Tau Fibril Formation in Cultured Cells Compatible with a Mouse Model of Tauopathy

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Generation of Stable Cell Lines with Intracellular Tau Aggregates
2.2. Tau Biochemistry in Tau Fibril Cell Lines
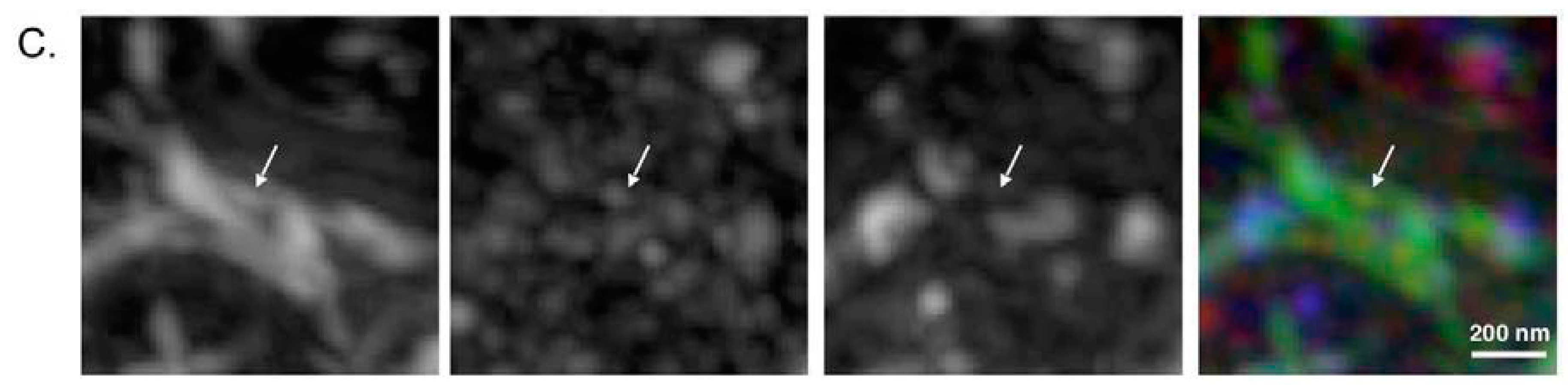
2.3. Super-Resolution Microscopic Analysis for Detecting Filamentous Tau Aggregates
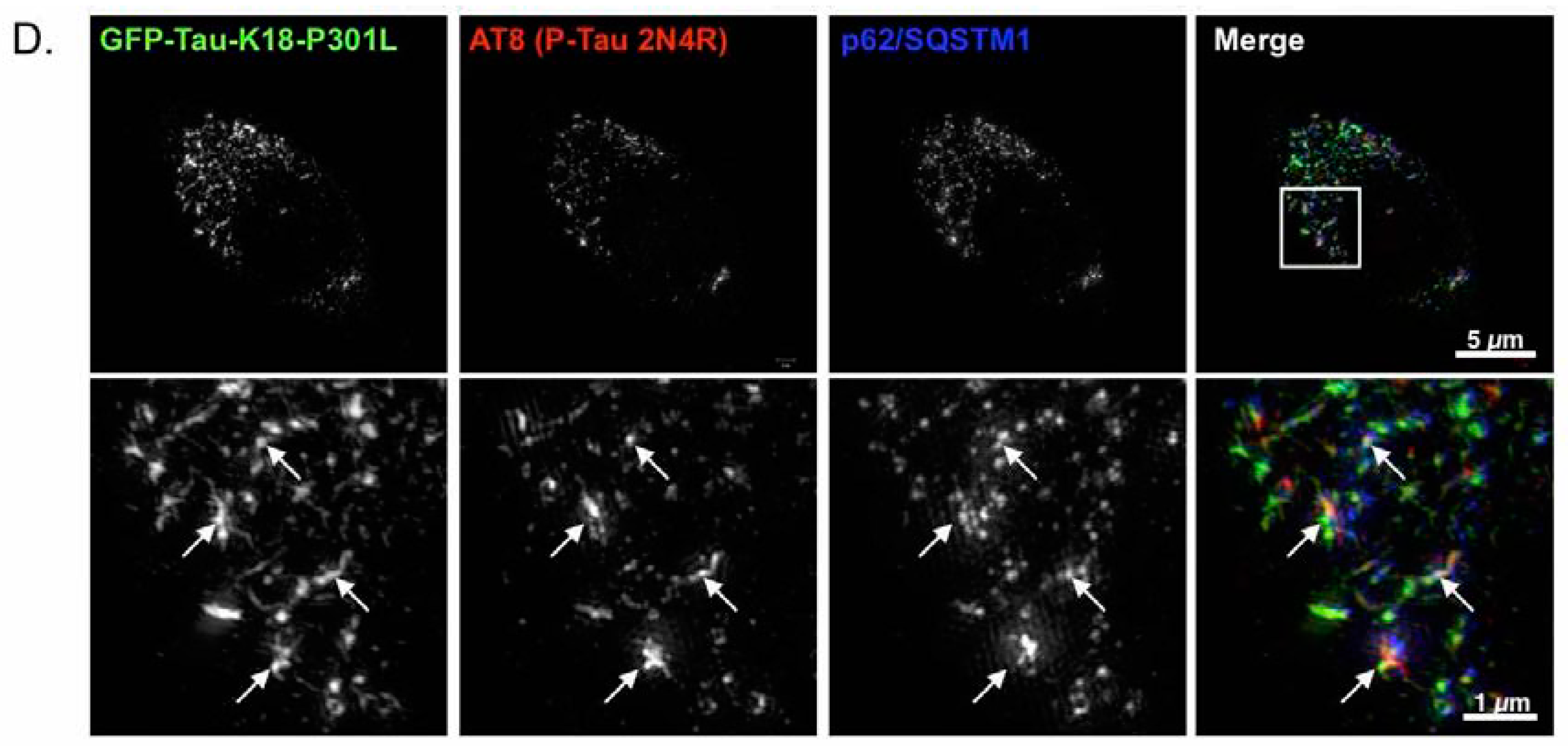
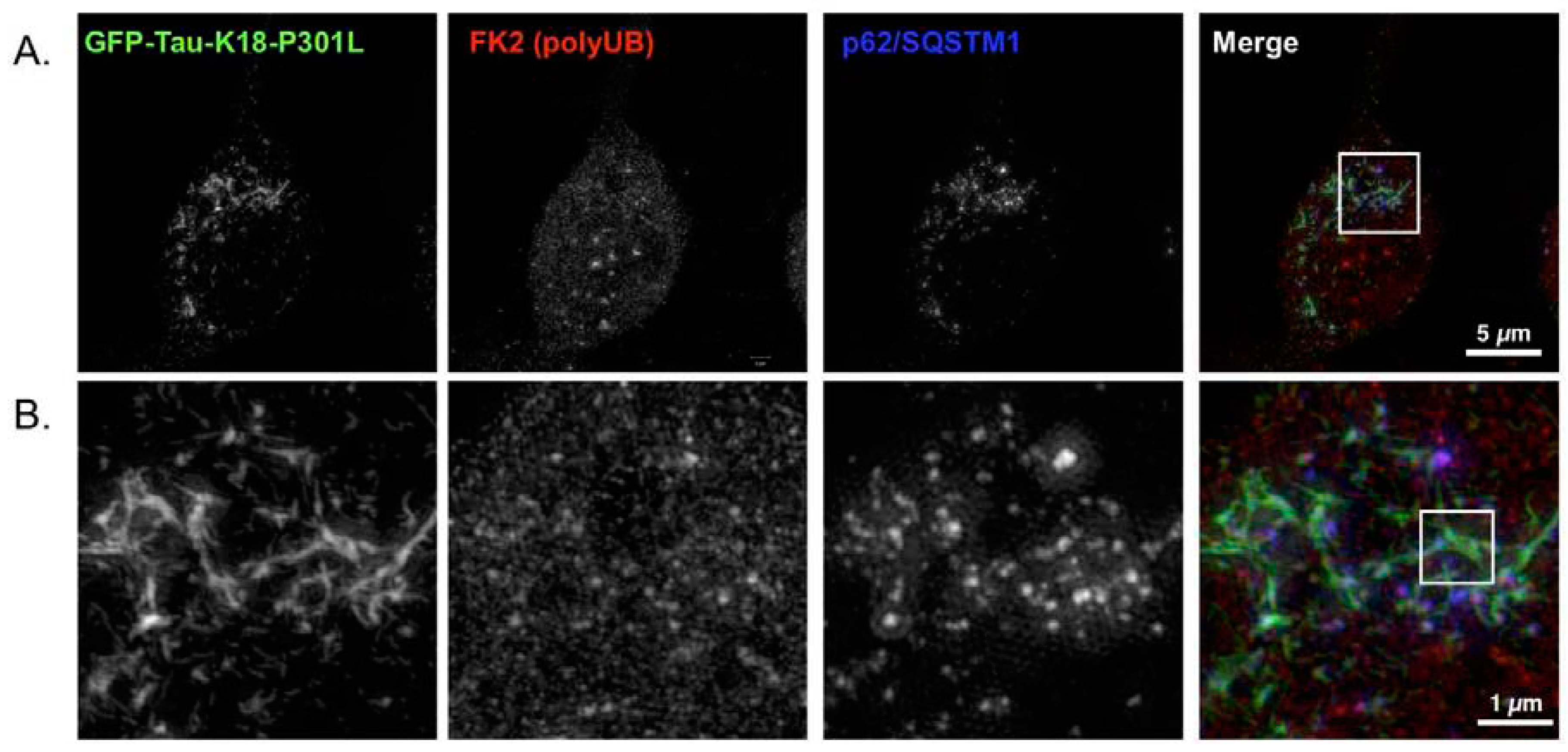
2.4. p62 and Polyubiquitin Localization in Filamentous Tau Aggregates
3. Discussion
4. Materials and Methods
4.1. Construction of Tau Fibril Cell Lines
4.2. Immunofluorescence Microscopy
4.3. Mice
4.4. Tissue and Cell Lysate Extraction
4.5. Western Blotting
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Lee, V.M.; Goedert, M.; Trojanowski, J.Q. Neurodegenerative tauopathies. Annu. Rev. Neurosci. 2001, 24, 1121–1159. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, K.; Liu, F.; Gong, C.X. Tau and neurodegenerative disease: The story so far. Nat. Rev. Neurol. 2016, 12, 15–27. [Google Scholar] [CrossRef] [PubMed]
- Goedert, M.; Jakes, R.; Spillantini, M.G.; Hasegawa, M.; Smith, M.J.; Crowther, R.A. Assembly of microtubule-associated protein tau into alzheimer-like filaments induced by sulphated glycosaminoglycans. Nature 1996, 383, 550–553. [Google Scholar] [CrossRef] [PubMed]
- Perez, M.; Valpuesta, J.M.; Medina, M.; Montejo de Garcini, E.; Avila, J. Polymerization of tau into filaments in the presence of heparin: The minimal sequence required for tau-tau interaction. J. Neurochem. 1996, 67, 1183–1190. [Google Scholar] [CrossRef] [PubMed]
- DeTure, M.; Ko, L.W.; Easson, C.; Yen, S.H. Tau assembly in inducible transfectants expressing wild-type or ftdp-17 tau. Am. J. Pathol. 2002, 161, 1711–1722. [Google Scholar] [CrossRef]
- Vogelsberg-Ragaglia, V.; Bruce, J.; Richter-Landsberg, C.; Zhang, B.; Hong, M.; Trojanowski, J.Q.; Lee, V.M. Distinct ftdp-17 missense mutations in tau produce tau aggregates and other pathological phenotypes in transfected cho cells. Mol. Biol. Cell 2000, 11, 4093–4104. [Google Scholar] [CrossRef] [PubMed]
- Ebneth, A.; Godemann, R.; Stamer, K.; Illenberger, S.; Trinczek, B.; Mandelkow, E. Overexpression of tau protein inhibits kinesin-dependent trafficking of vesicles, mitochondria, and endoplasmic reticulum: Implications for Alzheimer’s disease. J. Cell Biol. 1998, 143, 777–794. [Google Scholar] [CrossRef] [PubMed]
- Dayanandan, R.; Van Slegtenhorst, M.; Mack, T.G.; Ko, L.; Yen, S.H.; Leroy, K.; Brion, J.P.; Anderton, B.H.; Hutton, M.; Lovestone, S. Mutations in tau reduce its microtubule binding properties in intact cells and affect its phosphorylation. FEBS Lett. 1999, 446, 228–232. [Google Scholar] [CrossRef]
- Ko, L.W.; Rush, T.; Sahara, N.; Kersh, J.S.; Easson, C.; Deture, M.; Lin, W.L.; Connor, Y.D.; Yen, S.H. Assembly of filamentous tau aggregates in human neuronal cells. J. Alzheimer’s Dis. 2004, 6, 605–622; discussion 673–681. [Google Scholar] [CrossRef]
- Frost, B.; Jacks, R.L.; Diamond, M.I. Propagation of tau misfolding from the outside to the inside of a cell. J. Biol. Chem. 2009, 284, 12845–12852. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.L.; Buist, A.; Soares, A.; Callaerts, K.; Calafate, S.; Stevenaert, F.; Daniels, J.P.; Zoll, B.E.; Crowe, A.; Brunden, K.R.; et al. The dynamics and turnover of tau aggregates in cultured cells: Insights into therapies for tauopathies. J. Biol. Chem. 2016, 291, 13175–13193. [Google Scholar] [CrossRef] [PubMed]
- Varghese, M.; Santa-Maria, I.; Ho, L.; Ward, L.; Yemul, S.; Dubner, L.; Ksiezak-Reding, H.; Pasinetti, G.M. Extracellular tau paired helical filaments differentially affect tau pathogenic mechanisms in mitotic and post-mitotic cells: Implications for mechanisms of tau propagation in the brain. J. Alzheimer's Dis. 2016, 54, 477–496. [Google Scholar] [CrossRef] [PubMed]
- Woerman, A.L.; Aoyagi, A.; Patel, S.; Kazmi, S.A.; Lobach, I.; Grinberg, L.T.; McKee, A.C.; Seeley, W.W.; Olson, S.H.; Prusiner, S.B. Tau prions from Alzheimer’s disease and chronic traumatic encephalopathy patients propagate in cultured cells. Proc. Natl. Acad. Sci. USA 2016, 113, E8187–E8196. [Google Scholar] [CrossRef] [PubMed]
- Falcon, B.; Cavallini, A.; Angers, R.; Glover, S.; Murray, T.K.; Barnham, L.; Jackson, S.; O’Neill, M.J.; Isaacs, A.M.; Hutton, M.L.; et al. Conformation determines the seeding potencies of native and recombinant tau aggregates. J. Biol. Chem. 2015, 290, 1049–1065. [Google Scholar] [CrossRef] [PubMed]
- Kfoury, N.; Holmes, B.B.; Jiang, H.; Holtzman, D.M.; Diamond, M.I. Trans-cellular propagation of tau aggregation by fibrillar species. J. Biol. Chem. 2012, 287, 19440–19451. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.W.; Hussaini, S.A.; Bastille, I.M.; Rodriguez, G.A.; Mrejeru, A.; Rilett, K.; Sanders, D.W.; Cook, C.; Fu, H.; Boonen, R.A.; et al. Neuronal activity enhances tau propagation and tau pathology in vivo. Nat. Neurosci. 2016, 19, 1085–1092. [Google Scholar] [CrossRef] [PubMed]
- Santacruz, K.; Lewis, J.; Spires, T.; Paulson, J.; Kotilinek, L.; Ingelsson, M.; Guimaraes, A.; DeTure, M.; Ramsden, M.; McGowan, E.; et al. Tau suppression in a neurodegenerative mouse model improves memory function. Science 2005, 309, 476–481. [Google Scholar] [CrossRef] [PubMed]
- Sahara, N.; Deture, M.; Ren, Y.; Ebrahim, A.S.; Kang, D.; Knight, J.; Volbracht, C.; Pedersen, J.T.; Dickson, D.W.; Yen, S.H.; et al. Characteristics of tbs-extractable hyperphosphorylated tau species: Aggregation intermediates in rtg4510 mouse brain. J. Alzheimer’s Dis. 2013, 33, 249–263. [Google Scholar]
- Sanders, D.W.; Kaufman, S.K.; DeVos, S.L.; Sharma, A.M.; Mirbaha, H.; Li, A.; Barker, S.J.; Foley, A.C.; Thorpe, J.R.; Serpell, L.C.; et al. Distinct tau prion strains propagate in cells and mice and define different tauopathies. Neuron 2014, 82, 1271–1288. [Google Scholar] [CrossRef] [PubMed]
- Holmes, B.B.; Furman, J.L.; Mahan, T.E.; Yamasaki, T.R.; Mirbaha, H.; Eades, W.C.; Belaygorod, L.; Cairns, N.J.; Holtzman, D.M.; Diamond, M.I. Proteopathic tau seeding predicts tauopathy in vivo. Proc. Natl. Acad. Sci. USA 2014, 111, E4376–4385. [Google Scholar] [CrossRef] [PubMed]
- Krammer, C.; Schatzl, H.M.; Vorberg, I. Prion-like propagation of cytosolic protein aggregates: Insights from cell culture models. Prion 2009, 3, 206–212. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, S.G.; Davies, P. A preparation of alzheimer paired helical filaments that displays distinct tau proteins by polyacrylamide gel electrophoresis. Proc. Natl. Acad. Sci. USA 1990, 87, 5827–5831. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.; Sahara, N. Characteristics of tau oligomers. Front. Neurol. 2013, 4, 102. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, M.; Shimada, H.; Suhara, T.; Shinotoh, H.; Ji, B.; Maeda, J.; Zhang, M.R.; Trojanowski, J.Q.; Lee, V.M.; Ono, M.; et al. Imaging of tau pathology in a tauopathy mouse model and in alzheimer patients compared to normal controls. Neuron 2013, 79, 1094–1108. [Google Scholar] [CrossRef] [PubMed]
- Overbye, A.; Fengsrud, M.; Seglen, P.O. Proteomic analysis of membrane-associated proteins from rat liver autophagosomes. Autophagy 2007, 3, 300–322. [Google Scholar] [CrossRef] [PubMed]
- Knaevelsrud, H.; Simonsen, A. Fighting disease by selective autophagy of aggregate-prone proteins. FEBS Lett. 2010, 584, 2635–2645. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, G.; Wada, K.; Okuno, M.; Kurosawa, M.; Nukina, N. Serine 403 phosphorylation of p62/sqstm1 regulates selective autophagic clearance of ubiquitinated proteins. Mol. Cell 2011, 44, 279–289. [Google Scholar] [CrossRef] [PubMed]
- Grober, E.; Dickson, D.; Sliwinski, M.J.; Buschke, H.; Katz, M.; Crystal, H.; Lipton, R.B. Memory and mental status correlates of modified braak staging. Neurobiol. Aging 1999, 20, 573–579. [Google Scholar] [CrossRef]
- Wittmann, C.W.; Wszolek, M.F.; Shulman, J.M.; Salvaterra, P.M.; Lewis, J.; Hutton, M.; Feany, M.B. Tauopathy in drosophila: Neurodegeneration without neurofibrillary tangles. Science 2001, 293, 711–714. [Google Scholar] [CrossRef] [PubMed]
- Kimura, T.; Sharma, G.; Ishiguro, K.; Hisanaga, S.I. Phospho-tau bar code: Analysis of phosphoisotypes of tau and its application to tauopathy. Front. Neurosci. 2018, 12, 44. [Google Scholar] [CrossRef] [PubMed]
- Fitzpatrick, A.W.P.; Falcon, B.; He, S.; Murzin, A.G.; Murshudov, G.; Garringer, H.J.; Crowther, R.A.; Ghetti, B.; Goedert, M.; Scheres, S.H.W. Cryo-em structures of tau filaments from Alzheimer’s disease. Nature 2017, 547, 185–190. [Google Scholar] [CrossRef] [PubMed]
- Von Bergen, M.; Friedhoff, P.; Biernat, J.; Heberle, J.; Mandelkow, E.M.; Mandelkow, E. Assembly of tau protein into alzheimer paired helical filaments depends on a local sequence motif ((306)vqivyk(311)) forming beta structure. Proc. Natl. Acad. Sci. USA 2000, 97, 5129–5134. [Google Scholar] [CrossRef] [PubMed]
- Sahara, N.; Maeda, S.; Murayama, M.; Suzuki, T.; Dohmae, N.; Yen, S.H.; Takashima, A. Assembly of two distinct dimers and higher-order oligomers from full-length tau. Eur. J. Neurosci. 2007, 25, 3020–3029. [Google Scholar] [CrossRef] [PubMed]
- Kuusisto, E.; Salminen, A.; Alafuzoff, I. Ubiquitin-binding protein p62 is present in neuronal and glial inclusions in human tauopathies and synucleinopathies. Neuroreport 2001, 12, 2085–2090. [Google Scholar] [CrossRef] [PubMed]
- Scott, I.S.; Lowe, J.S. The ubiquitin-binding protein p62 identifies argyrophilic grain pathology with greater sensitivity than conventional silver stains. Acta Neuropathol. 2007, 113, 417–420. [Google Scholar] [CrossRef] [PubMed]
- Schaeffer, V.; Lavenir, I.; Ozcelik, S.; Tolnay, M.; Winkler, D.T.; Goedert, M. Stimulation of autophagy reduces neurodegeneration in a mouse model of human tauopathy. Brain J. Neurol. 2012, 135, 2169–2177. [Google Scholar] [CrossRef] [PubMed]
- Mori, H.; Kondo, J.; Ihara, Y. Ubiquitin is a component of paired helical filaments in Alzheimer’s disease. Science 1987, 235, 1641–1644. [Google Scholar] [CrossRef] [PubMed]
- Perry, G.; Friedman, R.; Shaw, G.; Chau, V. Ubiquitin is detected in neurofibrillary tangles and senile plaque neurites of alzheimer disease brains. Proc. Natl. Acad. Sci. USA 1987, 84, 3033–3036. [Google Scholar] [CrossRef] [PubMed]
- Cripps, D.; Thomas, S.N.; Jeng, Y.; Yang, F.; Davies, P.; Yang, A.J. Alzheimer disease-specific conformation of hyperphosphorylated paired helical filament-tau is polyubiquitinated through lys-48, lys-11, and lys-6 ubiquitin conjugation. J. Biol. Chem. 2006, 281, 10825–10838. [Google Scholar] [CrossRef] [PubMed]







© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Matsumoto, G.; Matsumoto, K.; Kimura, T.; Suhara, T.; Higuchi, M.; Sahara, N.; Mori, N. Tau Fibril Formation in Cultured Cells Compatible with a Mouse Model of Tauopathy. Int. J. Mol. Sci. 2018, 19, 1497. https://doi.org/10.3390/ijms19051497
Matsumoto G, Matsumoto K, Kimura T, Suhara T, Higuchi M, Sahara N, Mori N. Tau Fibril Formation in Cultured Cells Compatible with a Mouse Model of Tauopathy. International Journal of Molecular Sciences. 2018; 19(5):1497. https://doi.org/10.3390/ijms19051497
Chicago/Turabian StyleMatsumoto, Gen, Kazuki Matsumoto, Taeko Kimura, Tetsuya Suhara, Makoto Higuchi, Naruhiko Sahara, and Nozomu Mori. 2018. "Tau Fibril Formation in Cultured Cells Compatible with a Mouse Model of Tauopathy" International Journal of Molecular Sciences 19, no. 5: 1497. https://doi.org/10.3390/ijms19051497