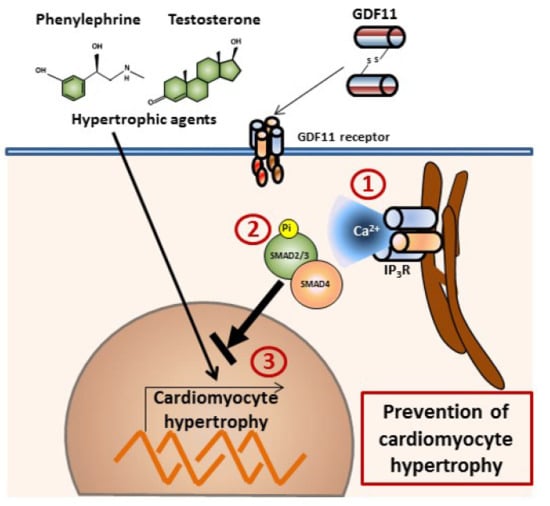
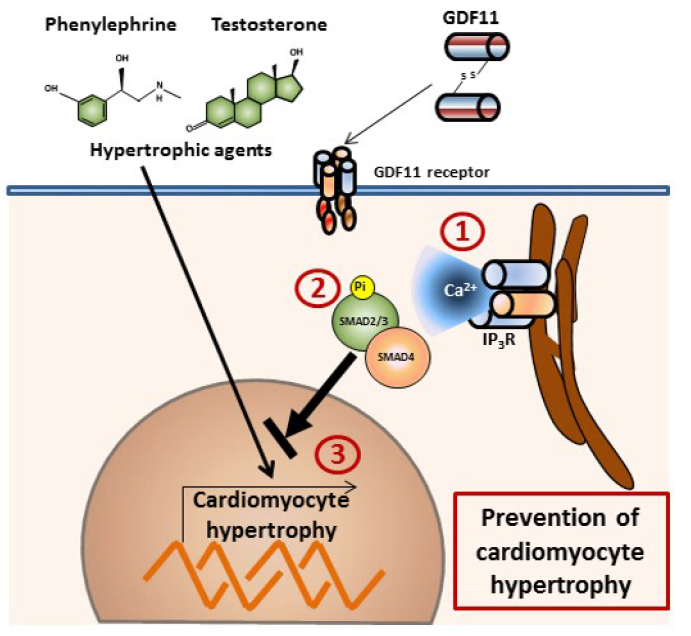
GDF11 Modulates Ca2+-Dependent Smad2/3 Signaling to Prevent Cardiomyocyte Hypertrophy


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
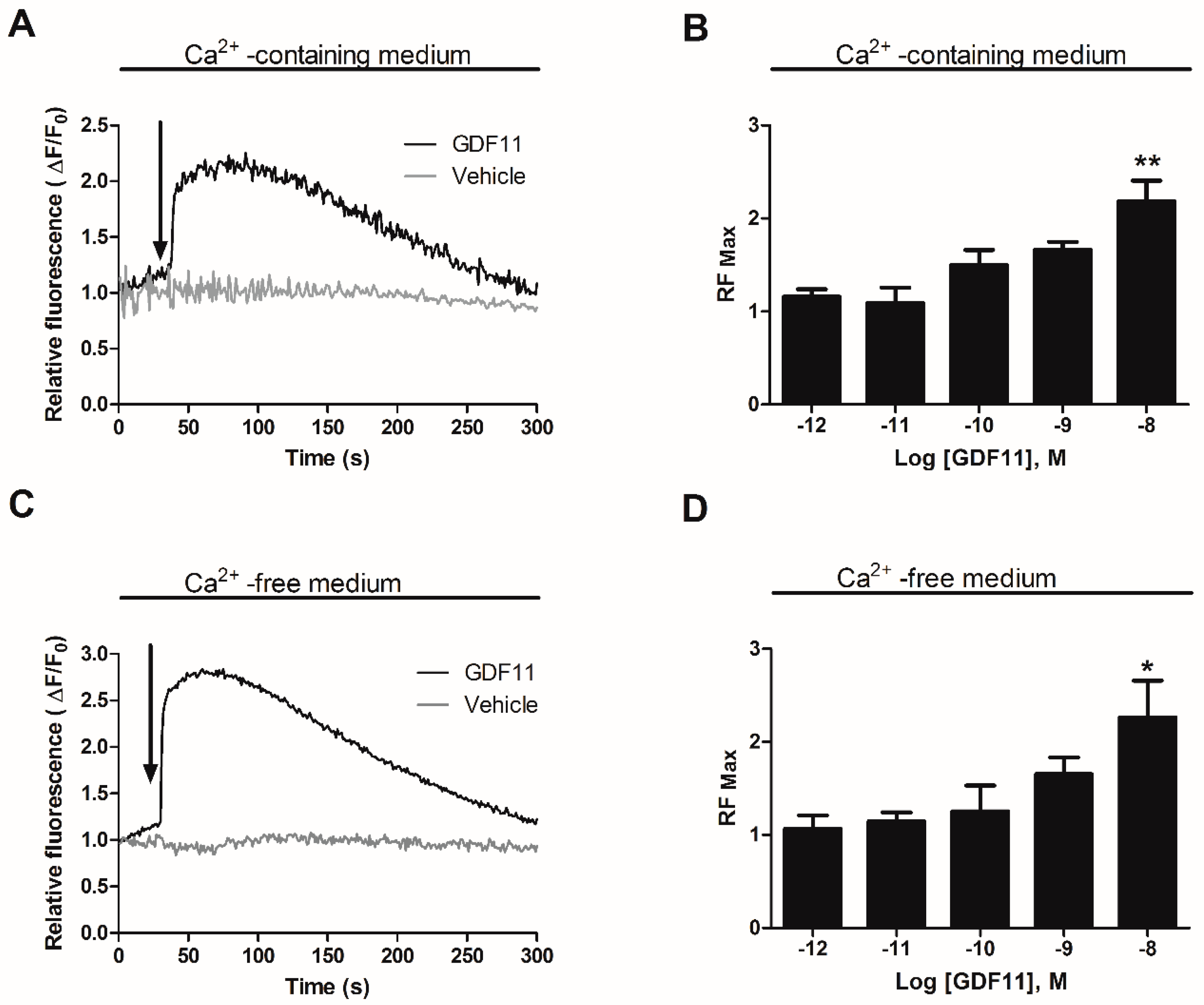
2.1. GDF11 Increased Intracellular Ca2+ in Neonatal Cultured Cardiomyocytes
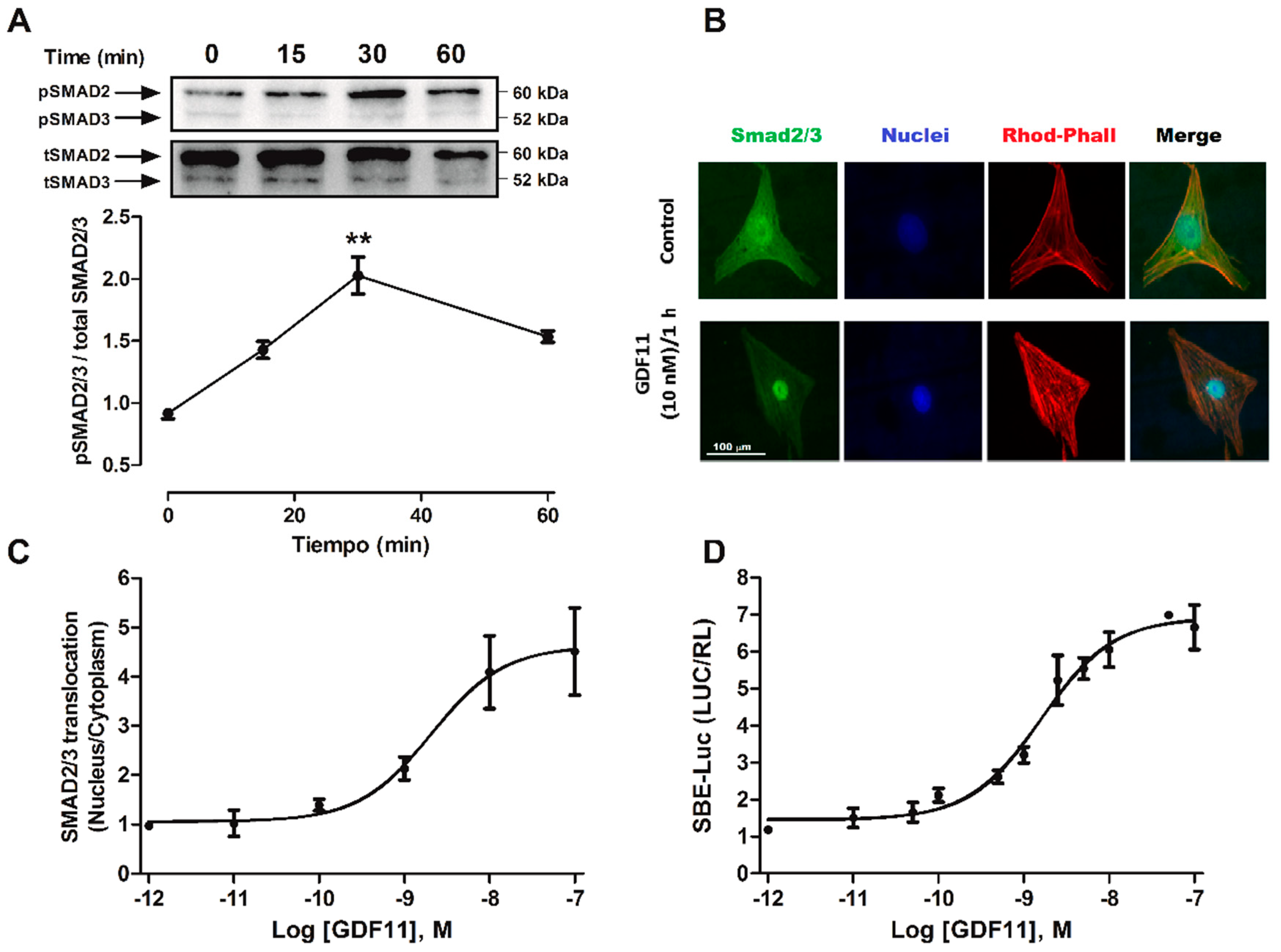
2.2. GDF11 Activated Smad2/3 in Cardiomyocytes
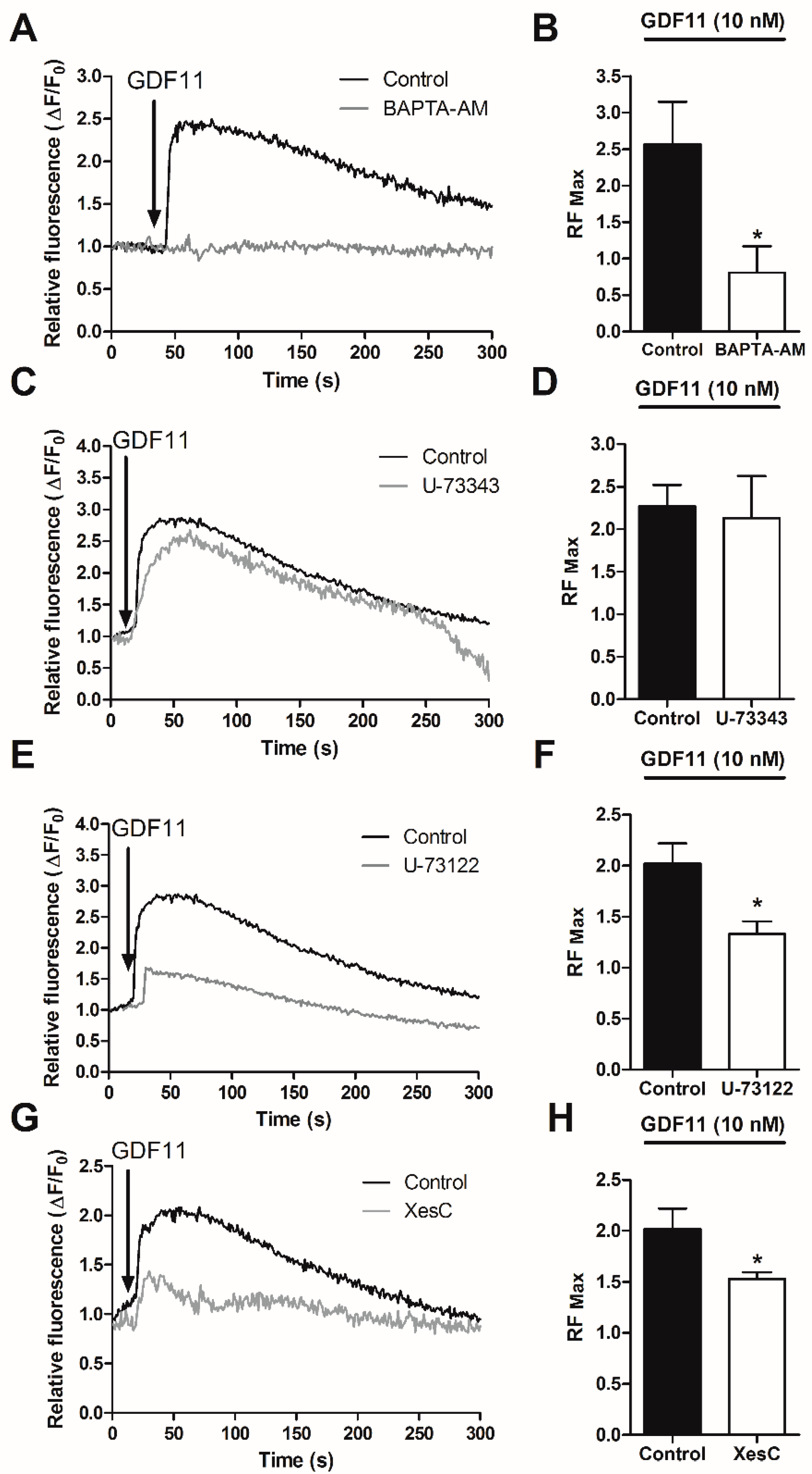
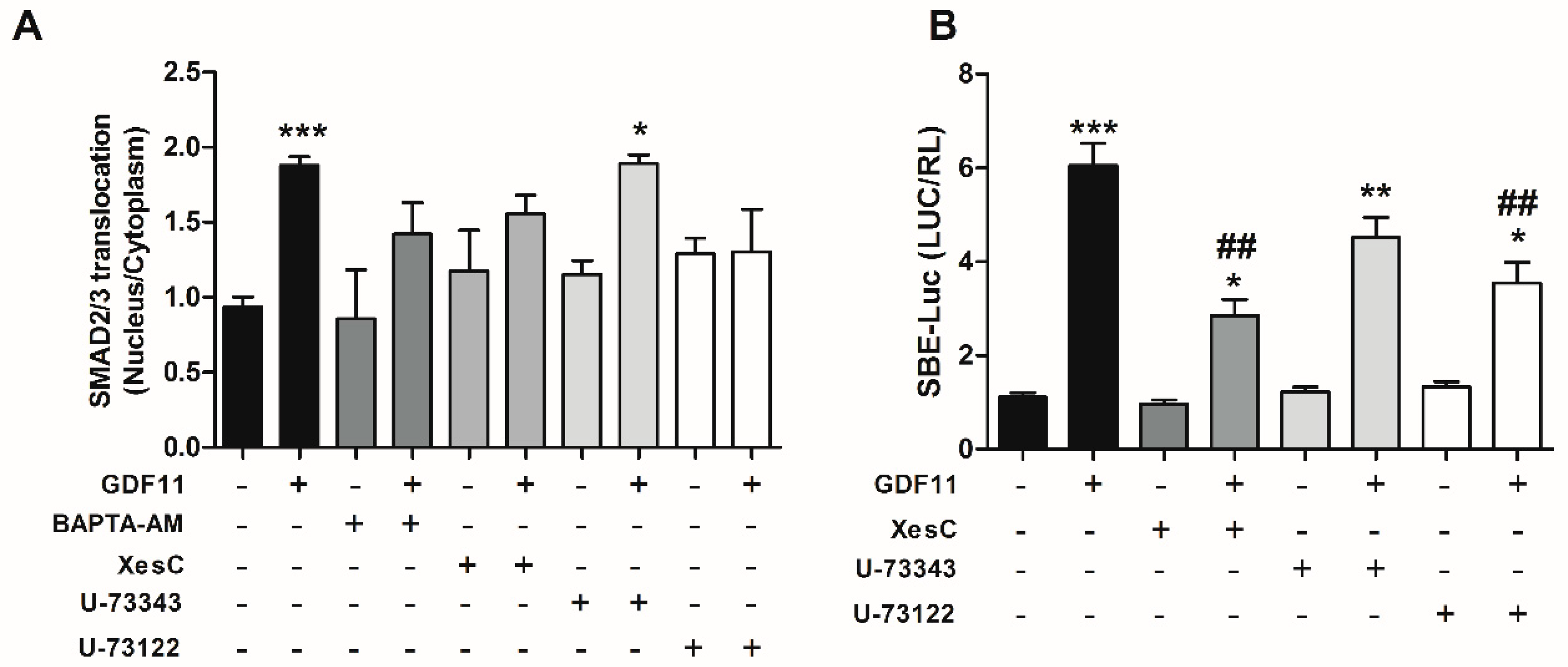
2.3. Effect of Ca2+-Dependent Pathways on Smad2/3 Activity Induced by GDF11 in Cardiomyocytes
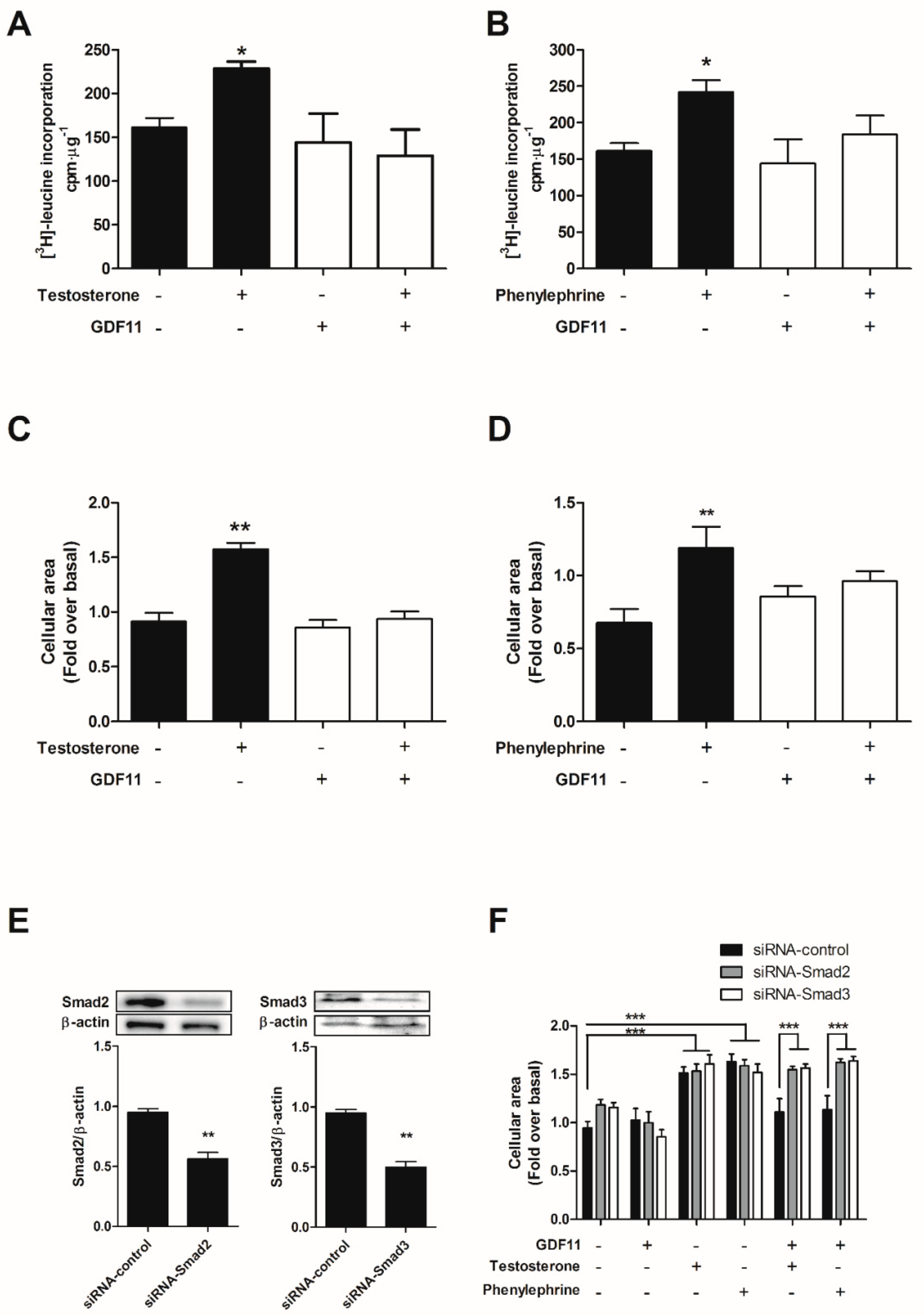
2.4. GDF11 Prevented Cardiomyocyte Hypertrophy Induced by Testosterone and Phenylephrine
3. Discussion
4. Materials and Methods
4.1. Isolation and Culture of Cardiomyocytes from Neonatal Rats
4.2. Measurement of Intracellular Ca2+
4.3. Transfections and Reporter Assay
4.4. Western Blotting
4.5. Immunocytochemistry
4.6. Incorporation of Amino Acids
4.7. Cell Size
4.8. Statistical Analysis
5. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Xie, J.; Tu, T.; Zhou, S.; Liu, Q. Transforming growth factor (TGF)-beta1 signal pathway: A promising therapeutic target for attenuating cardiac fibrosis. Int. J. Cardiol. 2017, 239, 9. [Google Scholar] [CrossRef] [PubMed]
- Yue, Y.; Meng, K.; Pu, Y.; Zhang, X. Transforming growth factor beta (TGF-beta) mediates cardiac fibrosis and induces diabetic cardiomyopathy. Diabetes Res. Clin. Pract. 2017, 133, 124–130. [Google Scholar] [CrossRef] [PubMed]
- Du, G.Q.; Shao, Z.B.; Wu, J.; Yin, W.J.; Li, S.H.; Wu, J.; Weisel, R.D.; Tian, J.W.; Li, R.K. Targeted myocardial delivery of GDF11 gene rejuvenates the aged mouse heart and enhances myocardial regeneration after ischemia-reperfusion injury. Basic Res. Cardiol. 2017, 112, 7. [Google Scholar] [CrossRef] [PubMed]
- Zimmers, T.A.; Jiang, Y.; Wang, M.; Liang, T.W.; Rupert, J.E.; Au, E.D.; Marino, F.E.; Couch, M.E.; Koniaris, L.G. Exogenous GDF11 induces cardiac and skeletal muscle dysfunction and wasting. Basic Res. Cardiol. 2017, 112, 48. [Google Scholar] [CrossRef] [PubMed]
- Olson, K.A.; Beatty, A.L.; Heidecker, B.; Regan, M.C.; Brody, E.N.; Foreman, T.; Kato, S.; Mehler, R.E.; Singer, B.S.; Hveem, K.; et al. Association of growth differentiation factor 11/8, putative anti-ageing factor, with cardiovascular outcomes and overall mortality in humans: Analysis of the Heart and Soul and HUNT3 cohorts. Eur. Heart J. 2015, 36, 3426–3434. [Google Scholar] [CrossRef] [PubMed]
- Loffredo, F.S.; Steinhauser, M.L.; Jay, S.M.; Gannon, J.; Pancoast, J.R.; Yalamanchi, P.; Sinha, M.; Dall’Osso, C.; Khong, D.; Shadrach, J.L.; et al. Growth differentiation factor 11 is a circulating factor that reverses age-related cardiac hypertrophy. Cell 2013, 153, 828–839. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.C.; Zhang, X.; Gross, P.; Starosta, T.; Mohsin, S.; Franti, M.; Gupta, P.; Hayes, D.; Myzithras, M.; Kahn, J.; et al. GDF11 does not rescue aging-related pathological hypertrophy. Circ. Res. 2015, 117, 926–932. [Google Scholar] [CrossRef] [PubMed]
- Egerman, M.A.; Cadena, S.M.; Gilbert, J.A.; Meyer, A.; Nelson, H.N.; Swalley, S.E.; Mallozzi, C.; Jacobi, C.; Jennings, L.L.; Clay, I.; et al. GDF11 Increases with Age and Inhibits Skeletal Muscle Regeneration. Cell Metab. 2015, 22, 164–174. [Google Scholar] [CrossRef] [PubMed]
- Dewenter, M.; von der Lieth, A.; Katus, H.A.; Backs, J. Calcium Signaling and Transcriptional Regulation in Cardiomyocytes. Circ. Res. 2017, 121, 1000–1020. [Google Scholar] [CrossRef] [PubMed]
- Fearnley, C.J.; Roderick, H.L.; Bootman, M.D. Calcium signaling in cardiac myocytes. Cold Spring Harb. Perspect. Biol. 2011, 3, a004242. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.M.; Wang, C.Y.; Hu, C.; Fang, Y.J.; Mei, Y.A. GDF-15 enhances intracellular Ca2+ by increasing Cav1.3 expression in rat cerebellar granule neurons. Biochem. J. 2016, 473, 1895–1904. [Google Scholar] [CrossRef] [PubMed]
- McGowan, T.A.; Madesh, M.; Zhu, Y.; Wang, L.; Russo, M.; Deelman, L.; Henning, R.; Joseph, S.; Hajnoczky, G.; Sharma, K. TGF-beta-induced Ca(2+) influx involves the type III IP(3) receptor and regulates actin cytoskeleton. Am. J. Physiol. Ren. Physiol. 2002, 282, F910–F920. [Google Scholar] [CrossRef] [PubMed]
- Molkentin, J.D.; Dorn, G.W., 2nd. Cytoplasmic signaling pathways that regulate cardiac hypertrophy. Annu. Rev. Physiol. 2001, 63, 391–426. [Google Scholar] [CrossRef] [PubMed]
- Heineke, J.; Molkentin, J.D. Regulation of cardiac hypertrophy by intracellular signalling pathways. Nat. Rev. Mol. Cell Biol. 2006, 7, 589–600. [Google Scholar] [CrossRef] [PubMed]
- Walker, R.G.; Czepnik, M.; Goebel, E.J.; McCoy, J.C.; Vujic, A.; Cho, M.; Oh, J.; Aykul, S.; Walton, K.L.; Schang, G.; et al. Structural basis for potency differences between GDF8 and GDF11. BMC Biol. 2017, 15, 19. [Google Scholar] [CrossRef] [PubMed]
- Gaunt, S.J. Gdf11/Smad signalling and Cdx proteins cooperate to activate the Hoxc8 early enhancer in HepG2 cells. Int. J. Dev. Biol. 2017, 61, 427–432. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.; Tu, M.L.; Li, C.J.; Zhang, L.; Jiang, T.J.; Liu, T.; Luo, X.H. GDF11 Inhibits Bone Formation by Activating Smad2/3 in Bone Marrow Mesenchymal Stem Cells. Calcif. Tissue Int. 2016, 99, 500–509. [Google Scholar] [CrossRef] [PubMed]
- Massague, J.; Seoane, J.; Wotton, D. Smad transcription factors. Genes Dev. 2005, 19, 2783–2810. [Google Scholar] [CrossRef] [PubMed]
- Schmierer, B.; Hill, C.S. TGFbeta-SMAD signal transduction: Molecular specificity and functional flexibility. Nat. Rev. Mol. Cell Biol. 2007, 8, 970–982. [Google Scholar] [CrossRef] [PubMed]
- Poggioli, T.; Vujic, A.; Yang, P.; Macias-Trevino, C.; Uygur, A.; Loffredo, F.S.; Pancoast, J.R.; Cho, M.; Goldstein, J.; Tandias, R.M.; et al. Circulating Growth Differentiation Factor 11/8 Levels Decline with Age. Circ. Res. 2016, 118, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Katsimpardi, L.; Litterman, N.K.; Schein, P.A.; Miller, C.M.; Loffredo, F.S.; Wojtkiewicz, G.R.; Chen, J.W.; Lee, R.T.; Wagers, A.J.; Rubin, L.L. Vascular and neurogenic rejuvenation of the aging mouse brain by young systemic factors. Science 2014, 344, 630–634. [Google Scholar] [CrossRef] [PubMed]
- Tando, T.; Hirayama, A.; Furukawa, M.; Sato, Y.; Kobayashi, T.; Funayama, A.; Kanaji, A.; Hao, W.; Watanabe, R.; Morita, M.; et al. Smad2/3 Proteins Are Required for Immobilization-induced Skeletal Muscle Atrophy. J. Biol. Chem. 2016, 291, 12184–12194. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Kimball, T.R.; Lorenz, J.N.; Brown, D.A.; Bauskin, A.R.; Klevitsky, R.; Hewett, T.E.; Breit, S.N.; Molkentin, J.D. GDF15/MIC-1 functions as a protective and antihypertrophic factor released from the myocardium in association with SMAD protein activation. Circ. Res. 2006, 98, 342–350. [Google Scholar] [CrossRef] [PubMed]
- Chow, J.Y.; Dong, H.; Quach, K.T.; Van Nguyen, P.N.; Chen, K.; Carethers, J.M. TGF-beta mediates PTEN suppression and cell motility through calcium-dependent PKC-alpha activation in pancreatic cancer cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 294, G899–G905. [Google Scholar] [CrossRef] [PubMed]
- Roach, K.M.; Feghali-Bostwick, C.; Wulff, H.; Amrani, Y.; Bradding, P. Human lung myofibroblast TGFbeta1-dependent Smad2/3 signalling is Ca(2+)-dependent and regulated by KCa3.1 K(+) channels. Fibrogenes. Tissue Repair 2015, 8, 5. [Google Scholar] [CrossRef] [PubMed]
- Walker, R.G.; Poggioli, T.; Katsimpardi, L.; Buchanan, S.M.; Oh, J.; Wattrus, S.; Heidecker, B.; Fong, Y.W.; Rubin, L.L.; Ganz, P.; et al. Biochemistry and Biology of GDF11 and Myostatin: Similarities, Differences, and Questions for Future Investigation. Circ. Res. 2016, 118, 1125–1141. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S.; Kolb, M.R.; Duan, F.; Janssen, L.J. Transforming growth factor-beta evokes Ca2+ waves and enhances gene expression in human pulmonary fibroblasts. Am. J. Respir. Cell Mol. Biol. 2012, 46, 757–764. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, H.; Bodi, I.; Maillet, M.; DeSantiago, J.; Domeier, T.L.; Mikoshiba, K.; Lorenz, J.N.; Blatter, L.A.; Bers, D.M.; Molkentin, J.D. The IP3 receptor regulates cardiac hypertrophy in response to select stimuli. Circ. Res. 2010, 107, 659–666. [Google Scholar] [CrossRef] [PubMed]
- Correll, R.N.; Makarewich, C.A.; Zhang, H.; Zhang, C.; Sargent, M.A.; York, A.J.; Berretta, R.M.; Chen, X.; Houser, S.R.; Molkentin, J.D. Caveolae-localized L-type Ca2+ channels do not contribute to function or hypertrophic signalling in the mouse heart. Cardiovasc. Res. 2017, 113, 749–759. [Google Scholar] [CrossRef] [PubMed]
- Takimoto, E.; Champion, H.C.; Li, M.; Belardi, D.; Ren, S.; Rodriguez, E.R.; Bedja, D.; Gabrielson, K.L.; Wang, Y.; Kass, D.A. Chronic inhibition of cyclic GMP phosphodiesterase 5A prevents and reverses cardiac hypertrophy. Nat. Med. 2005, 11, 214–222. [Google Scholar] [CrossRef] [PubMed]
- Moustafa, A.; Sakamoto, K.Q.; Habara, Y. Nitric oxide stimulates IP3 production via a cGMP/PKG-dependent pathway in rat pancreatic acinar cells. Jpn. J. Vet. Res. 2011, 59, 5–14. [Google Scholar] [PubMed]
- Feger, M.; Hase, P.; Zhang, B.; Hirche, F.; Glosse, P.; Lang, F.; Foller, M. The production of fibroblast growth factor 23 is controlled by TGF-beta2. Sci. Rep. 2017, 7, 4982. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Xu, N.; Feng, X.; Hou, N.; Zhang, J.; Cheng, X.; Chen, Y.; Zhang, Y.; Yang, X. Targeted disruption of Smad4 in cardiomyocytes results in cardiac hypertrophy and heart failure. Circ. Res. 2005, 97, 821–828. [Google Scholar] [CrossRef] [PubMed]
- Salvarani, N.; Maguy, A.; De Simone, S.A.; Miragoli, M.; Jousset, F.; Rohr, S. TGF-beta1 (Transforming Growth Factor-beta1) Plays a Pivotal Role in Cardiac Myofibroblast Arrhythmogenicity. Circ. Arrhythm. Electrophysiol. 2017, 10, e004567. [Google Scholar] [CrossRef] [PubMed]
- Sinha, M.; Jang, Y.C.; Oh, J.; Khong, D.; Wu, E.Y.; Manohar, R.; Miller, C.; Regalado, S.G.; Loffredo, F.S.; Pancoast, J.R.; et al. Restoring systemic GDF11 levels reverses age-related dysfunction in mouse skeletal muscle. Science 2014, 344, 649–652. [Google Scholar] [CrossRef] [PubMed]
- Schafer, M.J.; Atkinson, E.J.; Vanderboom, P.M.; Kotajarvi, B.; White, T.A.; Moore, M.M.; Bruce, C.J.; Greason, K.L.; Suri, R.M.; Khosla, S.; et al. Quantification of GDF11 and Myostatin in Human Aging and Cardiovascular Disease. Cell Metab. 2016, 23, 1207–1215. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.E.; Cadena, S.M.; Gong, C.; Wang, X.; Chen, Z.; Wang, S.X.; Vickers, C.; Chen, H.; Lach-Trifilieff, E.; Hadcock, J.R.; et al. Supraphysiologic Administration of GDF11 Induces Cachexia in Part by Upregulating GDF15. Cell Rep. 2018, 22, 1522–1530. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Li, Y.; Xiang, L.; Zhang, J.; Zhu, B.; Xiang, L.; Dong, J.; Liu, M.; Xiang, G. GDF11 Attenuates Development of Type 2 Diabetes via Improvement of Islet beta-Cell Function and Survival. Diabetes 2017, 66, 1914–1927. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Li, Q.; Liu, D.; Huang, Q.; Cai, G.; Cui, S.; Sun, X.; Chen, X. GDF11 improves tubular regeneration after acute kidney injury in elderly mice. Sci. Rep. 2016, 6, 34624. [Google Scholar] [CrossRef] [PubMed]
- Jing, Y.Y.; Li, D.; Wu, F.; Gong, L.L.; Li, R. GDF11 does not improve the palmitate induced insulin resistance in C2C12. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 1795–1802. [Google Scholar] [PubMed]
- Freitas-Rodriguez, S.; Rodriguez, F.; Folgueras, A.R. GDF11 administration does not extend lifespan in a mouse model of premature aging. Oncotarget 2016, 7, 55951–55956. [Google Scholar] [CrossRef] [PubMed]
- Duran, J.; Lagos, D.; Pavez, M.; Troncoso, M.F.; Ramos, S.; Barrientos, G.; Ibarra, C.; Lavandero, S.; Estrada, M. Ca(2+)/Calmodulin-Dependent Protein Kinase II and Androgen Signaling Pathways Modulate MEF2 Activity in Testosterone-Induced Cardiac Myocyte Hypertrophy. Front. Pharmacol. 2017, 8, 604. [Google Scholar] [CrossRef] [PubMed]
- Vicencio, J.M.; Ibarra, C.; Estrada, M.; Chiong, M.; Soto, D.; Parra, V.; Diaz-Araya, G.; Jaimovich, E.; Lavandero, S. Testosterone induces an intracellular calcium increase by a nongenomic mechanism in cultured rat cardiac myocytes. Endocrinology 2006, 147, 1386–1395. [Google Scholar] [CrossRef] [PubMed]
- Lin, A.H.; Luo, J.; Mondshein, L.H.; ten Dijke, P.; Vivien, D.; Contag, C.H.; Wyss-Coray, T. Global analysis of Smad2/3-dependent TGF-beta signaling in living mice reveals prominent tissue-specific responses to injury. J. Immunol. 2005, 175, 547–554. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Zawel, L.; Lengauer, C.; Kinzler, K.W.; Vogelstein, B. Characterization of human FAST-1, a TGF beta and activin signal transducer. Mol. Cell 1998, 2, 121–127. [Google Scholar] [CrossRef]





© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Duran, J.; Troncoso, M.F.; Lagos, D.; Ramos, S.; Marin, G.; Estrada, M. GDF11 Modulates Ca2+-Dependent Smad2/3 Signaling to Prevent Cardiomyocyte Hypertrophy. Int. J. Mol. Sci. 2018, 19, 1508. https://doi.org/10.3390/ijms19051508
Duran J, Troncoso MF, Lagos D, Ramos S, Marin G, Estrada M. GDF11 Modulates Ca2+-Dependent Smad2/3 Signaling to Prevent Cardiomyocyte Hypertrophy. International Journal of Molecular Sciences. 2018; 19(5):1508. https://doi.org/10.3390/ijms19051508
Chicago/Turabian StyleDuran, Javier, Mayarling Francisca Troncoso, Daniel Lagos, Sebastian Ramos, Gabriel Marin, and Manuel Estrada. 2018. "GDF11 Modulates Ca2+-Dependent Smad2/3 Signaling to Prevent Cardiomyocyte Hypertrophy" International Journal of Molecular Sciences 19, no. 5: 1508. https://doi.org/10.3390/ijms19051508