DNA Damage-Response Pathway Heterogeneity of Human Lung Cancer A549 and H1299 Cells Determines Sensitivity to 8-Chloro-Adenosine


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
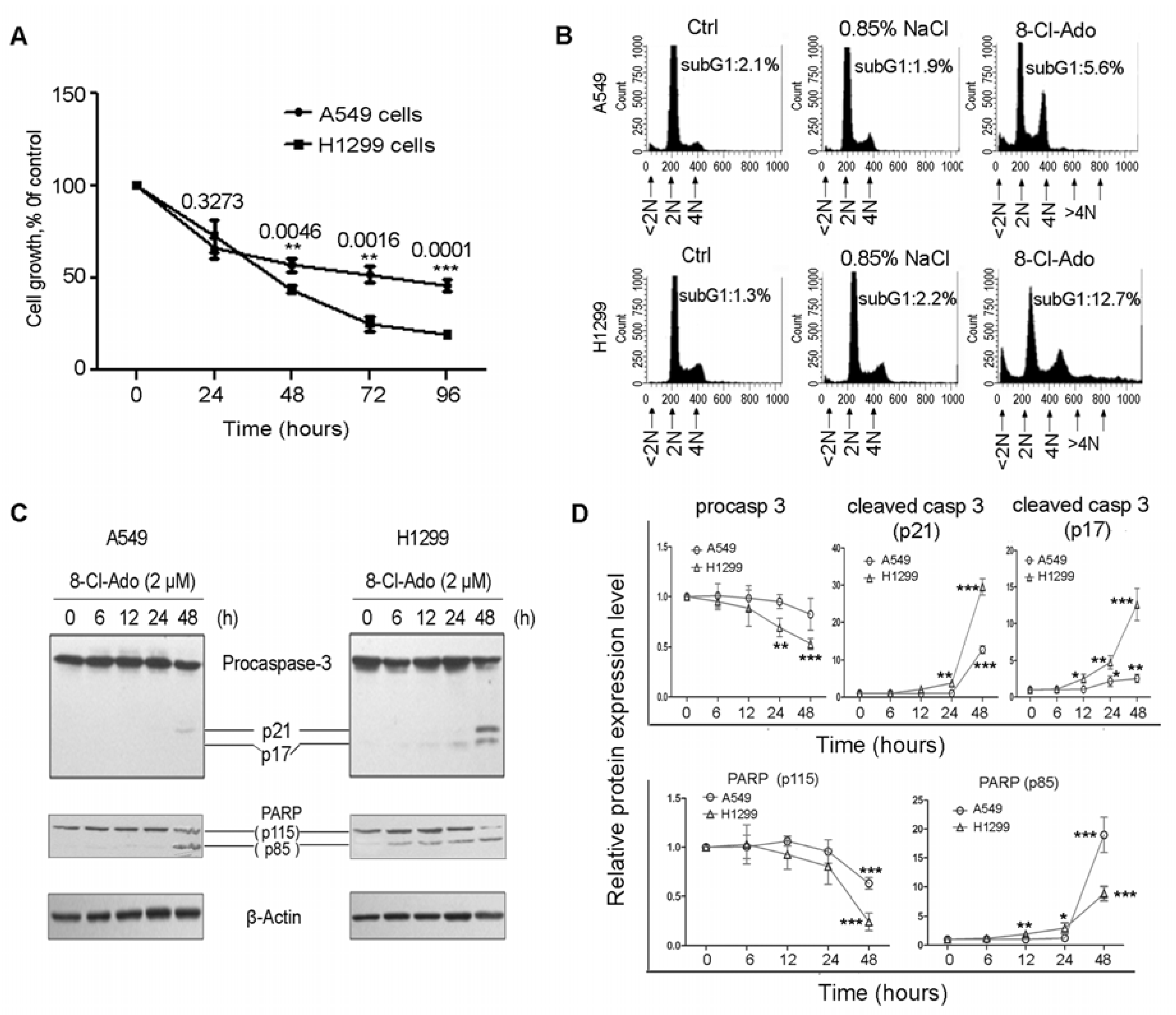
2.1. H1299 Cells Are More Sensitive to 8-Cl-Ado-Induced Growth Inhibition and Apoptosis than A549 Cells
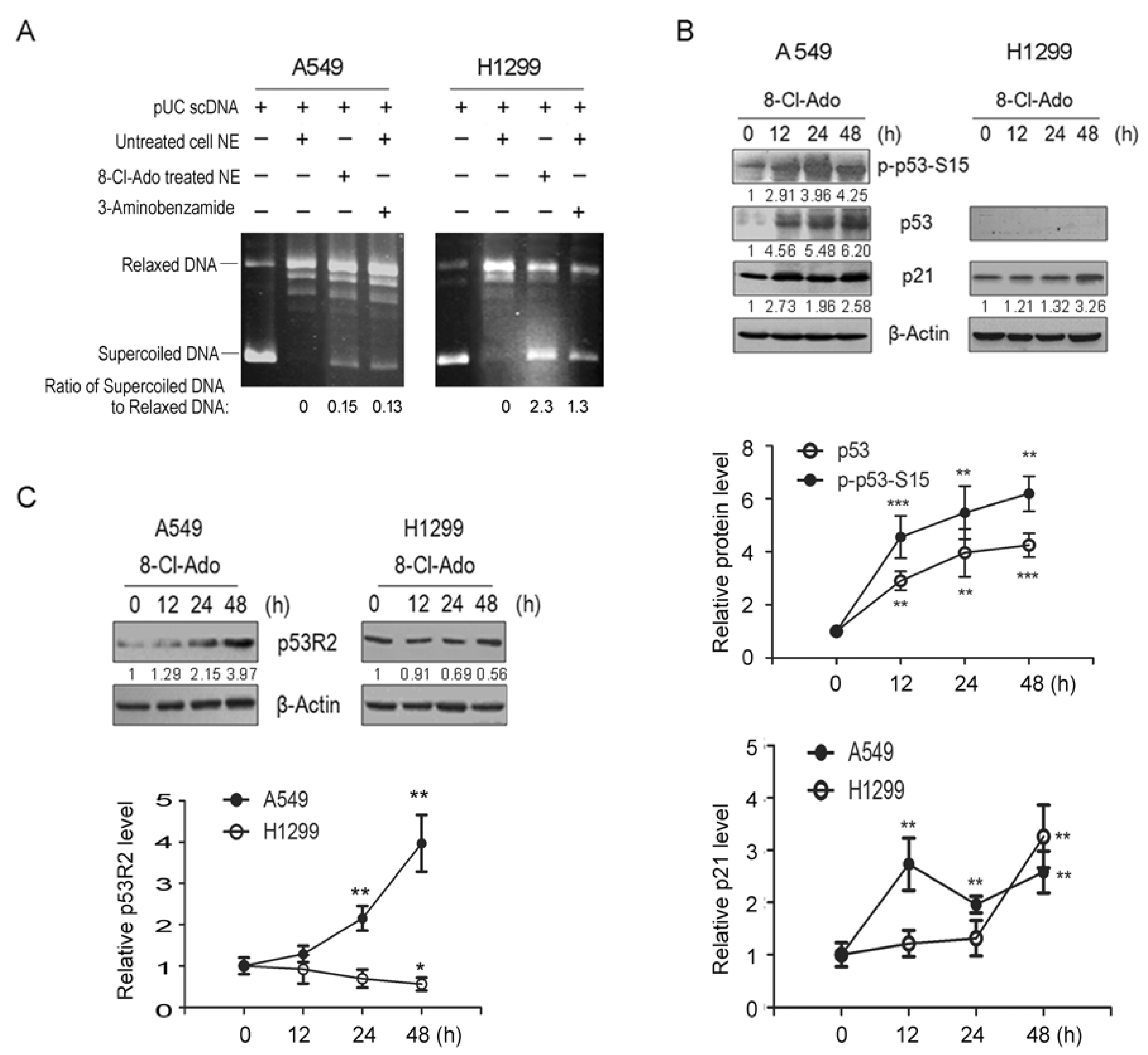
2.2. 8-Cl-Ado Diminishes PARP-1-Associated TOPO I Activity and p53R2 Expression in H1299 Cells More Greatly than A549 Cells
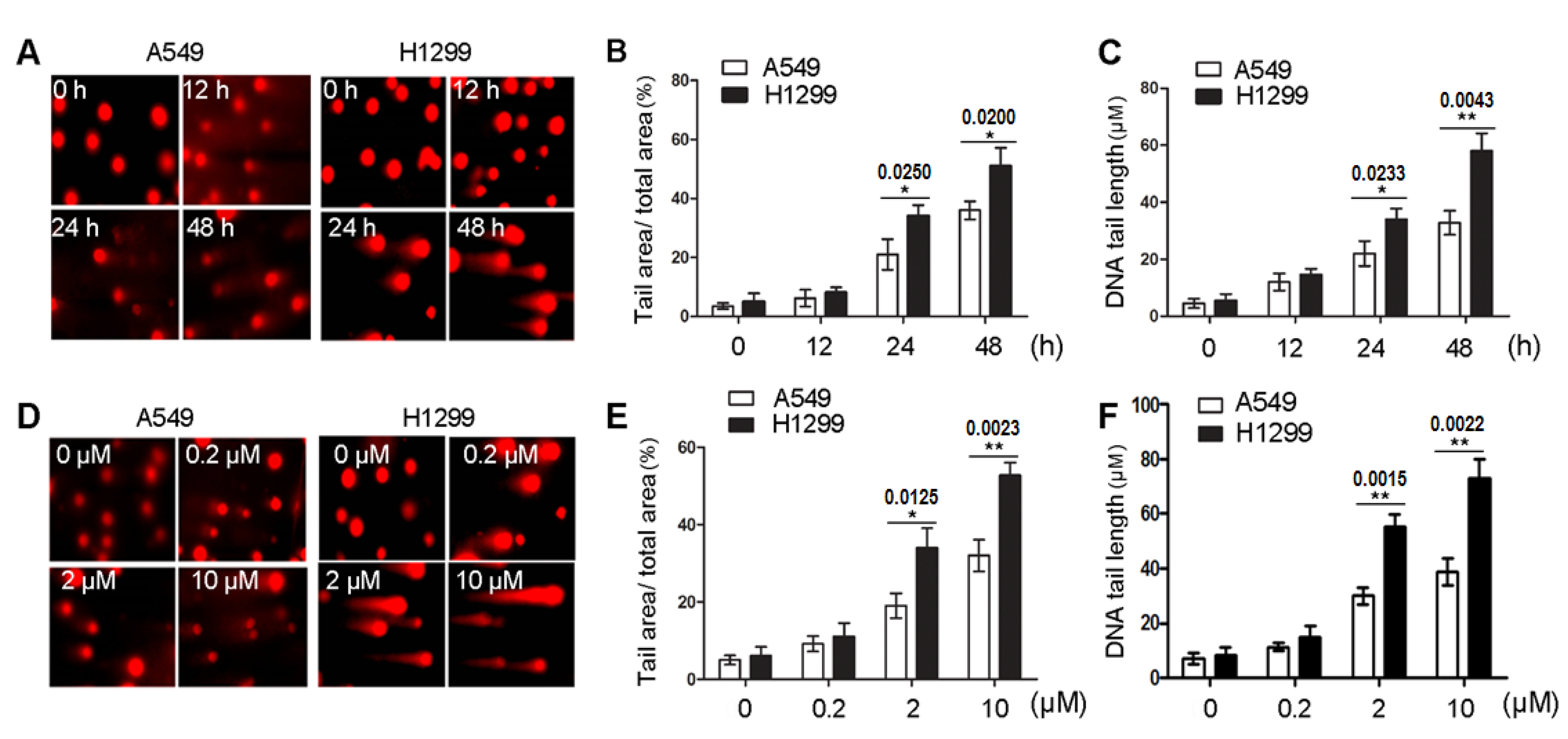
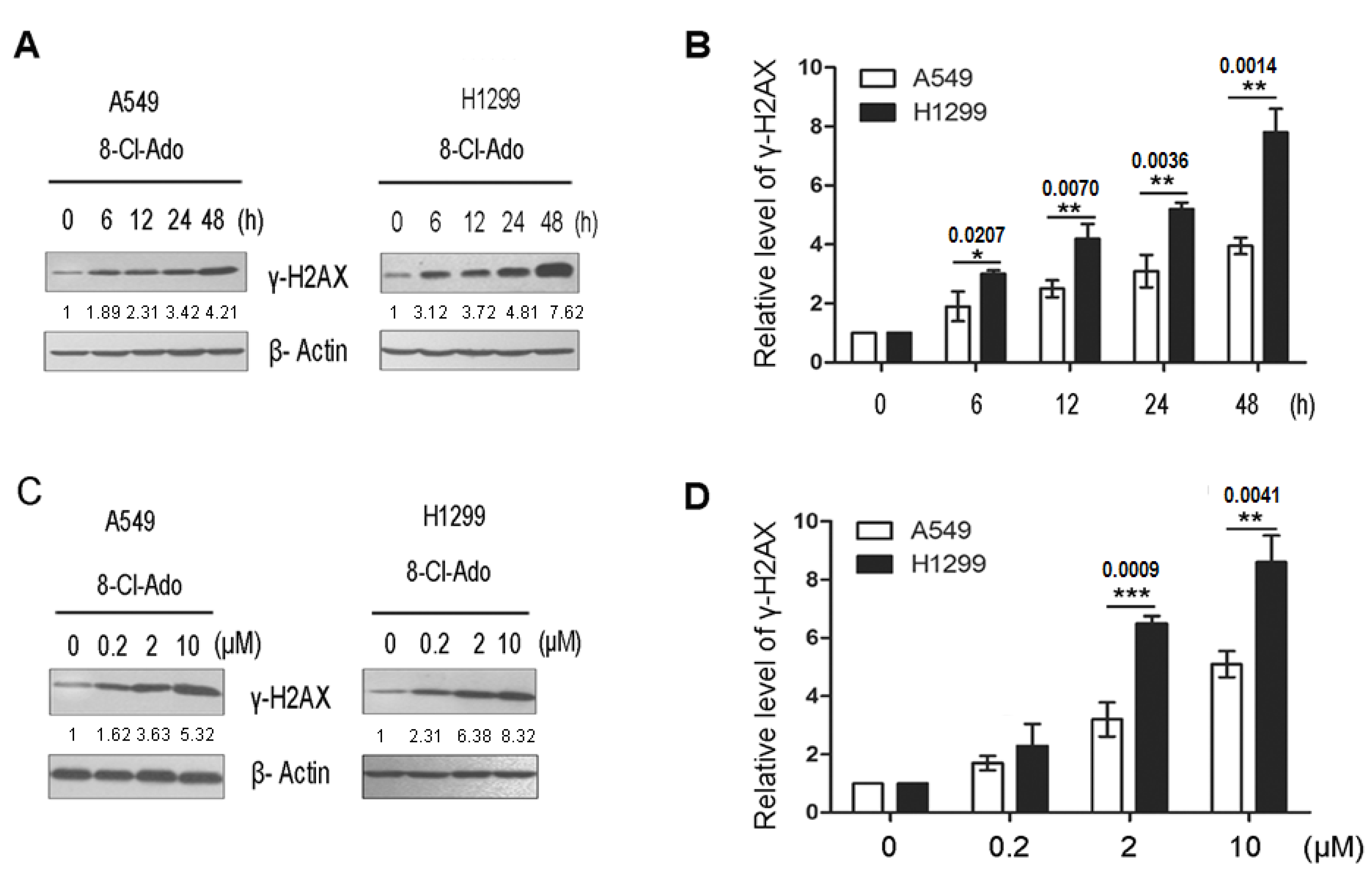
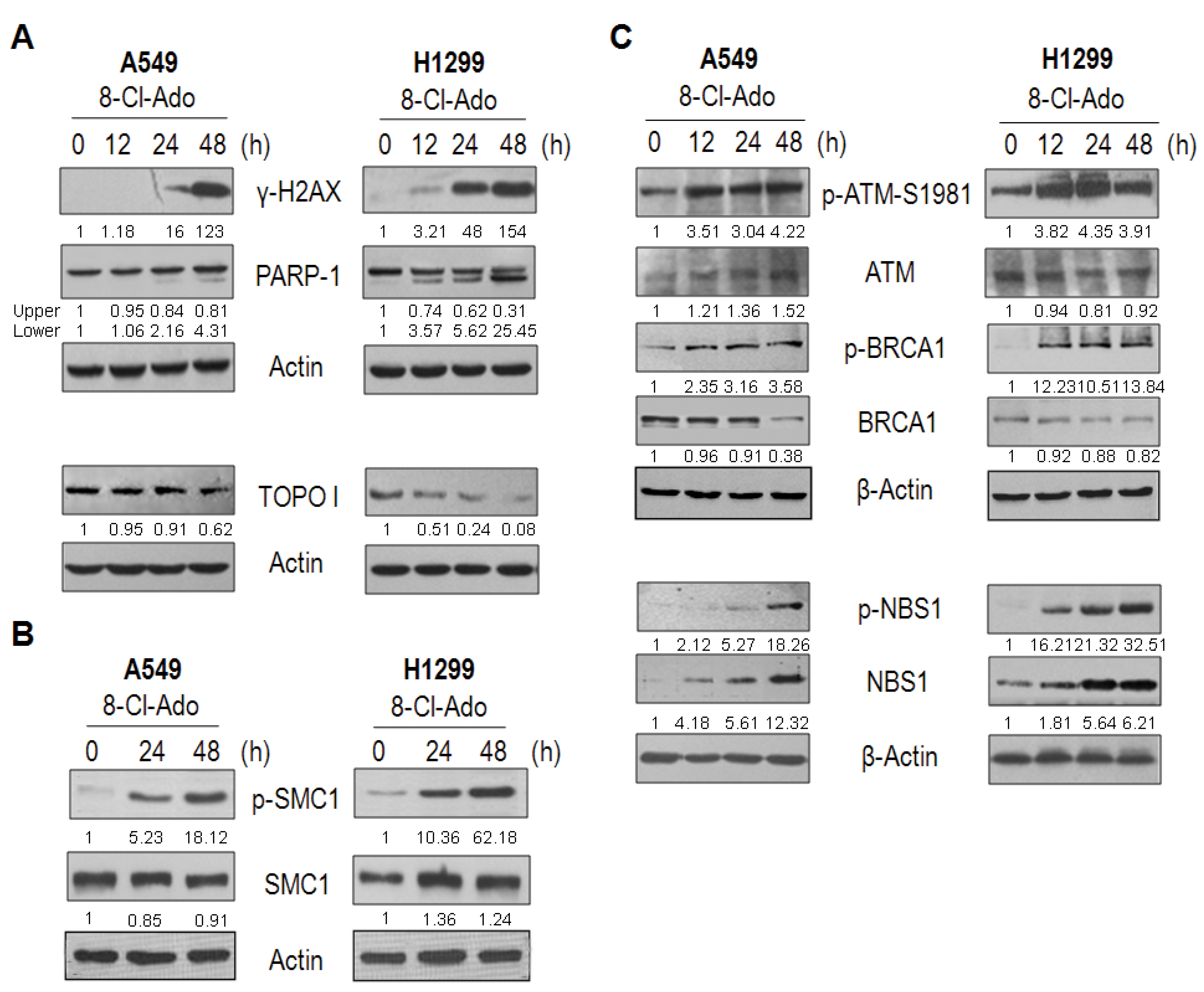
2.3. 8-Cl-Ado Induces More Accumulation of DSBs in H1299 Cells than in A549 Cells
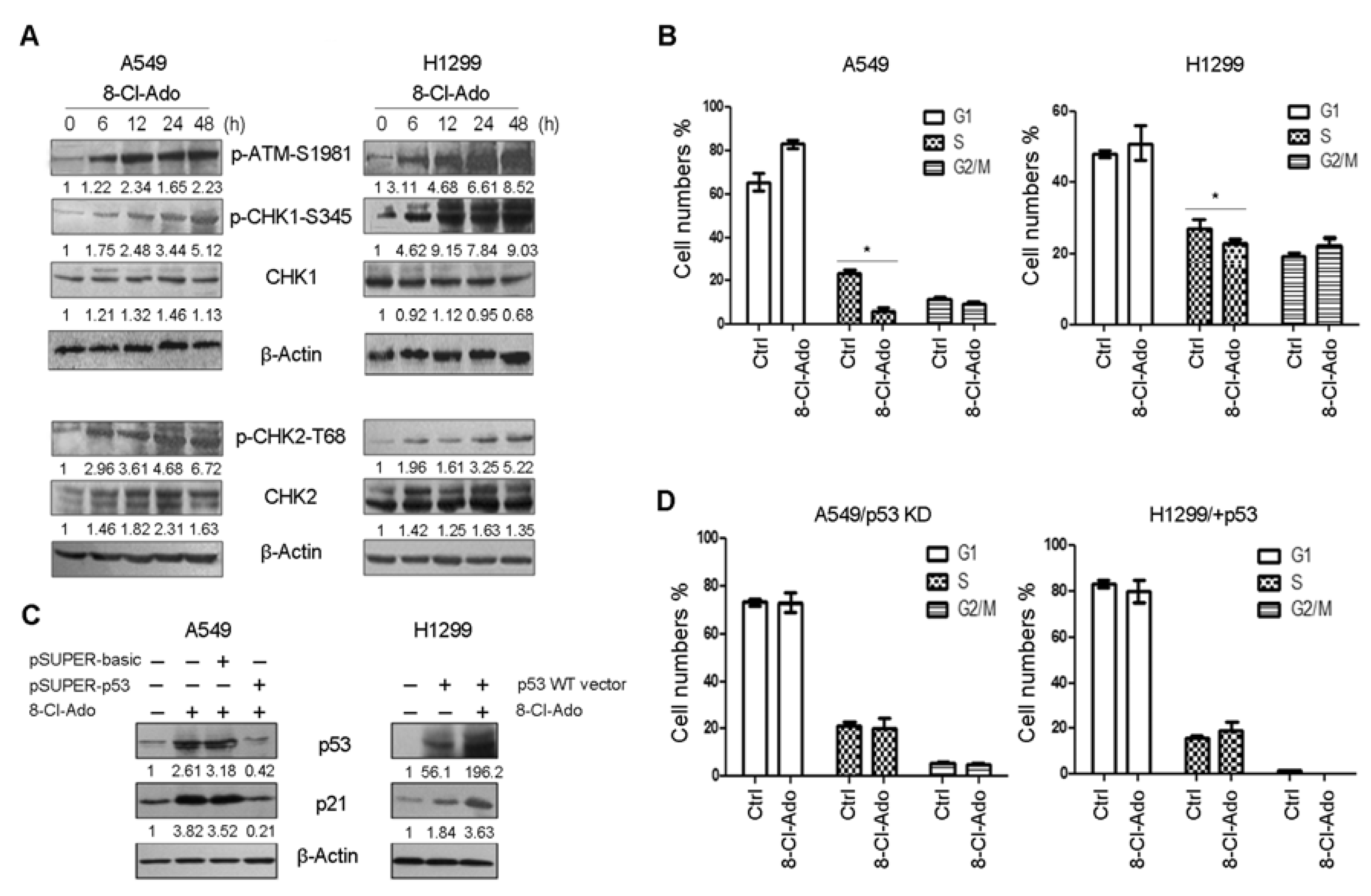
2.4. Defect in p53-p21 Signal in H1299 Cells Leads to Increased S Subpopulation upon DSBs
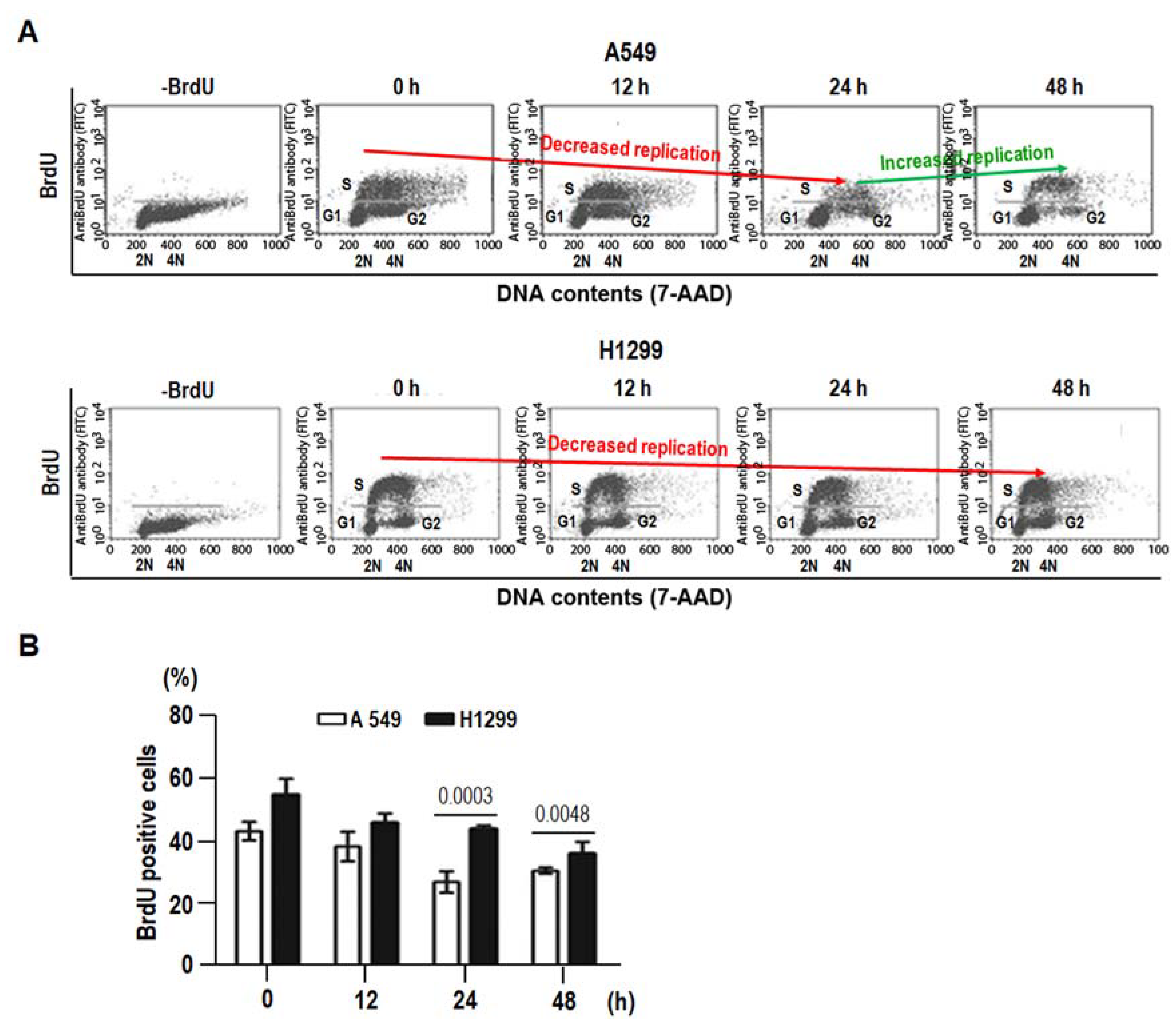
2.5. 8-Cl-Ado-Induced More Accumulation of DSBs in H1299 Is Associated with DNA Replication in S Phase
2.6. DNA Damage Response Proteins Are Time-Differentially Mobilized in H1299 and A549 Cells during DSBs
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Treatment
4.2. Cell Proliferation Assay
4.3. Flow Cytometry Analysis
4.4. DNA Relaxation
4.5. Comet Assay
4.6. Constructs and Transfection
4.7. Western Blotting
4.8. BrdU Incorporation Assay
4.9. Statistical Analysis
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Hoeijmakers, J.H. Genome maintenance mechanisms for preventing cancer. Nature 2001, 411, 366–374. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef] [PubMed]
- Basu, A.K. DNA Damage, Mutagenesis and Cancer. Int. J. Mol. Sci. 2018, 19, 970. [Google Scholar] [CrossRef] [PubMed]
- Helleday, T.; Lo, J.; van Gent, D.C.; Engelward, B.P. DNA double-strand break repair: From mechanistic understanding to cancer treatment. DNA Rep. 2007, 6, 923–935. [Google Scholar] [CrossRef] [PubMed]
- Polo, S.E.; Jackson, S.P. Dynamics of DNA damage response proteins at DNA breaks: A focus on protein modifications. Genes Dev. 2011, 25, 409–433. [Google Scholar] [CrossRef] [PubMed]
- Meek, D.W. The p53 response to DNA damage. DNA Rep. 2004, 3, 1049–1056. [Google Scholar] [CrossRef] [PubMed]
- Niculescu, A.B., 3rd; Chen, X.; Smeets, M.; Hengst, L.; Prives, C.; Reed, S.I. Effects of p21(Cip1/Waf1) at both the G1/S and the G2/M cell cycle transitions: pRb is a critical determinant in blocking DNA replication and in preventing endoreduplication. Mol. Cell. Biol. 1998, 18, 629–643. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, H.; Arakawa, H.; Yamaguchi, T.; Shiraishi, K.; Fukuda, S.; Matsui, K.; Takei, Y.; Nakamura, Y. A ribonucleotide reductase gene involved in a p53-dependent cell-cycle checkpoint for DNA damage. Nature 2000, 404, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Taylor, W.R.; Stark, G.R. Regulation of the G2/M transition by p53. Oncogene 2001, 20, 1803–1815. [Google Scholar] [CrossRef] [PubMed]
- Petermann, E.; Keil, C.; Oei, S.L. Importance of poly(ADP-ribose) polymerases in the regulation of DNA-dependent processes. Cell. Mol. Life Sci. 2005, 62, 731–738. [Google Scholar] [CrossRef] [PubMed]
- Yung, T.M.; Parent, M.; Ho, E.L.; Satoh, M.S. Camptothecin-sensitive relaxation of supercoiled DNA by the topoisomerase I-like activity associated with poly(ADP-ribose) polymerase-1. J. Biol. Chem. 2004, 279, 11992–11999. [Google Scholar] [CrossRef] [PubMed]
- Jensen, L.H.; Dejligbjerg, M.; Hansen, L.T.; Grauslund, M.; Jensen, P.B.; Sehested, M. Characterisation of cytotoxicity and DNA damage induced by the topoisomerase II-directed bisdioxopiperazine anti-cancer agent ICRF-187 (dexrazoxane) in yeast and mammalian cells. BMC Pharmacol. 2004, 4, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radhakrishna Pillai, G.; Srivastava, A.S.; Hassanein, T.I.; Chauhan, D.P.; Carrier, E. Induction of apoptosis in human lung cancer cells by curcumin. Cancer Lett. 2004, 208, 163–170. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.Y.; Gu, Y.Y.; Li, Z.G.; Jia, Y.H.; Yuan, L.; Li, S.Y.; An, G.S.; Ni, J.H.; Jia, H.T. Exposure of human lung cancer cells to 8-chloro-adenosine induces G2/M arrest and mitotic catastrophe. Neoplasia 2004, 6, 802–812. [Google Scholar] [CrossRef] [PubMed]
- Langeveld, C.H.; Jongenelen, C.A.; Theeuwes, J.W.; Baak, J.P.; Heimans, J.J.; Stoof, J.C.; Peters, G.J. The antiproliferative effect of 8-chloro-adenosine, an active metabolite of 8-chloro-cyclic adenosine monophosphate, and disturbances in nucleic acid synthesis and cell cycle kinetics. Biochem. Pharmacol. 1997, 53, 141–148. [Google Scholar] [CrossRef]
- Gandhi, V.; Ayres, M.; Halgren, R.G.; Krett, N.L.; Newman, R.A.; Rosen, S.T. 8-chloro-cAMP and 8-chloro-adenosine act by the same mechanism in multiple myeloma cells. Cancer Res. 2001, 61, 5474–5479. [Google Scholar] [PubMed]
- Dennison, J.B.; Balakrishnan, K.; Gandhi, V. Preclinical activity of 8-chloroadenosine with mantle cell lymphoma: Roles of energy depletion and inhibition of DNA and RNA synthesis. Br. J. Haematol. 2009, 147, 297–307. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.Y.; Jia, X.Z.; Feng, L.Y.; Li, S.Y.; An, G.S.; Ni, J.H.; Jia, H.T. Inhibition of topoisomerase II by 8-chloro-adenosine triphosphate induces DNA double-stranded breaks in 8-chloro-adenosine-exposed human myelocytic leukemia K562 cells. Biochem. Pharmacol. 2009, 77, 433–443. [Google Scholar] [CrossRef] [PubMed]
- Bauer, P.I.; Buki, K.G.; Comstock, J.A.; Kun, E. Activation of topoisomerase I by poly [ADP-ribose] polymerase. Int. J. Mol. Med. 2000, 5, 533–540. [Google Scholar] [CrossRef] [PubMed]
- Tomicic, M.T.; Kaina, B. Topoisomerase degradation, DSB repair, p53 and IAPs in cancer cell resistance to camptothecin-like topoisomerase I inhibitors. Biochim. Biophys. Acta 2013, 1835, 11–27. [Google Scholar] [CrossRef] [PubMed]
- Banerji, C.R.; Severini, S.; Caldas, C.; Teschendorff, A.E. Intra-tumour signalling entropy determines clinical outcome in breast and lung cancer. PLoS Comp. Biol. 2015, 11, e1004115. [Google Scholar] [CrossRef] [PubMed]
- Mirzayans, R.; Andrais, B.; Kumar, P.; Murray, D. Significance of wild-type p53 signaling in suppressing apoptosis in response to chemical genotoxic agents: Impact on chemotherapy outcome. Int. J. Mol. Sci. 2017, 18, 928. [Google Scholar] [CrossRef] [PubMed]
- Merritt, A.J.; Allen, T.D.; Potten, C.S.; Hickman, J.A. Apoptosis in small intestinal epithelial from p53-null mice: Evidence for a delayed, p53-independent G2/M-associated cell death after gamma-irradiation. Oncogene 1997, 14, 2759–2766. [Google Scholar] [CrossRef] [PubMed]
- Han, J.W.; Dionne, C.A.; Kedersha, N.L.; Goldmacher, V.S. p53 status affects the rate of the onset but not the overall extent of doxorubicin-induced cell death in rat-1 fibroblasts constitutively expressing c-Myc. Cancer Res. 1997, 57, 176–182. [Google Scholar] [PubMed]
- Tannock, I.F.; Lee, C. Evidence against apoptosis as a major mechanism for reproductive cell death following treatment of cell lines with anti-cancer drugs. Br. J. Cancer 2001, 84, 100–105. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.J.; Cai, Y.J.; Ding, J. Curcumin induces DNA damage and caffeine-insensitive cell cycle arrest in colorectal carcinoma HCT116 cells. Mol. Cell. Biochem. 2011, 354, 247–252. [Google Scholar] [CrossRef] [PubMed]
- Levine, A.J. p53, the cellular gatekeeper for growth and division. Cell 1997, 88, 323–331. [Google Scholar] [CrossRef]
- Frouin, I.; Maga, G.; Denegri, M.; Riva, F.; Savio, M.; Spadari, S.; Prosperi, E.; Scovassi, A.I. Human proliferating cell nuclear antigen, poly(ADP-ribose) polymerase-1, and p21waf1/cip1. A dynamic exchange of partners. J. Biol. Chem. 2003, 278, 39265–39268. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.T.; Xu, B.; Kastan, M.B. Involvement of the cohesin protein, Smc1, in Atm-dependent and independent responses to DNA damage. Genes Dev. 2002, 16, 560–570. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, W.K. The human intra-S checkpoint response to UVC-induced DNA damage. Carcinogenesis 2010, 31, 751–765. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, S.; Ballif, B.A.; Smogorzewska, A.; McDonald, E.R., 3rd; Hurov, K.E.; Luo, J.; Bakalarski, C.E.; Zhao, Z.; Solimini, N.; Lerenthal, Y.; et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 2007, 316, 1160–1166. [Google Scholar] [CrossRef] [PubMed]
- Gebow, D.; Miselis, N.; Liber, H.L. Homologous and nonhomologous recombination resulting in deletion: Effects of p53 status, microhomology, and repetitive DNA length and orientation. Mol. Cell. Biol. 2000, 20, 4028–4035. [Google Scholar] [CrossRef] [PubMed]
- MacLachlan, T.K.; Takimoto, R.; El-Deiry, W.S. BRCA1 directs a selective p53-dependent transcriptional response towards growth arrest and DNA repair targets. Mol. Cell. Biol. 2002, 22, 4280–4292. [Google Scholar] [CrossRef] [PubMed]
- Roninson, I.B. Oncogenic functions of tumour suppressor p21(Waf1/Cip1/Sdi1): Association with cell senescence and tumour-promoting activities of stromal fibroblasts. Cancer Lett. 2002, 179, 1–14. [Google Scholar] [CrossRef]
- La Thangue, N.B. The yin and yang of E2F-1: Balancing life and death. Nat. Cell Biol. 2003, 5, 587–589. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.Q.; An, G.S.; Ni, J.H.; Li, S.Y.; Jia, H.T. ATM-dependent E2F1 accumulation in the nucleolus is an indicator of ribosomal stress in early response to DNA damage. Cell Cycle 2014, 13, 1627–1638. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.X.; Li, S.Y.; An, G.S.; Mao, Z.B.; Jia, H.T.; Ni, J.H. E2F1-regulated DROSHA promotes miR-630 biosynthesis in cisplatin-exposed cancer cells. Biochem. Biophys. Res. Commun. 2014, 450, 470–475. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, D.J.; Lam, E.W.; Griffin, S.; Zhong, S.; Leighton, L.C.; Burbidge, S.A.; Lu, X. Physical and functional interactions between p53 and cell cycle co-operating transcription factors, E2F1 and DP1. EMBO J. 1995, 14, 6184–6192. [Google Scholar] [PubMed]
- Audebert, M.; Salles, B.; Calsou, P. Involvement of poly(ADP-ribose) polymerase-1 and XRCC1/DNA ligase III in an alternative route for DNA double-strand breaks rejoining. J. Biol. Chem. 2004, 279, 55117–55126. [Google Scholar] [CrossRef] [PubMed]
- D’Amours, D.; Sallmann, F.R.; Dixit, V.M.; Poirier, G.G. Gain-of-function of poly(ADP-ribose) polymerase-1 upon cleavage by apoptotic proteases: Implications for apoptosis. J. Cell Sci. 2001, 114, 3771–3778. [Google Scholar] [PubMed]
- Haince, J.F.; McDonald, D.; Rodrigue, A.; Dery, U.; Masson, J.Y.; Hendzel, M.J.; Poirier, G.G. PARP1-dependent kinetics of recruitment of MRE11 and NBS1 proteins to multiple DNA damage sites. J. Biol. Chem. 2008, 283, 1197–1208. [Google Scholar] [CrossRef] [PubMed]
- Malanga, M.; Althaus, F.R. Poly(ADP-ribose) reactivates stalled DNA topoisomerase I and Induces DNA strand break resealing. J. Biol. Chem. 2004, 279, 5244–5248. [Google Scholar] [CrossRef] [PubMed]
- Harris, S.; Levine, A. The p53 pathway: Positive and negative feedback loops. Oncogene 2005, 24, 2899–2908. [Google Scholar] [CrossRef] [PubMed]
- Tarasov, V.; Jung, P.; Verdoodt, B.; Lodygin, D.; Epanchintsev, A. Differential regulation of microRNAs by p53 revealed by massively parallel sequencing: miR-34a is a p53 target that induces apoptosis and G1-arrest. Cell Cycle 2007, 6, 1586–1593. [Google Scholar] [CrossRef] [PubMed]
- Issler, M.V.C.; Mombach, J.C.M. MicroRNA-16 feedback loop with p53 and Wip1 can regulate cell fate determination between apoptosis and senescence in DNA damage response. PLoS ONE 2017, 12, e0185794. [Google Scholar] [CrossRef] [PubMed]
- Anderson, D.; Yu, T.W.; Phillips, B.J.; Schmezer, P. The effect of various antioxidants and other modifying agents on oxygen-radical-generated DNA damage in human lymphocytes in the COMET assay. Mutat. Res. 1994, 307, 261–271. [Google Scholar] [CrossRef]







© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, S.-Y.; Li, Y.; An, G.-S.; Ni, J.-H.; Jia, H.-T.; Li, S.-Y. DNA Damage-Response Pathway Heterogeneity of Human Lung Cancer A549 and H1299 Cells Determines Sensitivity to 8-Chloro-Adenosine. Int. J. Mol. Sci. 2018, 19, 1587. https://doi.org/10.3390/ijms19061587
Yang S-Y, Li Y, An G-S, Ni J-H, Jia H-T, Li S-Y. DNA Damage-Response Pathway Heterogeneity of Human Lung Cancer A549 and H1299 Cells Determines Sensitivity to 8-Chloro-Adenosine. International Journal of Molecular Sciences. 2018; 19(6):1587. https://doi.org/10.3390/ijms19061587
Chicago/Turabian StyleYang, Sheng-Yong, Yi Li, Guo-Shun An, Ju-Hua Ni, Hong-Ti Jia, and Shu-Yan Li. 2018. "DNA Damage-Response Pathway Heterogeneity of Human Lung Cancer A549 and H1299 Cells Determines Sensitivity to 8-Chloro-Adenosine" International Journal of Molecular Sciences 19, no. 6: 1587. https://doi.org/10.3390/ijms19061587