, M. Endothelial dysfunction and preeclampsia: Role of oxidative stress. Front. Physiol. 2014, 5, 372. [Google Scholar] [CrossRef] [PubMed]
- Bilodeau, J.F. Review: Maternal and placental antioxidant response to preeclampsia—Impact on vasoactive eicosanoids. Placenta 2014, 35, S32–S38. [Google Scholar] [CrossRef] [PubMed]
- Weissgerber, T.L.; Gandley, R.E.; McGee, P.L.; Spong, C.Y.; Myatt, L.; Leveno, K.J.; Thorp, J.M., Jr.; Mercer, B.M.; Peaceman, A.M.; Ramin, S.M.; et al. Haptoglobin phenotype, preeclampsia risk and the efficacy of vitamin c and e supplementation to prevent preeclampsia in a racially diverse population. PLoS ONE 2013, 8, e60479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hofmeyr, G.J.; Seuc, A.H.; Betran, A.P.; Purnat, T.D.; Ciganda, A.; Munjanja, S.P.; Manyame, S.; Singata, M.; Fawcus, S.; Frank, K.; et al. The effect of calcium supplementation on blood pressure in non-pregnant women with previous pre-eclampsia: An exploratory, randomized placebo controlled study. Pregnancy Hypertens. 2015, 5, 273–279. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Guideline: Calcium Supplementation in Pregnant Women; World Health Organization: Geneva, Switzerland, 2013. [Google Scholar]
- Bodnar, L.M.; Catov, J.M.; Simhan, H.N.; Holick, M.F.; Powers, R.W.; Roberts, J.M. Maternal vitamin d deficiency increases the risk of preeclampsia. J. Clin. Endocrinol. Metab. 2007, 92, 3517–3522. [Google Scholar] [CrossRef] [PubMed]
- Purswani, J.M.; Gala, P.; Dwarkanath, P.; Larkin, H.M.; Kurpad, A.; Mehta, S. The role of vitamin d in pre-eclampsia: A systematic review. BMC Pregnancy Childbirth 2017, 17, 231. [Google Scholar] [CrossRef] [PubMed]
- Ertek, S.; Akgul, E.; Cicero, A.F.; Kutuk, U.; Demirtas, S.; Cehreli, S.; Erdogan, G. 25-hydroxy vitamin d levels and endothelial vasodilator function in normotensive women. Arch. Med. Sci. 2012, 8, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Jia, X.; Xu, J.; Gu, Y.; Gu, X.; Li, W.; Wang, Y. Vitamin d suppresses oxidative stress-induced microparticle release by human umbilical vein endothelial cells. Biol. Reprod. 2017, 96, 199–210. [Google Scholar] [PubMed]
- Onda, K.; Tong, S.; Nakahara, A.; Kondo, M.; Monchusho, H.; Hirano, T.; Kaitu’u-Lino, T.; Beard, S.; Binder, N.; Tuohey, L.; et al. Sofalcone upregulates the nuclear factor (erythroid-derived 2)-like 2/heme oxygenase-1 pathway, reduces soluble fms-like tyrosine kinase-1, and quenches endothelial dysfunction: Potential therapeutic for preeclampsia. Hypertension 2015, 65, 855–862. [Google Scholar] [CrossRef] [PubMed]
- Onda, K.; Tong, S.; Beard, S.; Binder, N.; Muto, M.; Senadheera, S.N.; Parry, L.; Dilworth, M.; Renshall, L.; Brownfoot, F.; et al. Proton pump inhibitors decrease soluble fms-like tyrosine kinase-1 and soluble endoglin secretion, decrease hypertension, and rescue endothelial dysfunction. Hypertension 2017, 69, 457–468. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, C.; Kenny, L.C. Therapeutically targeting mitochondrial redox signalling alleviates endothelial dysfunction in preeclampsia. Sci. Rep. 2016, 6, 32683. [Google Scholar] [CrossRef] [PubMed]
- Centlow, M.; Carninci, P.; Nemeth, K.; Mezey, E.; Brownstein, M.; Hansson, S.R. Placental expression profiling in preeclampsia: Local overproduction of hemoglobin may drive pathological changes. Fertil. Steril. 2008, 90, 1834–1843. [Google Scholar] [CrossRef] [PubMed]
- May, K.; Rosenlof, L.; Olsson, M.G.; Centlow, M.; Morgelin, M.; Larsson, I.; Cederlund, M.; Rutardottir, S.; Siegmund, W.; Schneider, H.; et al. Perfusion of human placenta with hemoglobin introduces preeclampsia-like injuries that are prevented by α1-microglobulin. Placenta 2011, 32, 323–332. [Google Scholar] [CrossRef] [PubMed]
- Hansson, S.R.; Naav, A.; Erlandsson, L. Oxidative stress in preeclampsia and the role of free fetal hemoglobin. Front. Physiol. 2014, 5, 516. [Google Scholar] [CrossRef] [PubMed]
- Hannan, N.J.; Brownfoot, F.C.; Cannon, P.; Deo, M.; Beard, S.; Nguyen, T.V.; Palmer, K.R.; Tong, S.; Kaitu’u-Lino, T.J. Resveratrol inhibits release of soluble fms-like tyrosine kinase (sFLT-1) and soluble endoglin and improves vascular dysfunction—Implications as a preeclampsia treatment. Sci. Rep. 2017, 7, 1819. [Google Scholar] [CrossRef] [PubMed]
- Ueki, N.; Takeda, S.; Koya, D.; Kanasaki, K. The relevance of the renin-angiotensin system in the development of drugs to combat preeclampsia. Int. J. Endocrinol. 2015, 2015, 572713. [Google Scholar] [CrossRef] [PubMed]
- Burke, S.D.; Zsengeller, Z.K.; Khankin, E.V.; Lo, A.S.; Rajakumar, A.; DuPont, J.J.; McCurley, A.; Moss, M.E.; Zhang, D.; Clark, C.D.; et al. Soluble fms-like tyrosine kinase 1 promotes angiotensin ii sensitivity in preeclampsia. J. Clin. Invest. 2016, 126, 2561–2574. [Google Scholar] [CrossRef] [PubMed]
- Rolnik, D.L.; Wright, D.; Poon, L.C.; O’Gorman, N.; Syngelaki, A.; de Paco Matallana, C.; Akolekar, R.; Cicero, S.; Janga, D.; Singh, M.; et al. Aspirin versus placebo in pregnancies at high risk for preterm preeclampsia. N. Engl. J. Med. 2017, 377, 613–622. [Google Scholar] [CrossRef] [PubMed]
- Cadavid, A.P. Aspirin: The mechanism of action revisited in the context of pregnancy complications. Front. Immunol. 2017, 8, 261. [Google Scholar] [CrossRef] [PubMed]
- Vane, J.R.; Botting, R.M. The mechanism of action of aspirin. Thromb. Res. 2003, 110, 255–258. [Google Scholar] [CrossRef]
- Schrör, K. Acetylsalicylic Acid; John Wiley and Sons: Hoboken, NJ, USA, 2010. [Google Scholar]
- Taubert, D.; Berkels, R.; Grosser, N.; Schroder, H.; Grundemann, D.; Schomig, E. Aspirin induces nitric oxide release from vascular endothelium: A novel mechanism of action. Br. J. Pharmacol. 2004, 143, 159–165. [Google Scholar] [CrossRef] [PubMed]
- Grosser, N.; Abate, A.; Oberle, S.; Vreman, H.J.; Dennery, P.A.; Becker, J.C.; Pohle, T.; Seidman, D.S.; Schroder, H. Heme oxygenase-1 induction may explain the antioxidant profile of aspirin. Biochem. Biophys. Res. Commun. 2003, 308, 956–960. [Google Scholar] [CrossRef]
- Gilroy, D.W. The role of aspirin-triggered lipoxins in the mechanism of action of aspirin. Prostaglandins Leukot. Essent. Fat. Acids 2005, 73, 203–210. [Google Scholar] [CrossRef] [PubMed]
- Gilroy, D.W. New insights into the anti-inflammatory actions of aspirin-induction of nitric oxide through the generation of epi-lipoxins. Mem. Inst. Oswaldo Cruz 2005, 100, 49–54. [Google Scholar] [CrossRef] [PubMed]
- De La Cruz, J.P.; Guerrero, A.; Gonzalez-Correa, J.A.; Arrebola, M.M.; de la Cuesta, S.F. Antioxidant effect of acetylsalicylic and salicylic acid in rat brain slices subjected to hypoxia. J. Neurosci. Res. 2004, 75, 280–290. [Google Scholar] [CrossRef] [PubMed]
- Seki, H. Animal models of preeclampsia: An examination of usefulness and limitations based on the metabolic domino theory. Hypertens. Res. Pregnancy 2017, 5, 52–58. [Google Scholar] [CrossRef]




© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aouache, R.; Biquard, L.; Vaiman, D.; Miralles, F. Oxidative Stress in Preeclampsia and Placental Diseases. Int. J. Mol. Sci. 2018, 19, 1496. https://doi.org/10.3390/ijms19051496
Aouache R, Biquard L, Vaiman D, Miralles F. Oxidative Stress in Preeclampsia and Placental Diseases. International Journal of Molecular Sciences. 2018; 19(5):1496. https://doi.org/10.3390/ijms19051496
Chicago/Turabian StyleAouache, Rajaa, Louise Biquard, Daniel Vaiman, and Francisco Miralles. 2018. "Oxidative Stress in Preeclampsia and Placental Diseases" International Journal of Molecular Sciences 19, no. 5: 1496. https://doi.org/10.3390/ijms19051496
APA StyleAouache, R., Biquard, L., Vaiman, D., & Miralles, F. (2018). Oxidative Stress in Preeclampsia and Placental Diseases. International Journal of Molecular Sciences, 19(5), 1496. https://doi.org/10.3390/ijms19051496