Increased Phenacetin Oxidation upon the L382V Substitution in Cytochrome P450 1A2 is Associated with Altered Substrate Binding Orientation

Abstract
:1. Introduction
2. Results
2.1. Purification of CYP1A2 Enzymes
2.2. Interactions of Phenacetin with CYP1A2 Enzymes
2.3. Determination of Spin State in CYP1A2 Enzymes
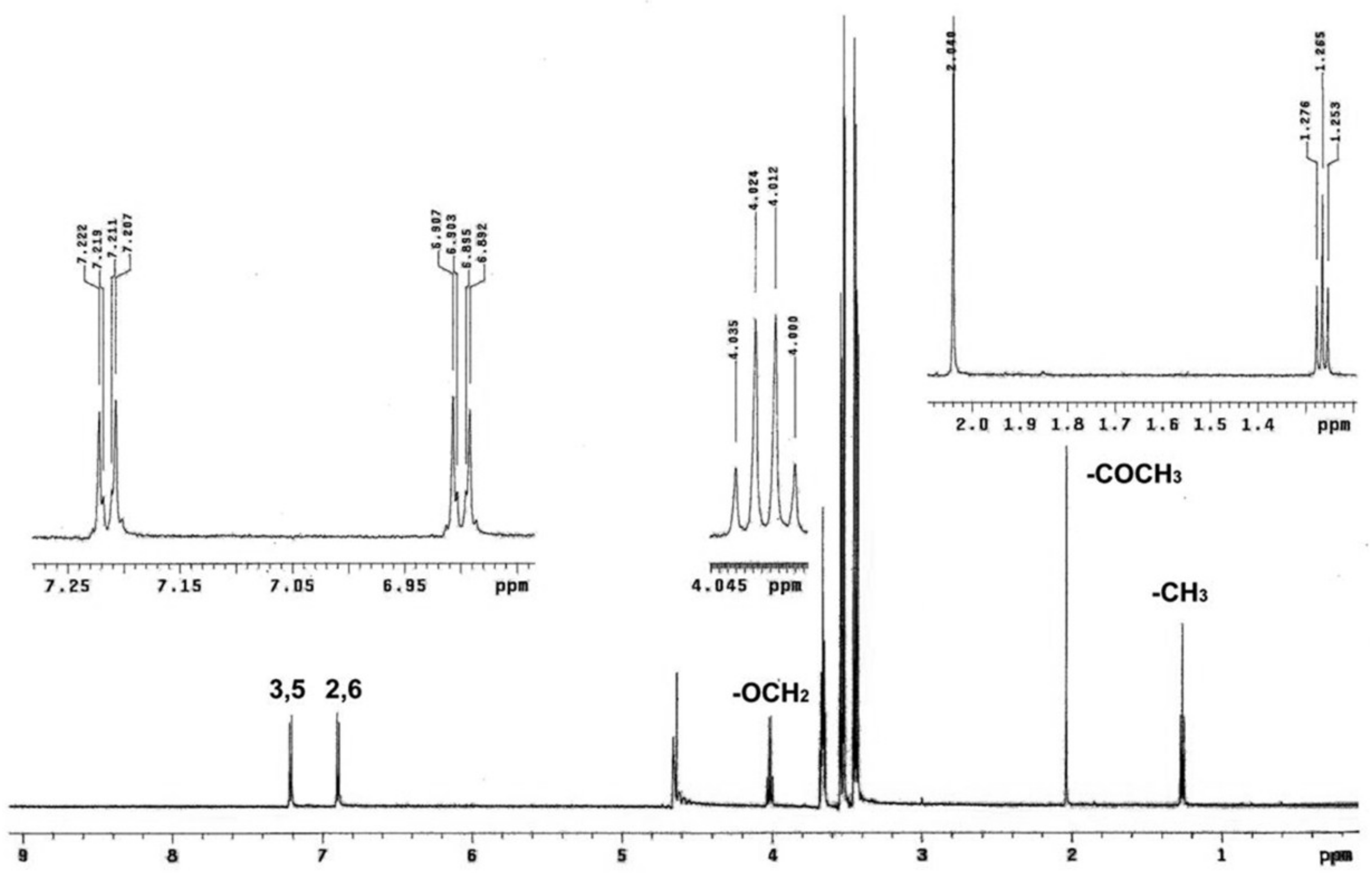
2.4. Nuclear Magnetic Resonance (NMR) T1 Experiments
2.5. Position of Phenacetin Relative to Heme in CYP1A2 Wild Type (WT) and Mutants
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Protein Expression and Purification
4.3. Binding Constant Determination
4.4. Spin State Determination
4.5. Enzyme Preparation for NMR
4.6. NMR Spectroscopy
4.7. Proton-Heme Iron Distance Calculations
4.8. Molecular Modeling: General
4.9. Substrate Docking with Distance Restraints
Author Contributions
Acknowledgments
Conflicts of Interest
Abbreviations
| CYP | Cytochrome P450 |
| WT | Wild Type |
| EDTA | Ethlenediaminetetraacetic Acid |
| 1H NMR | Proton Nuclear Magnetic Resonance |
References
- Guengerich, F.P. Mechanisms of cytochrome P450 substrate oxidation: Mini Review. J. Biochem. Mol. Toxicol. 2007, 21, 163–168. [Google Scholar] [CrossRef] [PubMed]
- Isin, E.M.; Guengerich, F.P. Substrate binding to cytochromes P450. Anal. Bioanal. Chem. 2008, 392, 1019–1030. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.F.; Chan, E.; Zhou, Z.W.; Xue, C.C.; Lai, X.; Duan, W. Insights into the structure, function, and regulation of human cytochrome P450 1A2. Curr. Drug Metab. 2009, 10, 1–17. [Google Scholar] [CrossRef]
- Distlerath, L.M.; Reilly, P.E.; Martin, M.V.; Davis, G.G.; Wilkinson, G.R.; Guengerich, F.P. Purification and characterization of the human liver cytochromes P-450 involved in debrisoquine 4-hydroxylation and phenacetin O-deethylation, two prototypes for genetic polymorphism in oxidative drug metabolism. J. Biol. Chem. 1985, 260, 9057–9067. [Google Scholar] [PubMed]
- Yuan, R.; Madani, S.; Wei, X.X.; Reynolds, K.; Huang, S.M. Evaluation of cytochrome P450 probe substrates commonly used by the pharmaceutical industry to study in vitro drug interactions. Drug Metab. Dispos. 2002, 30, 1311–1319. [Google Scholar] [CrossRef] [PubMed]
- Sansen, S.; Yano, J.K.; Reynald, R.L.; Schoch, G.A.; Griffin, K.J.; Stout, C.D.; Johnson, E.F. Adaptations for the oxidation of polycyclic aromatic hydrocarbons exhibited by the structure of human P450 1A2. J. Biol. Chem. 2007, 282, 14348–14355. [Google Scholar] [CrossRef] [PubMed]
- Yano, J.K.; Hsu, M.H.; Griffin, K.J.; Stout, C.D.; Johnson, E.F. Structures of human microsomal cytochrome P450 2A6 complexed with coumarin and methoxsalen. Nat. Struct. Mol. Biol. 2005, 12, 822–823. [Google Scholar] [CrossRef] [PubMed]
- Schoch, G.A.; Yano, J.K.; Wester, M.R.; Griffin, K.J.; Stout, C.D.; Johnson, E.F. Structure of human microsomal cytochrome P450 2C8. Evidence for a peripheral fatty acid binding site. J. Biol. Chem. 2004, 279, 9497–9503. [Google Scholar] [CrossRef] [PubMed]
- Williams, P.A.; Cosme, J.; Vinkovic, D.M.; Ward, A.; Angove, H.C.; Day, P.J.; Vonrhein, C.; Tickle, I.J.; Jhoti, H. Crystal structures of human cytochrome P450 3A4 bound to metyrapone and progesterone. Science 2004, 305, 683–686. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Ericksen, S.S.; Sivaneri, M.; Besspiata, D.; Fisher, C.W.; Szklarz, G.D. The effect of reciprocal active site mutations in human cytochromes P450 1A1 and 1A2 on alkoxyresorufin metabolism. Arch. Biochem. Biophys. 2004, 424, 33–43. [Google Scholar] [CrossRef] [PubMed]
- Tu, Y.; Deshmukh, R.; Sivaneri, M.; Szklarz, G.D. Application of molecular modeling for prediction of substrate specificity in cytochrome P450 1A2 mutants. Drug Metab. Dispos. 2008, 36, 2371–2380. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Szklarz, G.D. Significant increase in phenacetin oxidation upon Leu-382→Val substitution in human cytochrome P450 1A2. Drug Metab. Dispos. 2010, 38, 1039–1045. [Google Scholar] [CrossRef] [PubMed]
- Gay, S.C.; Roberts, A.G.; Halpert, J.R. Structural features of cytochromes P450 and ligands that affect drug metabolism as revealed by X-ray crystallography and NMR. Future Med. Chem. 2010, 2, 1451–1468. [Google Scholar] [CrossRef] [PubMed]
- Mak, P.J.; Denisov, I.G. Spectroscopic studies of the cytochrome P450 reaction mechanisms. Biochim. Biophys. Acta 2018, 1866, 178–204. [Google Scholar] [CrossRef] [PubMed]
- Regal, K.A.; Nelson, S.D. Orientation of caffeine within the active site of human cytochrome P450 1A2 based on NMR longitudinal (T1) relaxation measurements. Arch. Biochem. Biophys. 2000, 384, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Mildvan, A.S.; Gupta, R.K. Nuclear relaxation measurements of the geometry of enzyme-bound substrates and analogs. Methods Enzymol. 1978, 49, 322–359. [Google Scholar] [PubMed]
- Huang, Q.; Deshmukh, R.S.; Ericksen, S.S.; Tu, Y.; Szklarz, G.D. Preferred binding orientations of phenacetin in CYP 1A1 and 1A2 are associated with isoform-selective metabolism. Drug Metab. Dispos. 2012, 40, 2324–2331. [Google Scholar] [CrossRef] [PubMed]
- Roberts, A.G.; Sjögren, S.E.; Fomina, N.; Vu, K.T.; Almutairi, A.; Halpert, J.R. NMR-derived models of amidopyrine and its metabolites in complexes with rabbit cytochrome P450 2B4 reveal a structural mechanism of sequential N-dealkylation. Biochemistry 2011, 50, 2123–2134. [Google Scholar] [CrossRef] [PubMed]
- Dawson, J.H. Probing structure-function relations in heme-containing oxygenases and peroxidases. Science 1988, 240, 433–439. [Google Scholar] [CrossRef] [PubMed]
- Bertini, I.; Fragai, M.; Luchinat, C.; Parigi, G. Solvent (1)H NMRD study of hexaaquochromium(III): inferences on hydration and electron relaxation. Inorg. Chem. 2001, 40, 4030–4035. [Google Scholar] [CrossRef] [PubMed]
- Pintacuda, G.; Hohenthanner, K.; Otting, G.; Müller, N. Angular dependence of dipole-dipole-Curie-spin cross-correlation effects in high-spin and low-spin paramagnetic myoglobin. J. Biomol. NMR 2003, 27, 115–132. [Google Scholar] [CrossRef] [PubMed]
- Modi, S.; Primrose, W.U.; Boyle, J.M.; Gibson, C.F.; Lian, L.Y.; Roberts, G.C. NMR studies of substrate binding to cytochrome P450 BM3: Comparisons to cytochrome P450 cam. Biochemistry 1995, 34, 8982–8988. [Google Scholar] [CrossRef] [PubMed]
- Modi, S.; Paine, M.J.; Sutcliffe, M.J.; Lian, L.Y.; Primrose, W.U.; Wolf, C.R.; Roberts, G.C. A model for human cytochrome P450 2D6 based on homology modeling and NMR studies of substrate binding. Biochemistry 1996, 35, 4540–4550. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Poulos, T.L. The structure of the cytochrome P450BM3 heam domain complexed with the fatty acid substrate, palmitoleic acid. Nat. Struct. Biol. 1997, 4, 140–146. [Google Scholar] [CrossRef] [PubMed]
- Poli-Scaife, S.; Attias, R.; Dansette, P.M.; Mansuy, D. The substrate binding site of human liver cytochrome P450 2C9: An NMR study. Biochemistry 1997, 36, 12672–12682. [Google Scholar] [CrossRef] [PubMed]
- Estrada, D.F. The cytochrome P450 24A1 interaction with adrenodoxin relies on multiple recognition sites that vary among species. J. Biol. Chem. 2018, 293, 4167–4179. [Google Scholar] [CrossRef] [PubMed]
- Lelo, A.; Miners, J.O.; Robson, R.A.; Birkett, D.J. Quantitative assessment of caffeine partial clearances in man. Br. J. Clin. Pharmacol. 1986, 22, 183–186. [Google Scholar] [CrossRef] [PubMed]
- Gu, L.; Gonzalez, F.J.; Kalow, W.; Tang, B.K. Biotransformation of caffeine, paraxanthine, theobromine and theophylline by cDNA-expressed human CYP1A2 and CYP2E1. Pharmacogenetics 1992, 2, 73–77. [Google Scholar] [CrossRef] [PubMed]
- Hummel, M.A.; Gannett, P.M.; Aguilar, J.S.; Tracy, T.S. Substrate proton to heme distances in CYP2C9 allelic variants and alterations by the heterotropic activator, dapsone. Arch. Biochem. Biophys. 2008, 475, 175–183. [Google Scholar] [CrossRef] [PubMed]
- Hummel, M.A.; Gannett, P.M.; Aguilar, J.S.; Tracy, T.S. Effector-mediated alteration of substrate orientation in cytochrome P450 2C9. Biochemistry 2004, 43, 7207–7214. [Google Scholar] [CrossRef] [PubMed]
- Schoch, G.A.; Attias, R.; Belghazi, M.; Dansette, P.M.; Werck-Reichhart, D. Engineering of a water-soluble plant cytochrome P450, CYP73A1, and NMR-based orientation of natural and alternate substrates in the active site. Plant Physiol. 2003, 133, 1198–1208. [Google Scholar] [CrossRef] [PubMed]
- Locuson, C.W.; Hutzler, J.M.; Tracy, T.S. Visible spectra of type II cytochrome P450-drug complexes: Evidence that “incomplete” heme coordination is common. Drug Metab. Dispos. 2007, 35, 614–622. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Ericksen, S.S.; Besspiata, D.; Fisher, C.W.; Szklarz, G.D. Characterization of substrate binding to cytochrome P450 1A1 using molecular modeling and kinetic analyses: Case of residue 382. Drug Metab. Dispos. 2003, 31, 412–420. [Google Scholar] [CrossRef] [PubMed]
- Jefcoate, C.R. Measurement of substrate and inhibitor binding to microsomal cytochrome P-450 by optical-difference spectroscopy. Methods Enzymol. 1978, 52, 258–279. [Google Scholar] [PubMed]
- Paine, M.J.; Gilham, D.; Roberts, G.C.; Wolf, C.R. Functional high level expression of cytochrome P450 CYP2D6 using baculoviral expression systems. Arch. Biochem. Biophys. 1996, 328, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Paulsen, M.D.; Ornstein, R.L. A 175-psec molecular dynamics simulation of camphorbound cytochrome P-450cam. Proteins 1991, 11, 184–204. [Google Scholar] [CrossRef] [PubMed]
- Paulsen, M.D.; Ornstein, R.L. Predicting the product specificity and coupling of cytochrome P450cam. J. Comput. Aided Mol. Des. 1992, 6, 449–460. [Google Scholar] [CrossRef] [PubMed]
- Ericksen, S.S.; Szklarz, G.D. Regiospecificity of human cytochrome P450 1A1-mediated oxidations: The role of steric effects. J. Biomol. Struct. Dyn. 2005, 23, 243–256. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Enzyme | Spectrum Type () | Ks |
|---|---|---|
| (nm) | (μM) | |
| CYP1A2 WT | I (390–420) | 17.1 ± 0.6 |
| CYP1A2 N312L | I (390–420) | 10.2 ± 0.4 |
| CYP1A2 L382V | I (390–420) | 0.7 ± 0.1 |
| CYP1A2 L382V/N312L | I (390–420) | 3.5 ± 0.2 |
| Enzyme (+Substrate) | Low Spin | High Spin |
|---|---|---|
| % | ||
| CYP1A2 WT (no substrate) | 90 | 10 |
| +Phenacetin | 73 | 27 |
| CYP1A2 N312L (no substrate) | 94 | 6 |
| +Phenacetin | 86 | 14 |
| CYP1A2 L382V (no substrate) | 95 | 5 |
| +Phenacetin | 79 | 21 |
| CYP1A2 L382V/N312L (no substrate) | 94 | 6 |
| +Phenacetin | 83 | 17 |
| Proton a | CYP1A2 WT b | CYP1A2 L382V c | ||||||
|---|---|---|---|---|---|---|---|---|
| T1,Fe3+ | T1,Fe2+-CO | r f | R g | T1,Fe3+ | T1,Fe2+-CO | r f | R g | |
| Å | Å | |||||||
| 2,6 | 2.22 (0.11) | 2.78 (0.05) | 6.76 (0.34) | 8.25 | 2.28 (0.11) | 3.06 (0.04) | 6.34 (0.31) | 7.96 |
| 3.5 | 2.02 (0.04) | 2.50 (0.01) | 6.72 (0.43) | 8.32 | 2.39 (0.22) | 3.21 (0.08) | 6.39 (0.28) | 7.87 |
| –OCH2– | 1.54 (0.15) | 1.85 (0.18) | 6.55 (0.87) | 8.01 | 2.43 (0.18) | 4.10 (0.07) | 5.93 (0.31) | 6.95 |
| –COCH3 | 1.53 (0.07) | 1.62 (0.06) | 7.85 (0.81) | 9.44 | 1.64 (0.13) | 1.87 (0.11) | 6.79 (0.71) | 8.05 |
| –CH3 | 1.33 (0.07) | 1.53 (0.06) | 6.69 (0.45) | 8.64 | 2.03 (0.13) | 2.82 (0.11) | 6.13 (0.89) | 7.58 |
| Proton a | CYP1A2 N312L d | CYP1A2 L382V/N312Le | ||||||
| T1,Fe3+ | T1,Fe2+-CO | R f (Å) | R g(Å) | T1,Fe3+ | T1,Fe2+-CO | r f (Å) | R g (Å) | |
| Å | Å | |||||||
| 2,6 | 2.32 (0.10) | 2.45 (0.10) | 8.03 (0.42) | 9.11 | 2.43 (0.07) | 3.00 (0.06) | 6.72 (0.31) | 7.28 |
| 3.5 | 1.94 (0.09) | 2.04 (0.10) | 7.94 (0.43) | 9.38 | 2.95 (0.09) | 3.70 (0.11) | 6.87 (0.47) | 7.25 |
| –OCH2– | 1.62 (0.04) | 1.69 (0.04) | 7.84 (0.77) | 8.67 | 1.98 (0.12) | 2.90 (0.09) | 5.96 (0.35) | 7.24 |
| –COCH3 | 1.54 (0.12) | 1.60 (0.11) | 8.19 (1.12) | 9.34 | 2.12 (0.14) | 2.59 (0.12) | 6.62 (0.15) | 8.01 |
| –CH3 | 1.87 (0.12) | 1.97 (0.07) | 7.83 (0.52) | 8.94 | 2.23 (0.09) | 2.61 (0.11) | 6.92 (0.34) | 7.47 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, Q.; Szklarz, G.D. Increased Phenacetin Oxidation upon the L382V Substitution in Cytochrome P450 1A2 is Associated with Altered Substrate Binding Orientation. Int. J. Mol. Sci. 2018, 19, 1580. https://doi.org/10.3390/ijms19061580
Huang Q, Szklarz GD. Increased Phenacetin Oxidation upon the L382V Substitution in Cytochrome P450 1A2 is Associated with Altered Substrate Binding Orientation. International Journal of Molecular Sciences. 2018; 19(6):1580. https://doi.org/10.3390/ijms19061580
Chicago/Turabian StyleHuang, Qingbiao, and Grazyna D. Szklarz. 2018. "Increased Phenacetin Oxidation upon the L382V Substitution in Cytochrome P450 1A2 is Associated with Altered Substrate Binding Orientation" International Journal of Molecular Sciences 19, no. 6: 1580. https://doi.org/10.3390/ijms19061580