Palladacyclic Conjugate Group Promotes Hybridization of Short Oligonucleotides


Department of Chemistry, University of Turku, Vatselankatu 2, 20014 Turku, Finland
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2018, 19(6), 1588; https://doi.org/10.3390/ijms19061588
Submission received: 18 May 2018
/
Revised: 25 May 2018
/
Accepted: 26 May 2018
/
Published: 28 May 2018
(This article belongs to the Special Issue A Commemorative Issue in Honor of Professor Nick Hadjiliadis: Metal Complex Interactions with Nucleic Acids and/or DNA)
Abstract
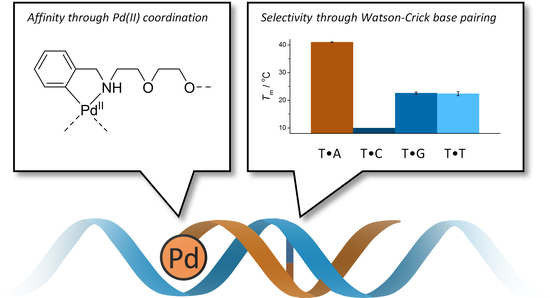
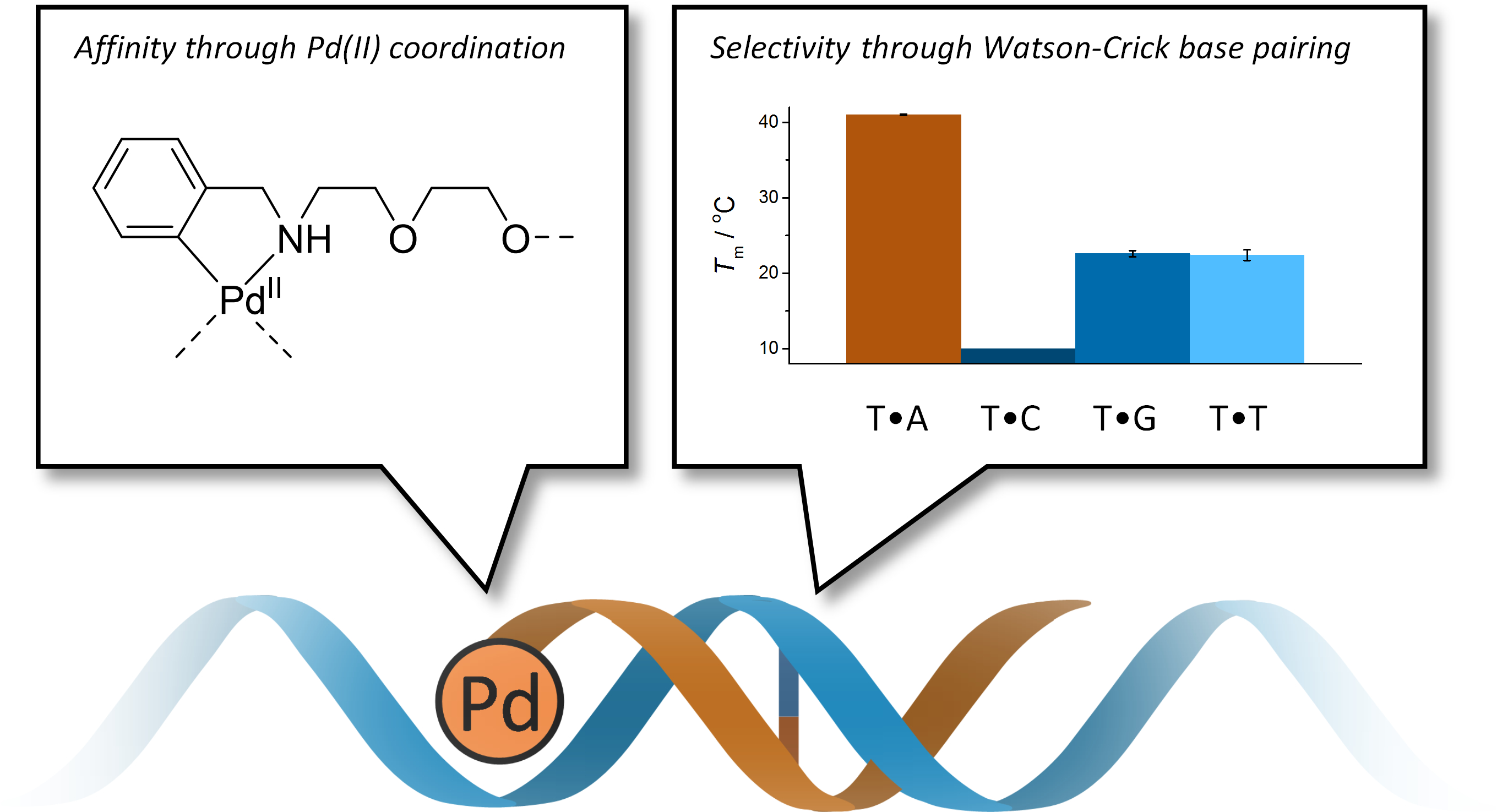
:Short oligonucleotides with cyclopalladated benzylamine moieties at their 5′-termini have been prepared to test the possibility of conferring palladacyclic anticancer agents sequence-selectivity by conjugation with a guiding oligonucleotide. Hybridization of these oligonucleotides with natural counterparts was studied by UV and CD (circular dichroism) melting experiments in the absence and presence of a competing ligand (2-mercaptoethanol). Cyclopalladated benzylamine proved to be strongly stabilizing relative to unmetalated benzylamine and modestly stabilizing relative to an extra A•T base pair. The stabilization was largely abolished in the presence of 2-mercaptoethanol, suggesting direct coordination of Pd(II) to a nucleobase of the complementary strand. In all cases, fidelity of Watson-Crick base pairing between the two strands was retained. Hybridization of the cyclopalladated oligonucleotides was characterized by relatively large negative enthalpy and entropy, consistent with stabilizing Pd(II) coordination partially offset by the entropic penalty of imposing conformational constraints on the flexible diethylene glycol linker between the oligonucleotide and the palladacyclic moiety.
1. Introduction
The groundbreaking discovery of the antitumor activity of cisplatin [1,2] has been followed by efforts to develop more potent anticancer agents based on transition metal complexes [3,4,5,6,7,8,9,10,11]. In particular, problems associated with the presently available platinum anticancer compounds, notably acquired or intrinsic resistance, limited spectrum of activity and relatively high degree of toxicity [12,13,14], have prompted interest in transition metals other than platinum for chemotherapeutic use [4,6,7,8,15]. Palladium is an attractive candidate because its coordination chemistry is similar to that of platinum [16,17]. Pd(II) complexes are, however, kinetically approximately five orders of magnitude more labile than the respective Pt(II) complexes [18]. While the relatively rapid ligand-exchange of Pd(II) should allow formation of thermodynamic (rather than kinetic) products and thus higher selectivity than attainable with Pt(II)-based drugs, it is also likely to result in a different mode of action, at least with simple analogues [19]. No clinically approved palladium-containing drugs are presently available.
The possibility of using palladacyclic complexes as anticancer agents to circumvent the problems caused by the kinetic lability of Pd(II) has received attention over the past decade. The high stability of palladacyclic compounds in physiological media and the resultant low toxicity to normal cells make them promising candidates for future therapeutic agents [9,20]. The selectivity of these agents could be further improved by conjugation to a guiding oligonucleotide. The feasibility of this approach has already been demonstrated with a number of Pt(II)-carrying DNA and PNA oligonucleotides [21,22,23,24,25,26] but with palladacyclic modifications we are only aware of a single recent example, a short DNA oligonucleotide incorporating a single cyclopalladated phenylpyridine residue in the middle of the sequence [27]. In that case, coordination of Pd(II) to the opposite base on a complementary strand was inferred from the abnormally high UV and CD (circular dichroism) signals but the expected stabilization of the double helix by such coordination could not be demonstrated unambiguously. Possibly a stable Pd(II)-mediated base pair was formed but could not be readily accommodated within the base stack, leading to disruption of the double helix.
Herein we describe the synthesis and hybridization properties of short oligonucleotides incorporating cyclopalladated benzylamine “warheads” at their 5′-termini. At monomer level, palladacyclic benzylamine derivatives have already been found to exhibit antitumor activity [28,29]. The 5′-terminal position was chosen to avoid disruption of the double helix by suboptimal coordination geometry. For the same reason, a relatively long and flexible diethylene glycol spacer was used between the cyclopalladated benzylamine and the oligonucleotide.
2. Results
2.1. Synthesis of the Benzylamine Phosphoramidite Building Block
Synthesis of the protected phosphoramidite building block of benzylamine (1) is outlined in Scheme 1. First, benzyl bromide was allowed to react with an excess of 2-(2-aminoethoxy)ethanol to give 2-[2-(benzylamino)ethoxy]ethanol (2). The secondary amino function was then protected as a trifluoroacetamide by treatment with ethyl trifluoroacetate. Finally, the protected intermediate 3 was phosphitylated by conventional methods to afford the phosphoramidite building block 1.
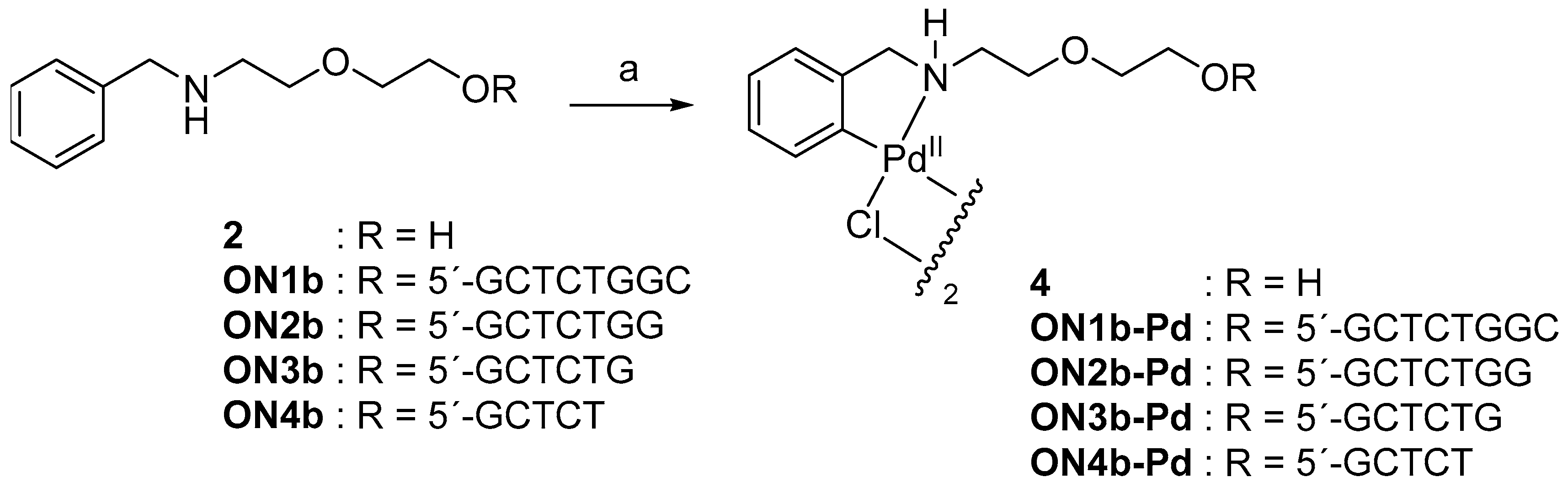
2.2. Cyclopalladation of 2-[2-(benzylamino)ethoxy]ethanol
Cyclopalladation was first tested at monomer level with 2-[2-(benzylamino)ethoxy]ethanol (2) by treatment with an equimolar amount of lithium tetrachloropalladate in a mixture of water and acetonitrile. Near-quantitative conversion of the starting material was achieved overnight. 1H NMR (nuclear magnetic resonance) spectrum of the product revealed loss of one ortho proton of the phenyl ring and 13C NMR spectrum a downfield shift of the respective carbon signal. Both results are consistent with replacement of the ortho proton with Pd(II). The most likely structure of the product is the chlorido-bridged dimer 4 (Scheme 2), as reported previously on related compounds [30,31,32]. While only mononuclear Pd(II) species could be unambiguously identified in the mass spectrum, the splitting of several peaks in both 1H and 13C NMR is consistent with formation of a dimer, present in both cisoid and transoid forms.
2.3. Oligonucleotide Synthesis
The sequences of the oligonucleotides used in the present study are summarised in Table 1. Synthesis of the modified oligonucleotides ON1b, ON2b, ON3b and ON4b, having a 5′-terminal benzylamine moiety, was carried out on an automated DNA synthesizer using conventional phosphoramidite strategy. Treatment with concentrated aq. ammonia was employed for removal of the base and phosphate protections and release of the oligonucleotides from the solid support. Cyclopalladation of oligonucleotides ON1b, ON2b, ON3b and ON4b was carried out as described above for the monomer 2 (Scheme 2), except that 2.0 equivalents of lithium tetrachloropalladate was used. All modified oligonucleotides were purified by reversed-phase high performance liquid chromatography (RP-HPLC), characterized by electrospray ionization mass spectrometry (ESI-MS) and quantified by UV spectrophotometry.
2.4. Hybridization Studies
The impact of the 5′-terminal palladacyclic “warheads” on the hybridization properties of short oligonucleotides was assessed by recording melting temperatures of duplexes formed by oligonucleotides ON1b-Pd, ON2b-Pd, ON3b-Pd and ON4b-Pd with the natural counterparts ON2a, ON2c, ON2g and ON2t. For reference, similar experiments were also carried out on respective duplexes formed by oligonucleotides ON1b, ON2b, ON3b, ON4b, ON1a, ON2a, ON3a and ON4a, having either an unmetalated benzylamine or an adenine residue at their 5′-termini. In all assemblies, the 5′-terminal residue was placed opposite to a thymine residue of a trinucleotide overhang of the complementary oligonucleotide (Figure 1). A single base pair within the double helical region, on the other hand, was varied to test the sensitivity of hybridization to a single-nucleotide mismatch. All experiments were performed at pH 7.4 (20 mM cacodylate buffer) and ionic strength of 0.10 M (adjusted with sodium perchlorate) and each sample was first annealed by heating to 90 °C and then slowly cooling down to room temperature.
The longest matched duplexes ON1x•ON5a all exhibited sigmoidal melting profiles, with Tm (melting temperature) values ranging from 36 to 41 °C (Figure 2A–C). The shorter duplexes did not fully hybridize even at the lowest temperature applicable (10 °C) but their Tm values could still be determined with reasonable accuracy as inflection points of the melting curves. The melting temperatures of the mismatched duplexes, on the other hand, were high enough to be determined reliably only in the case of the longest duplexes ON1x•ON5y. The A•C mismatch was particularly destabilizing and precluded determination of the Tm in all cases, regardless of the length of the duplex. Melting temperatures are summarized in Figure 2D for the longest duplexes and in the Supplementary Materials for all duplexes.
Melting temperatures of the longest matched duplexes ON1a•ON5a, ON1b•ON5a and ON1b-Pd•ON5a, were 40.4 ± 0.7 °C, 35.8 ± 0.6 °C and 41.0 ± 0.6 °C, respectively. In other words, the cyclopalladated benzylamine residue was modestly stabilizing relative to an adenine residue and strongly stabilizing relative to the unmetalated benzylamine residue. To explore the origin of this stabilization, the UV melting experiments were repeated in the presence of 2-mercaptoethanol (100 µM). 2-Mercaptoethanol is a strong ligand for soft transition metal ions and would, hence, be expected to disrupt coordination of Pd(II) to nucleobases. If such coordination is important for duplex stability, a decrease in Tm on addition of 2-mercaptoethanol should be observed.
Melting temperatures of the longest matching duplexes ON1x•ON5a in the absence and presence of 2-mercaptoethanol are presented in Figure 3 (all melting temperatures are presented in the Supporting Information). As expected, melting temperatures of duplexes ON1a•ON5a and ON1b•ON5a did not change appreciably on addition of 2-mercaptoethanol. With ON1b-Pd•ON5a, on the other hand, a clear drop in Tm was observed, consistent with stabilizing coordination of Pd(II) in the absence of competing ligands.
To further elucidate the role of the palladacyclic “warhead”, a detailed thermodynamic analysis of the hybridization of ON1a•ON5a, ON1b•ON5a and ON1b-Pd•ON5a was carried out as described in the literature [33]. The resultant enthalpies and entropies of hybridization are presented in Table 2. With ON1b-Pd•ON5a, both values were significantly more negative than with ON1a•ON5a or ON1b•ON5a.
Secondary structure of duplexes formed by the cyclopalladated oligonucleotides was studied CD spectropolarimetrically over a temperature range of 10–90 °C at 10 °C intervals. All the other conditions were identical to those of the UV melting experiments. With the longest matched duplexes, the spectra obtained at 10 °C were clearly characteristic of a B-type double helix, with prominent negative and positive signals at 260 and 280 nm, respectively (Figure 4 for ON1b-Pd•ON5a, all spectra are presented in the Supplementary Materials). Similar, but weaker, signals were observed with the shorter and/or mismatched duplexes, reflecting their lower melting temperatures. In all cases, the signals diminished on increasing temperature, consistent with unwinding of the double helix.
3. Discussion
3.1. Duplex Stabilization by the Palladacyclic “Warhead”
The sensitivity of the duplex stabilization by the cyclopalladated benzylamine moiety to the presence of 2-mercaptoethanol suggests direct coordination of Pd(II) to a base moiety of the complementary strand. The thymine base directly opposite to the palladacyclic residue appears as the most likely candidate but, given the length and flexibility of the diethylene glycol linker, coordination to other bases of the 3′-overhang cannot be ruled out. The most likely donor atoms within these bases are the N3 of cytosine and thymine and the N1 of guanine [34,35,36].
Duplex stabilization by the cyclopalladated benzylamine moiety is rather modest when compared to stabilizations achieved previously with metal mediated base pairing [37,38,39,40,41,42,43,44,45,46]. One should, however, bear in mind that within a double helix the most stable metal mediated base pairs are formed between two nucleosides or nucleoside analogues, preorganized to place the donor atoms at appropriate positions. In the present case the flexible diethylene glycol linker first has to adopt a conformation conducive to Pd(II) coordination, consistent with the observed highly negative entropy of hybridization. As a result, the stabilizing enthalpic contribution by Pd(II) coordination is almost entirely offset by the entropic penalty.
3.2. Impact of the Palladacyclic “Warhead” on Sequence Selectivity
Highly stabilizing modifications are known to be able to override sequence information and allow hybridization of even highly mismatched oligonucleotides [47]. Even in the present case, the mismatched duplexes were actually stabilized more than the matched ones by the cyclopalladated benzylamine moiety. However, the difference in Tm between the matched and the most stable mismatched duplex (ON1b-Pd•ON5a and ON1b-Pd•ON5g, respectively) was still nearly 18 °C, translating into a 104-fold preference of ON5a over ON5g in hybridization with ON1b-Pd.
4. Materials and Methods
4.1. General Methods
NMR spectra were recorded on Bruker 500 NMR spectrometers (Bruker, Billerica, MA, USA) and chemical shifts (δ, ppm) are quoted relative to the residual solvent peak as an internal standard. Mass spectra were recorded on a Bruker Daltonics microTOF-Q mass spectrometer (Bruker, Billerica, MA, USA). The solvents for organic synthesis were of reagent grade and dried over 4 Å molecular sieves. For preparation of HPLC elution buffers, freshly distilled triethylamine was used. The other chemicals, including unmodified oligonucleotides, were commercial products that were used as received.
4.2. N-benzyl-2,2,2-trifluoro-N-[2-(2-hydroxyethoxy)ethyl]acetamide (3)
2-[2-(benzylamino)ethoxy]ethanol (2, 2.00 g, 10.2 mmol) was dissolved in MeOH (10 mL). Et3N (2.90 mL, 20.4 mmol) was added and the resulting mixture stirred for 30 min at 25 °C. Ethyl trifluoroacetate (1.46 g, 10.2 mmol) was then added and the reaction mixture stirred for 16 h at 25 °C, after which it was evaporated to dryness to afford the desired product 3 as a mixture of two slowly interconverting rotamers (2.98 g, near quantitative yield). 1H NMR (CDCl3, 500 MHz, major rotamer): δ 7.19-7.48 (m, 5H), 4.80 (s, 2H), 3.40-3.70 (m, 8H), 2.82 (br, 1H). 1H NMR (CDCl3, 500 MHz, minor rotamer): δ 7.19-7.48 (m, 5H), 4.82 (s, 2H), 3.40-3.70 (m, 8H), 2.82 (br, 1H). 13C NMR (CDCl3, 125 MHz, major rotamer): δ 157.1 (q, J = 35.8 Hz), 136.3, 128.7, 127.7, 127.6, 116.8 (q, J = 285.9 Hz), 72.5, 67.6, 60.9, 50.3, 46.8 (q, J = 2.9 Hz). (CDCl3, 125 MHz, minor rotamer): δ 156.9 (q, J = 35.8 Hz), 135.8, 128.8, 127.9, 127.2, 116.8 (q, J = 285.9 Hz), 72.4, 68.9, 61.0, 51.4 (q, J = 3.0 Hz), 46.0. HRMS (ESI+) m/z calcd 314.0974 obsd 314.0972 [M + Na]+.
4.3. 2-[2-(N-benzyl-2,2,2-trifluoroacetamido)ethoxy]ethyl 2-cyanoethyl N,N-diisopropylphosphoramidite (1)
N-benzyl-2,2,2-trifluoro-N-(2-(2-hydroxyethoxy)ethyl)acetamide (3, 670 mg, 2.30 mmol) and Et3N (1.93 mL, 13.8 mmol) were dissolved in anhydrous CH2Cl2 (7 mL) under N2 atmosphere. 2-Cyanoethyl N,N-diisopropylchlorophosphoramidite (0.616 mL, 2.76 mmol) was added and the resulting mixture stirred for 3 h at 25 °C, after which the reaction was quenched by addition of saturated aq. NaHCO3 (100 mL). The phases were separated and the aqueous phase was extracted with CH2Cl2 (100 mL). The combined organic phases were washed with saturated aq. NaHCO3 (100 mL), dried over anhydrous Na2SO4 and evaporated to dryness. The crude product thus obtained was passed through a silica gel column eluting with a mixture of EtOAc and hexane (1:1, v/v) to afford the desired product 1 containing a major impurity of 2-Cyanoethyl N,N-diisopropylphosphonamidate (859 mg, 72% yield based on 31P NMR). This material was used in oligonucleotide synthesis without further purification. 1H NMR (CDCl3, 500 MHz, major rotamer): δ 7.22-7.48 (m, 5H), 4.80 (s, 2H), 3.36-3.89 (m, 12H), 2.66 (m, 2H), 1.20 (d, J = 6.8 Hz, 6H), 1.19 (d, J = 6.8 Hz, 6H). 1H NMR (CDCl3, 500 MHz, minor rotamer): δ 7.22-7.48 (m, 5H), 4.82 (s, 2H), 3.36-3.89 (m, 12H), 2.66 (m, 2H), 1.20 (d, J = 6.8 Hz, 6H), 1.19 (d, J = 6.8 Hz, 6H). 13C NMR (CDCl3, 125 MHz, major rotamer): δ 157.1 (q, J = 36.0 Hz), 136.3, 128.7, 127.7, 127.6, 118.6, 116.8 (q, J = 288.2 Hz), 71.1 (d, J = 7.6 Hz), 67.8, 62.6 (d, J = 16.8 Hz), 58.4 (d, J = 19.2 Hz), 50.3, 46.7 (q, J = 2.9 Hz), 42.9 (d, J = 12.6 Hz), 24.0 (d, J = 7.2 Hz), 20.0. 13C NMR (CDCl3, 125 MHz, minor rotamer): δ 156.7 (q, J = 35.4 Hz), 135.7, 129.0, 127.9, 127.2, 118.0, 116.8 (q, J = 288.7 Hz), 71.0 (d, J = 7.7 Hz), 69.1, 62.7 (d, J = 17.1 Hz), 58.4 (d, J = 19.2 Hz), 51.5 (q, J = 3.2 Hz), 45.9, 42.9 (d, J = 12.6 Hz), 24.0 (d, J = 7.2 Hz), 20.0 (d, J = 6.9 Hz). 31P NMR (CDCl3, 202 MHz, major rotamer): δ 148.1. 31P NMR (CDCl3, 202 MHz, minor rotamer): δ 148.0. HRMS (ESI+) m/z calcd 514.2053 obsd 514.2034 [M + Na]+.
4.4. Bis{N-[2-(2-hydroxyethoxy)ethyl]benzylaminato-C2,N} bis(µ-chloro) dipalladium(II) (4)
2-[2-(benzylamino)ethoxy]ethanol (2, 240 mg, 1.28 mmol) was dissolved in a mixture of H2O (3 mL) and MeCN (3 mL). Li2PdCl4 (336 mg, 1.28 mmol) was dissolved in a mixture of H2O (10 mL) and MeCN (10 mL) and the resulting solution was added to the solution of 2. After stirring for 16 h at 25 °C, the reaction mixture was evaporated to dryness. The residue was purified by silica gel column chromatography eluting with a mixture of EtOAc and hexane (8:2, v/v), affording the desired product 4 as a mixture of cisoid and transoid stereoisomers (189 mg, 44% yield). 1H NMR (CDCl3, 500 MHz, major stereoisomer): δ 7.34-7.47 (m, 4H), 4.46 (ddd, J1 = 10.6 Hz, J2 = 10.4 Hz, J3 = 2.5 Hz, 1H), 4.28 (dd, J1 = 13.1 Hz, J2 = 6.7 Hz, 1H), 4.03 (m, 1H), 3.44-3.80 (m, 6 H), 3.02 (m, 1H), 2.38 (m, 1H). 1H NMR (CDCl3, 500 MHz, minor stereoisomer): δ 7.34-7.47 (m, 4H), 4.33 (m, 1H), 4.24 (dd, J1 = 13.0 Hz, J2 = 7.6 Hz, 1H), 4.03 (m, 1H), 3.44-3.80 (m, 6 H), 3.02 (m, 1H), 2.38 (m, 1H). 13C NMR (CDCl3, 125 MHz, major stereoisomer): δ 147.5, 135.0, 129.9, 129.7, 128.9, 128.6, 72.8, 67.8, 61.6, 57.35, 50.8. 13C NMR (CDCl3, 125 MHz, minor stereoisomer): δ 142.7, 135.2, 129.9, 129.7, 128.9, 128.6, 72.8, 67.6, 61.4, 57.26, 51.5. HRMS (ESI+) m/z calcd 300.0216 obsd 300.0152 [M/2 − Cl]+.
4.5. Oligonucleotide Synthesis
The modified oligonucleotides ON1b, ON2b, ON3b and ON4b were assembled on an Applied Biosystems 3400 (Applied Biosystems, Waltham, MA, USA) automated DNA/RNA synthesizer using conventional phosphoramidite strategy. For the benzylamine building block 1, an extended coupling time (600 s) was used. Removal of the base and phosphate protections and release of the oligonucleotides from the solid support was accomplished by treatment with 25% aq. NH3 for 16 h at 55 °C. The cyclopalladated oligonucleotides ON1b-Pd, ON2b-Pd, ON3b-Pd and ON4b-Pd were prepared by incubating ON1b, ON2b, ON3b and ON4b (192, 260, 290 and 173 nmol, respectively) and Li2PdCl4 (384, 520, 580 and 346 nmol, respectively) in a mixture of H2O (530 µL) and MeCN (30 µL) for 16 h at 25 °C. All modified oligonucleotides were purified by reversed-phase high performance liquid chromatography (RP-HPLC) on a Hypersil ODS C18 column (250 × 4.6 mm, 5 µm, Thermo Fisher Scientific, Waltham, MA, USA) eluting with a linear gradient (0 to 30% over 25 min) of MeCN in 50 mM aqueous triethylammonium acetate. The flow rate was 1.0 mL·min−1 and the detection wavelength 260 nm. The purified oligonucleotides were characterized by electrospray ionization mass spectrometry (ESI-MS) and quantified UV spectrophotometrically using molar absorptivities calculated by an implementation of the nearest-neighbors method. Molar absorptivity of both free and cyclopalladated benzylamine was assumed to be negligible.
4.6. Melting Temperature Measurements
Melting profiles were recorded on a PerkinElmer Lambda 35 UV-Vis spectrometer equipped with a Peltier temperature control unit (PerkinElmer, Waltham, MA, USA). Samples were prepared by mixing the appropriate oligonucleotides (3.0 µM) in 20 mM cacodylate buffer (pH 7.4), the ionic strength of which was adjusted to 0.10 M with NaClO4. When applicable, 2-mercaptoethanol was used in 100 µM concentration and added after mixing of the oligonucleotides. Before measurement, the samples were annealed by heating to 90 °C and gradually cooling to room temperature. UV melting curves were acquired by monitoring the absorbance at λ = 260 nm over a temperature range of 10–90 °C, sampling at 10 °C intervals. The melting temperatures were determined as inflection points on the UV melting curves.
4.7. CD Measurements
CD spectra were recorded on an Applied Photophysics Chirascan spectropolarimeter equipped with a Peltier temperature control unit (Applied Photophysics, Leatherhead, UK). Samples used in the CD measurements were identical to those used in the UV melting temperature measurements. CD spectra were acquired between λ = 200 and 400 nm over a temperature range of 10–90 °C, sampling at 10 °C intervals. At each temperature, samples were allowed to equilibrate for 600 s before acquisition.
5. Conclusions
Cyclopalladated benzylamine, tethered at a terminal position of a short oligonucleotide by a flexible linker, promotes hybridization despite a significant entropy penalty for “freezing” the linker in an appropriate conformation. Sensitivity to the presence of competing ligands suggests direct coordination of Pd(II) to a nucleobase of the complementary strand as the origin of stabilization by the palladacyclic moiety. In light of these results, oligonucleotides furnished with palladacyclic “warheads” could prove useful as a sequence-selective alternative to current platinum-based anticancer agents.
Supplementary Materials
Supplementary materials can be found at https://www.mdpi.com/1422-0067/19/6/1588/s1.
Author Contributions
Conceptualization, T.L.; Methodology, T.L. and S.M.; Investigation, M.H. and S.M.; Resources, T.L.; Data Curation, T.L.; Writing-Original Draft Preparation, M.H. and T.L.; Writing-Review and Editing, T.L.; Visualization, T.L.; Supervision, T.L. and S.M.; Project Administration, T.L.; Funding Acquisition, T.L.
Funding
This project has received funding from the European Union’s Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement No. 721613 and from the Academy of Finland (decisions No. 286478 and No. 294008).
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| CD | circular dichroism |
| DNA | deoxyribonucleic acid |
| PNA | peptide nucleic acid |
| RP-HPLC | reversed-phase high performance liquid chromatography |
| ESI-MS | electrospray ionization mass spectrometry |
References
- Dasari, S.; Tchounwou, P.B. Cisplatin in cancer therapy: Molecular mechanisms of action. Eur. J. Pharmacol. 2014, 740, 364–378. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, B.; Van Camp, L.; Krigas, T. Inhibition of cell division in Escherichia coli by electrolysis products from a platinum electrode. Nature 1965, 205, 698–699. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Cano, C.; Hannon, M.J. Novel and emerging approaches for the delivery of metallo-drugs. Dalton Trans. 2009, 10702–10711. [Google Scholar] [CrossRef] [PubMed]
- Ott, I.; Gust, R. Non platinum metal complexes as anti-cancer drugs. Arch. Pharm. 2007, 340, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Bruijnincx, P.C.A.; Sadler, P.J. New trends for metal complexes with anticancer activity. Curr. Opin. Chem. Biol. 2008, 12, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Ott, I. On the medicinal chemistry of gold complexes as anticancer drugs. Coord. Chem. Rev. 2009, 253, 1670–1681. [Google Scholar] [CrossRef]
- Levina, A.; Mitra, A.; Lay, P.A. Recent developments in ruthenium anticancer drugs. Metallomics 2009, 1, 458–470. [Google Scholar] [CrossRef] [PubMed]
- Berners-Price, S.J.; Filipovska, A. Gold compounds as therapeutic agents for human diseases. Metallomics 2011, 3, 863–873. [Google Scholar] [CrossRef] [PubMed]
- Cutillas, N.; Yellol, G.S.; de Haro, C.; Vicente, C.; Rodríguez, V.; Ruiz, J. Anticancer cyclometalated complexes of platinum group metals and gold. Coord. Chem. Rev. 2013, 257, 2784–2797. [Google Scholar] [CrossRef]
- Johnstone, T.C.; Suntharalingam, K.; Lippard, S.J. The next generation of platinum drugs: Targeted Pt(II) agents, nanoparticle delivery, and Pt(IV) prodrugs. Chem. Rev. 2016, 116, 3436–3486. [Google Scholar] [CrossRef] [PubMed]
- Bindoli, A.; Rigobello, M.P.; Scutari, G.; Gabbiani, C.; Casini, A.; Messori, L. Thioredoxin reductase: A target for gold compounds acting as potential anticancer drugs. Coord. Chem. Rev. 2009, 253, 1692–1707. [Google Scholar] [CrossRef]
- Kelland, L. The resurgence of platinum-based cancer chemotherapy. Nat. Rev. Cancer 2007, 7, 573–584. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, J.T.; Lipp, H.-P. Toxicity of platinum compounds. Expert Opin. Pharmacother. 2003, 4, 889–901. [Google Scholar] [CrossRef] [PubMed]
- Daugaard, G.; Abildgaard, U. Cisplatin nephrotoxicity. A review. Cancer Chemother. Pharmacol. 1989, 25, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Johnstone, T.C.; Suntharalingam, K.; Lippard, S.J. Third row transition metals for the treatment of cancer. Philos. Trans. A Math. Phys. Eng. Sci. 2015, 373, 20140185. [Google Scholar] [CrossRef] [PubMed]
- Carreira, M.; Calvo-Sanjuán, R.; Sanaú, M.; Marzo, I.; Contel, M. Organometallic palladium complexes with a water-soluble iminophosphorane ligand as potential anticancer agents. Organometallics 2012, 31, 5772–5781. [Google Scholar] [CrossRef] [PubMed]
- Caires, A.C.F. Recent advances involving palladium (II) complexes for the cancer therapy. Anticancer Agents Med. Chem. 2007, 7, 484–491. [Google Scholar] [CrossRef] [PubMed]
- Taube, H. Rates and mechanisms of substitution in inorganic complexes in solution. Chem. Rev. 1952, 50, 69–126. [Google Scholar] [CrossRef]
- Lippert, B.; Miguel, P.J.S. Comparing PtII- and PdII-nucleobase coordination chemistry: Why PdII not always is a good substitute for PtII. Inorg. Chim. Acta 2018, 472, 207–213. [Google Scholar] [CrossRef]
- Serrano, F.A.; Matsuo, A.L.; Monteforte, P.T.; Bechara, A.; Smaili, S.S.; Santana, D.P.; Rodrigues, T.; Pereira, F.V.; Silva, L.S.; Machado, J.; et al. A cyclopalladated complex interacts with mitochondrial membrane thiol-groups and induces the apoptotic intrinsic pathway in murine and cisplatin-resistant human tumor cells. BMC Cancer 2011, 11, 296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marchán, V.; Grandas, A. Platinated oligonucleotides: Synthesis and applications for the control of gene expression. In Metal Complex–DNA Interactions; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2009; pp. 273–300. [Google Scholar] [CrossRef]
- Colombier, C.; Lippert, B.; Leng, M. Interstrand cross-linking reaction in triplexes containing a monofunctional transplatin-adduct. Nucleic Acids Res. 1996, 24, 4519–4524. [Google Scholar] [CrossRef] [PubMed]
- Brabec, V.; Reedijk, J.; Leng, M. Sequence-dependent distortions induced in DNA by monofunctional platinum(II) binding. Biochemistry 1992, 31, 12397–12402. [Google Scholar] [CrossRef] [PubMed]
- Algueró, B.; Pedroso, E.; Marchán, V.; Grandas, A. Incorporation of two modified nucleosides allows selective platination of an oligonucleotide making it suitable for duplex cross-linking. J. Biol. Inorg. Chem. 2007, 12, 901–911. [Google Scholar] [CrossRef] [PubMed]
- Algueró, B.; López de la Osa, J.; González, C.; Pedroso, E.; Marchán, V.; Grandas, A. Selective platination of modified oligonucleotides and duplex cross-links. Angew. Chem. Int. Ed. 2006, 45, 8194–8197. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, K.S.; Boudvillain, M.; Schwartz, A.; van der Marel, G.A.; van Boom, J.H.; Reedijk, J.; Lippert, B. Monofunctionally trans-diammine platinum(II)-modified peptide nucleic acid oligomers: A new generation of potential antisense drugs. Chem. Eur. J. 2002, 8, 5566–5570. [Google Scholar] [CrossRef]
- Maity Sajal, K.; Lönnberg, T. Oligonucleotides incorporating palladacyclic nucleobase surrogates. Chem. Eur. J. 2018, 24, 1274–1277. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, J.; Cutillas, N.; Vicente, C.; Villa, M.D.; López, G.; Lorenzo, J.; Avilés, F.X.; Moreno, V.; Bautista, D. New palladium(II) and platinum(II) complexes with the model nucleobase 1-methylcytosine: Antitumor activity and interactions with DNA. Inorg. Chem. 2005, 44, 7365–7376. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, J.; Lorenzo, J.; Vicente, C.; López, G.; López-de-Luzuriaga, J.M.; Monge, M.; Avilés, F.X.; Bautista, D.; Moreno, V.; Laguna, A. New palladium(II) and platinum(II) complexes with 9-aminoacridine: Structures, luminiscence, theoretical calculations, and antitumor activity. Inorg. Chem. 2008, 47, 6990–7001. [Google Scholar] [CrossRef] [PubMed]
- Fuchita, Y.; Tsuchiya, H. Synthesis of ortho-palladated complexes of n-methylbenzylamine: The first example of ortho-palladation of α-unsubstituted secondary benzylamine. Inorg. Chim. Acta 1993, 209, 229–230. [Google Scholar] [CrossRef]
- Fuchita, Y.; Tsuchiya, H.; Miyafuji, A. Cyclopalladation of secondary and primary benzylamines. Inorg. Chim. Acta 1995, 233, 91–96. [Google Scholar] [CrossRef]
- Selvakumar, K.; Vancheesan, S. Synthesis and characterization of cyclopalladated complexes of secondary benzylamines. Polyhedron 1997, 16, 2405–2411. [Google Scholar] [CrossRef]
- Mergny, J.-L.; Lacroix, L. Analysis of thermal melting curves. Oligonucleotides 2003, 13, 515–537. [Google Scholar] [CrossRef] [PubMed]
- Golubev, O.; Lönnberg, T.; Lönnberg, H. Interaction of Pd2+ complexes of 2,6-disubstituted pyridines with nucleoside 5′-monophosphates. J. Inorg. Biochem. 2014, 139, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Martin, R.B. Nucleoside sites for transition metal ion binding. Acc. Chem. Res. 1985, 18, 32–38. [Google Scholar] [CrossRef]
- Pneumatikakis, G.; Hadjiliadis, N.; Theophanides, T. Complexes of inosine, cytidine, and guanosine with palladium(II). Inorg. Chem. 1978, 17, 915–922. [Google Scholar] [CrossRef]
- Scharf, P.; Müller, J. Nucleic acids with metal-mediated base pairs and their applications. ChemPlusChem 2013, 78, 20–34. [Google Scholar] [CrossRef]
- Takezawa, Y.; Shionoya, M. Metal-mediated DNA base pairing: Alternatives to hydrogen-bonded Watson–Crick base pairs. Acc. Chem. Res. 2012, 45, 2066–2076. [Google Scholar] [CrossRef] [PubMed]
- Clever, G.H.; Kaul, C.; Carell, T. DNA–metal base pairs. Angew. Chem. Int. Ed. 2007, 46, 6226–6236. [Google Scholar] [CrossRef] [PubMed]
- Clever, G.H.; Shionoya, M. Metal–base pairing in DNA. Coord. Chem. Rev. 2010, 254, 2391–2402. [Google Scholar] [CrossRef]
- Müller, J. Chemistry: Metals line up for DNA. Nature 2006, 444, 698. [Google Scholar] [CrossRef] [PubMed]
- Müller, J. Metal-ion-mediated base pairs in nucleic acids. Eur. J. Inorg. Chem. 2008, 2008, 3749–3763. [Google Scholar] [CrossRef]
- Mandal, S.; Müller, J. Metal-mediated DNA assembly with ligand-based nucleosides. Curr. Opin. Chem. Biol. 2017, 37, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Takezawa, Y.; Müller, J.; Shionoya, M. Artificial DNA base pairing mediated by diverse metal ions. Chem. Lett. 2017, 46, 622–633. [Google Scholar] [CrossRef]
- Lippert, B.; Sanz Miguel, P.J. The renaissance of metal–pyrimidine nucleobase coordination chemistry. Acc. Chem. Res. 2016, 49, 1537–1545. [Google Scholar] [CrossRef] [PubMed]
- Taherpour, S.; Golubev, O.; Lönnberg, T. On the feasibility of recognition of nucleic acid sequences by metal-ion-carrying oligonucleotides. Inorg. Chim. Acta 2016, 452, 43–49. [Google Scholar] [CrossRef]
- Clever, G.H.; Söltl, Y.; Burks, H.; Spahl, W.; Carell, T. Metal–salen-base-pair complexes inside DNA: Complexation overrides sequence information. Chem. Eur. J. 2006, 12, 8708–8718. [Google Scholar] [CrossRef] [PubMed]
Scheme 1.
Synthesis of the benzylamine phosphoramidite 1. Reagents and conditions: (a) 2-(2-aminoethoxy)ethanol, MeCN, 25 °C, 16 h; (b) ethyl trifluoroacetate, Et3N, MeOH, 25 °C, 16 h; (c) 2-cyanoethyl-N,N-diisopropylchlorophosphoramidite, Et3N, CH2Cl2, N2 atmosphere, 25 °C, 3 h.
Scheme 1.
Synthesis of the benzylamine phosphoramidite 1. Reagents and conditions: (a) 2-(2-aminoethoxy)ethanol, MeCN, 25 °C, 16 h; (b) ethyl trifluoroacetate, Et3N, MeOH, 25 °C, 16 h; (c) 2-cyanoethyl-N,N-diisopropylchlorophosphoramidite, Et3N, CH2Cl2, N2 atmosphere, 25 °C, 3 h.

Scheme 2.
Cyclopalladation of 2-[2-(benzylamino)ethoxy]ethanol (2) and the corresponding modified oligonucleotides ON1b, ON2b, ON3b and ON4b. Reagents and conditions: a) Li2PdCl4, MeCN, H2O, 25 °C, 16 h.
Scheme 2.
Cyclopalladation of 2-[2-(benzylamino)ethoxy]ethanol (2) and the corresponding modified oligonucleotides ON1b, ON2b, ON3b and ON4b. Reagents and conditions: a) Li2PdCl4, MeCN, H2O, 25 °C, 16 h.
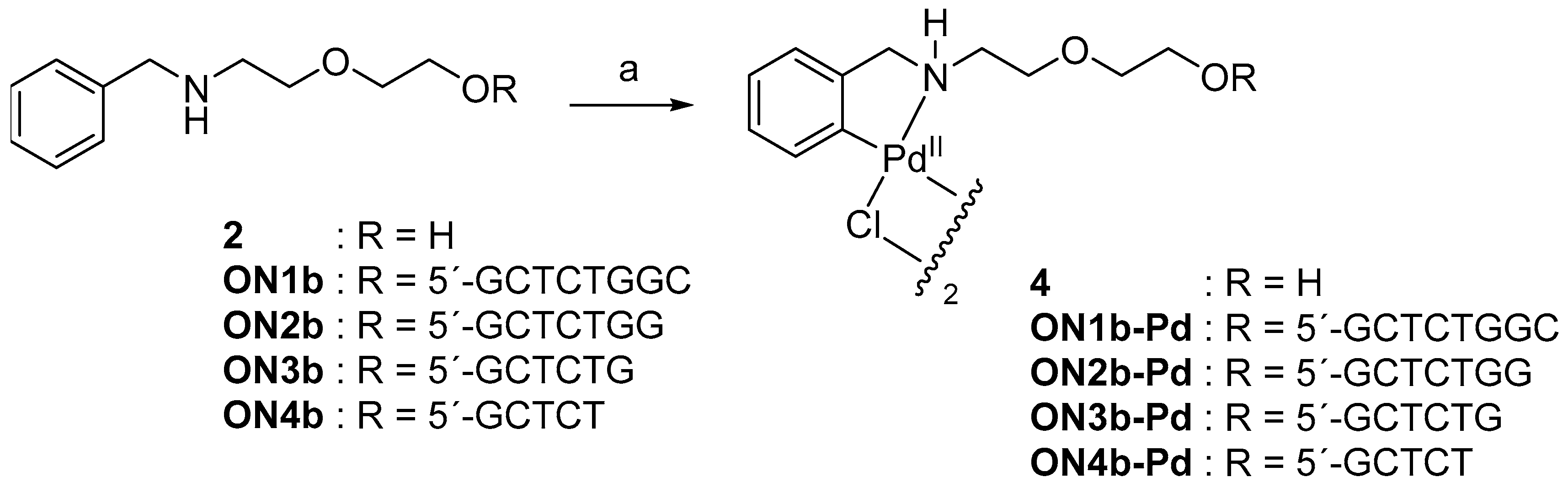
Figure 1.
Outline of the hybridization assays used. X is either adenine or unmetalated or cyclopalladated benzylamine and Y is any canonical nucleobase. The bullets indicate Watson-Crick base pairing.
Figure 1.
Outline of the hybridization assays used. X is either adenine or unmetalated or cyclopalladated benzylamine and Y is any canonical nucleobase. The bullets indicate Watson-Crick base pairing.

Figure 2.
UV melting profiles for duplexes formed by ON5a with (A) ON1a (cyan circles), ON2a (magenta triangles), ON3a (yellow squares) and ON4a (black diamonds); (B) ON1b (cyan circles), ON2b (magenta triangles), ON3b (yellow squares) and ON4b (black diamonds) and (C) ON1b-Pd (cyan circles), ON2b-Pd (magenta triangles), ON3b-Pd (yellow squares) and ON4b-Pd (black diamonds); (D) melting temperatures of duplexes formed by ON1a, ON1b and ON1b-Pd with ON5a (cyan), ON5g (magenta) and ON5t (yellow); pH = 7.4 (20 mM cacodylate buffer); [oligonucleotides] = 3.0 µM; I(NaClO4) = 0.10 M. The error bars represent standard deviations of three independent measurements.
Figure 2.
UV melting profiles for duplexes formed by ON5a with (A) ON1a (cyan circles), ON2a (magenta triangles), ON3a (yellow squares) and ON4a (black diamonds); (B) ON1b (cyan circles), ON2b (magenta triangles), ON3b (yellow squares) and ON4b (black diamonds) and (C) ON1b-Pd (cyan circles), ON2b-Pd (magenta triangles), ON3b-Pd (yellow squares) and ON4b-Pd (black diamonds); (D) melting temperatures of duplexes formed by ON1a, ON1b and ON1b-Pd with ON5a (cyan), ON5g (magenta) and ON5t (yellow); pH = 7.4 (20 mM cacodylate buffer); [oligonucleotides] = 3.0 µM; I(NaClO4) = 0.10 M. The error bars represent standard deviations of three independent measurements.

Figure 3.
Melting temperatures of duplexes ON1a•ON5a, ON1b•ON5a and ON1b-Pd•ON5a in the absence (cyan) and presence (magenta) of 2-mercaptoethanol; pH = 7.4 (20 mM cacodylate buffer); [oligonucleotides] = 3.0 µM; [2-mercaptoethanol] = 0/100 µM; I(NaClO4) = 0.10 M. The error bars represent standard deviations of three independent measurements.
Figure 3.
Melting temperatures of duplexes ON1a•ON5a, ON1b•ON5a and ON1b-Pd•ON5a in the absence (cyan) and presence (magenta) of 2-mercaptoethanol; pH = 7.4 (20 mM cacodylate buffer); [oligonucleotides] = 3.0 µM; [2-mercaptoethanol] = 0/100 µM; I(NaClO4) = 0.10 M. The error bars represent standard deviations of three independent measurements.
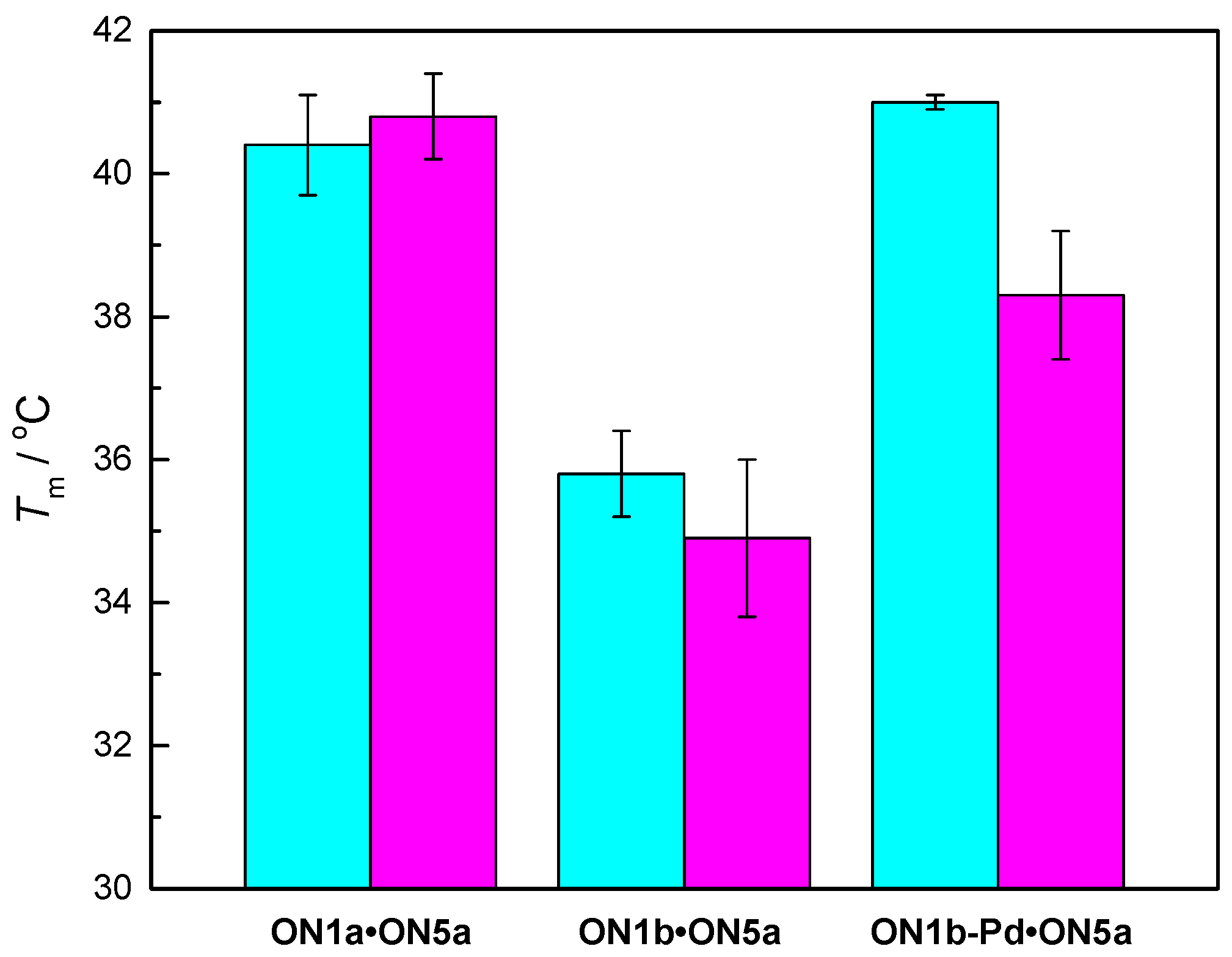
Figure 4.
CD (circular dichroism) spectra of ON1b-Pd•ON5a, recorded at 10 °C intervals between 10 and 90 °C; pH = 7.4 (20 mM cacodylate buffer); [oligonucleotides] = 3.0 µM; I(NaClO4) = 0.10 M. Spectra acquired at extreme temperatures are indicated by thicker lines and thermal shifts of the minima and maxima by arrows.
Figure 4.
CD (circular dichroism) spectra of ON1b-Pd•ON5a, recorded at 10 °C intervals between 10 and 90 °C; pH = 7.4 (20 mM cacodylate buffer); [oligonucleotides] = 3.0 µM; I(NaClO4) = 0.10 M. Spectra acquired at extreme temperatures are indicated by thicker lines and thermal shifts of the minima and maxima by arrows.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Oligonucleotides used in this study.
| Sequence 1 | Oligonucleotide |
|---|---|
| ON1a | 5′-AGCTCTGGC-3′ |
| ON2a | 5′-AGCTCTGG-3′ |
| ON3a | 5′-AGCTCTG-3′ |
| ON4a | 5′-AGCTCT-3′ |
| ON1b | 5′-BGCTCTGGC-3′ |
| ON2b | 5′-BGCTCTGG-3′ |
| ON3b | 5′-BGCTCTG-3′ |
| ON4b | 5′-BGCTCT-3′ |
| ON1b-Pd | 5′-BPdGCTCTGGC-3′ |
| ON2b-Pd | 5′-BPdGCTCTGG-3′ |
| ON3b-Pd | 5′-BPdGCTCTG-3′ |
| ON4b-Pd | 5′-BPdGCTCT-3′ |
| ON5a | 5′-GCCAGAGCTCG-3′ |
| ON5c | 5′-GCCAGCGCTCG-3′ |
| ON5g | 5′-GCCAGGGCTCG-3′ |
| ON5t | 5′-GCCAGTGCTCG-3′ |
1 B refers to unmetalated and BPd to cyclopalladated benzylamine residue. The residues varied in the hybridization studies have been underlined.
Table 2.
Thermodynamic parameters of hybridization for ON1a•ON5a, ON1b•ON5a and ON1b-Pd•ON5a; pH = 7.4 (20 mM cacodylate buffer); [oligonucleotides] = 3.0 µM; I(NaClO4) = 0.10 M.
Table 2.
Thermodynamic parameters of hybridization for ON1a•ON5a, ON1b•ON5a and ON1b-Pd•ON5a; pH = 7.4 (20 mM cacodylate buffer); [oligonucleotides] = 3.0 µM; I(NaClO4) = 0.10 M.
| Duplex | ΔH°/kJ·mol−1 | ΔS°/J·mol−1·K−1 |
|---|---|---|
| ON1a•ON5a | −200 ± 20 | −530 ± 50 |
| ON1b•ON5a | −200 ± 10 | −550 ± 40 |
| ON1b-Pd•ON5a | −250 ± 20 | −670 ± 50 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Hande, M.; Maity, S.; Lönnberg, T. Palladacyclic Conjugate Group Promotes Hybridization of Short Oligonucleotides. Int. J. Mol. Sci. 2018, 19, 1588. https://doi.org/10.3390/ijms19061588
AMA Style
Hande M, Maity S, Lönnberg T. Palladacyclic Conjugate Group Promotes Hybridization of Short Oligonucleotides. International Journal of Molecular Sciences. 2018; 19(6):1588. https://doi.org/10.3390/ijms19061588
Chicago/Turabian StyleHande, Madhuri, Sajal Maity, and Tuomas Lönnberg. 2018. "Palladacyclic Conjugate Group Promotes Hybridization of Short Oligonucleotides" International Journal of Molecular Sciences 19, no. 6: 1588. https://doi.org/10.3390/ijms19061588
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.