Persistent Infiltration and Impaired Response of Peripherally-Derived Monocytes after Traumatic Brain Injury in the Aged Brain

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
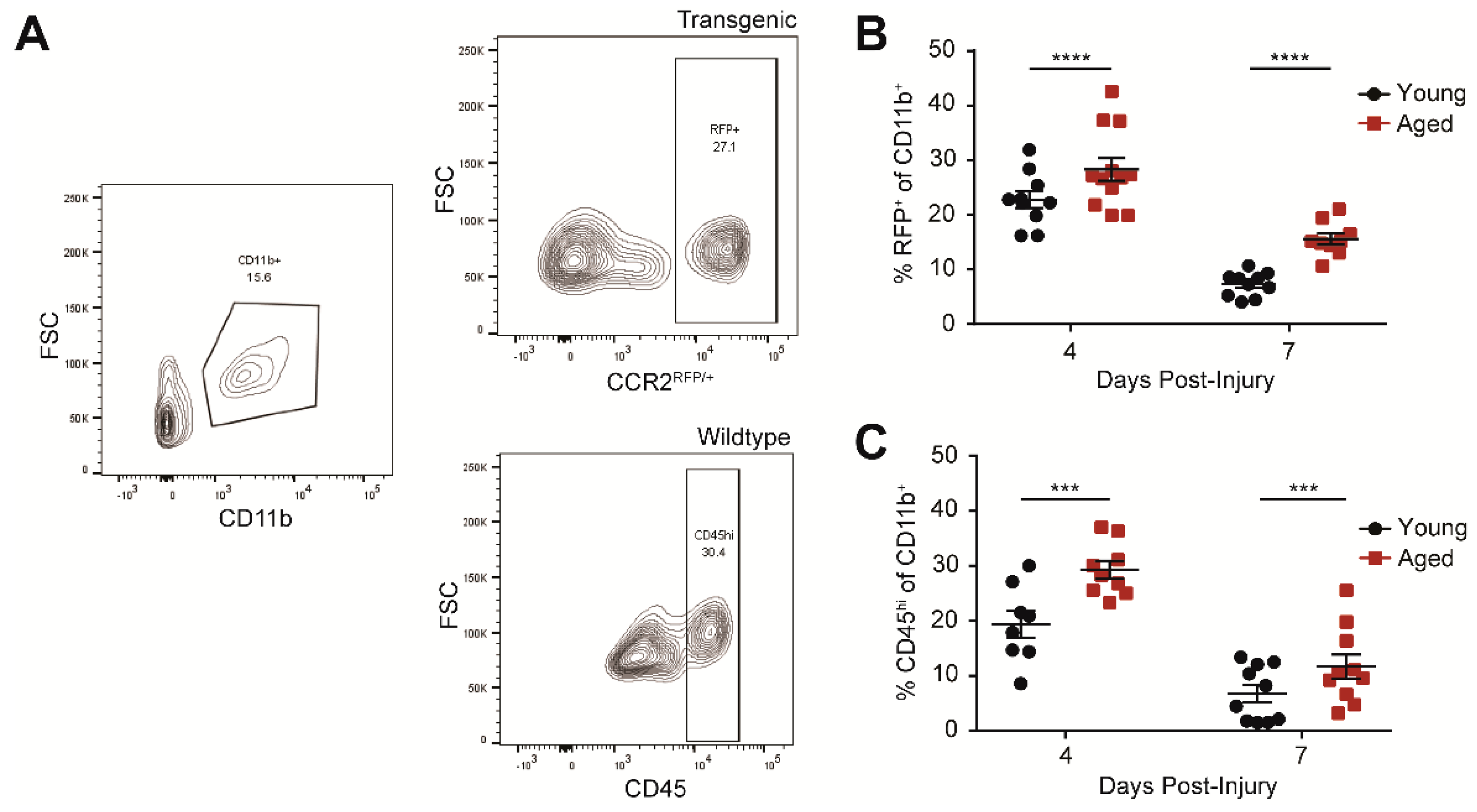
2.1. Age Increases Peripherally-Derived Monocytes in the Injured Brain at 4 and 7 Days Post-Injury
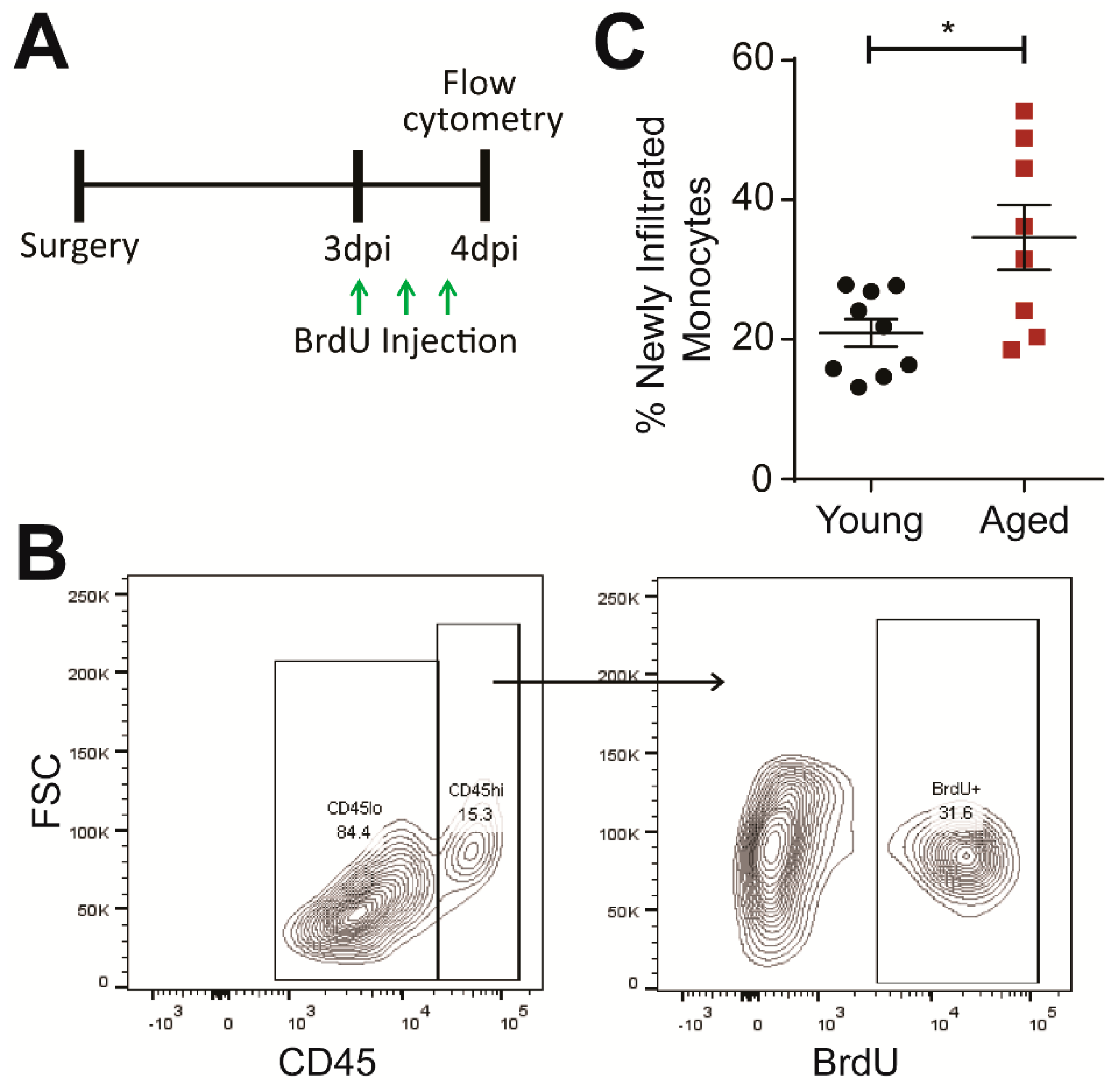
2.2. Age Exacerbates Subchronic Infiltration of Peripherally-Derived Monocytes to the Injured Brain
2.3. Age Increases CCR2 Signaling in the Injured Brain
2.4. Age Increases CCR2+ Monocytes in the Blood Population after Injury
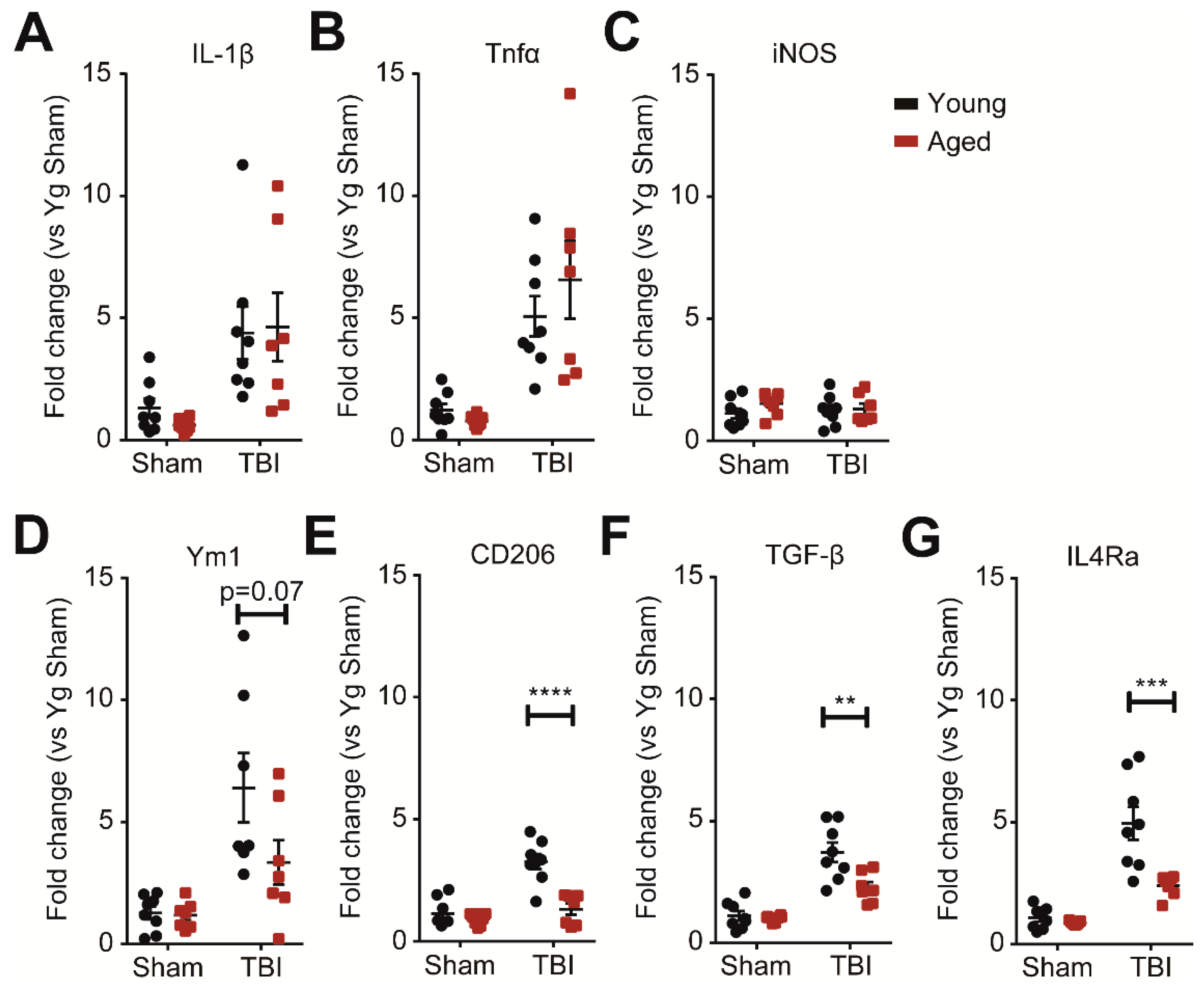
2.5. Age Modifies the Subchronic Anti-Inflammatory Response to TBI
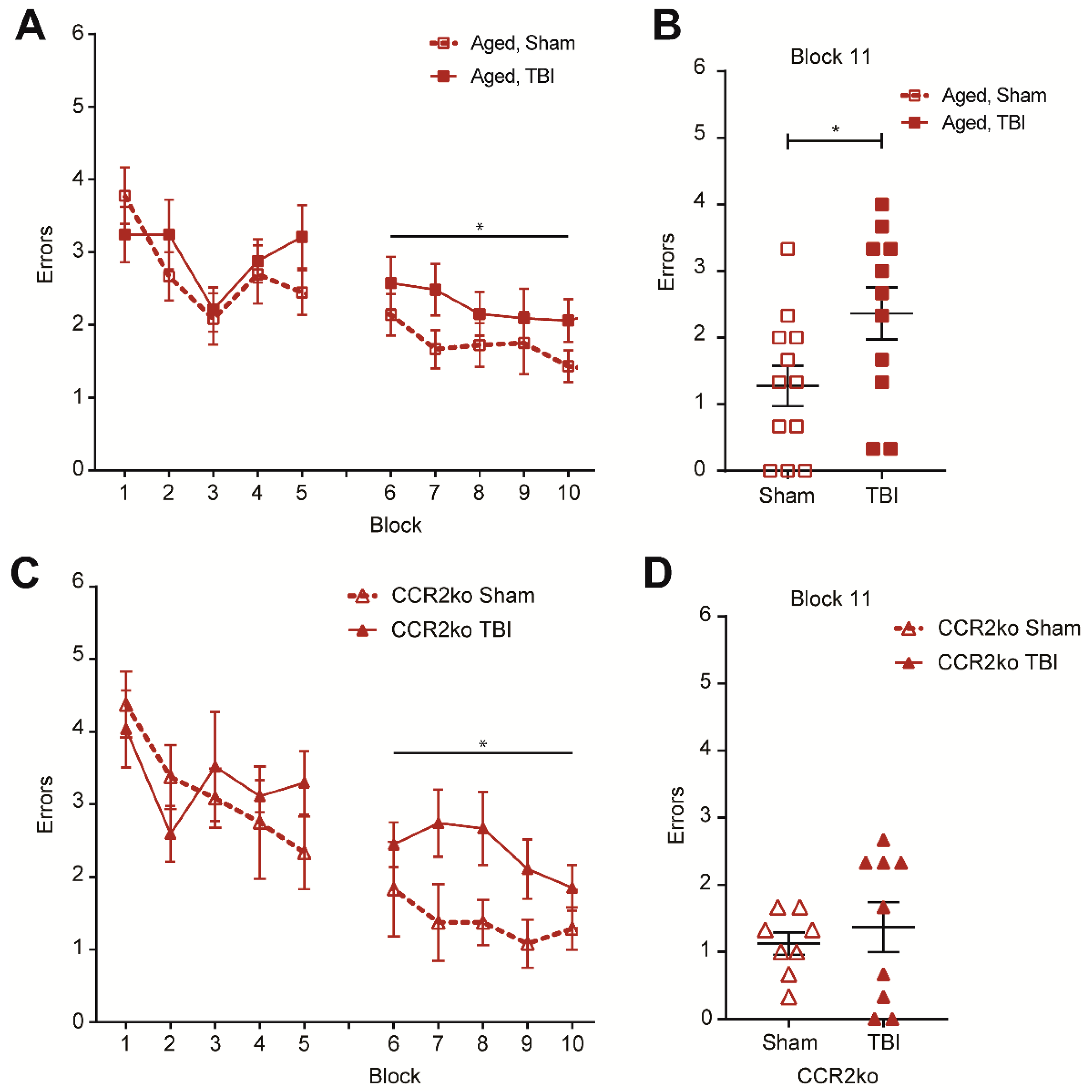
2.6. CCR2 Knockout Prevents TBI-Induced Memory Deficits in Aged Animals
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. TBI Surgical Procedure
4.3. BrdU/EdU Injection
4.4. Tissue Collection
4.5. Flow Cytometry
4.6. qRT-PCR
4.7. Radial Arm Water Maze
4.8. Statistical Analysis
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Johnson, E.V.; Stewart, W.; Smith, D.H. Traumatic brain injury and amyloid-beta Pathology: A link to alzheimer’s disease? Nat. Rev. Neurosci. 2010, 11, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Corrigan, D.J.; Selassie, A.W.; Orman, J.A. The Epidemiology of traumatic brain injury. J. Head Trauma Rehabil. 2010, 25, 72–80. [Google Scholar] [CrossRef] [PubMed]
- Stocchetti, N.; Paterno, R.; Citerio, G.; Beretta, L.; Colombo, A. Traumatic brain injury in an aging population. J. Neurotrauma 2012, 29, 1119–1125. [Google Scholar] [CrossRef] [PubMed]
- Himanen, L.; Portin, R.; Isoniemi, H.; Helenius, H.; Kurki, T.; Tenovuo, O. Longitudinal cognitive changes in traumatic brain Injury: A 30-year follow-up study. Neurology 2006, 66, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Hukkelhoven, C.W.; Steyerberg, E.W.; Rampen, A.J.; Farace, E.; Habbema, J.D.; Marshall, L.F.; Murray, G.D.; Maas, A.I. Patient Age and outcome following severe traumatic brain Injury: An analysis of 5600 patients. J. Neurosurg. 2003, 99, 666–673. [Google Scholar] [CrossRef] [PubMed]
- Schonberger, M.; Ponsford, J.; Reutens, D.; Beare, R.; O’Sullivan, R. The Relationship between age, injury severity, mri findings after traumatic brain injury. J. Neurotrauma 2009, 26, 2157–2167. [Google Scholar] [CrossRef] [PubMed]
- Susman, M.S.; Di Russo, M.; Sullivan, T.; Risucci, D.; Nealon, P.; Cuff, S.; Haider, A.; Benzil, D. Traumatic Brain injury in the Elderly: Increased mortality and worse functional outcome at discharge despite lower injury severity. J. Trauma 2002, 53, 219–223. [Google Scholar] [CrossRef] [PubMed]
- Mushkudiani, N.A.; Engel, D.C.; yerberg, E.W.S.; Butcher, I.; Lu, J.; Marmarou, A.; Slieker, F.; McHugh, G.S.; Murray, G.D.; Maas, A.I. Prognostic Value of demographic characteristics in traumatic brain Injury: Results from the impact study. J. Neurotrauma 2007, 24, 259–269. [Google Scholar] [CrossRef] [PubMed]
- Itoh, T.; Imano, M.; Nishida, S.; Tsubaki, M.; Mizuguchi, N.; Hashimoto, S.; Ito, A.; Satou, T. Increased Apoptotic neuronal cell death and cognitive impairment at early phase after traumatic brain injury in aged rats. Brain Struct. Funct. 2013, 218, 209–220. [Google Scholar] [CrossRef] [PubMed]
- Onyszchuk, G.; He, Y.Y.; Berman, N.E.; Brooks, W.M. Detrimental Effects of aging on outcome from traumatic brain Injury: A behavioral, magnetic resonance imaging, and histological study in mice. J. Neurotrauma 2008, 25, 153–171. [Google Scholar] [CrossRef] [PubMed]
- Timaru-Kast, R.; Luh, C.; Gotthardt, P.; Huang, C.; Schafer, M.K.; Engelhard, K.; Thal, S.C. Influence of Age on brain edema formation, secondary brain damage and inflammatory response after brain trauma in mice. PLoS ONE 2012, 7, e43829. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Loane, D.J. Neuroinflammation after Traumatic brain Injury: Opportunities for therapeutic intervention. Brain Behav. Immun. 2012, 26, 1191–1201. [Google Scholar] [CrossRef] [PubMed]
- Sandhir, R.; Puri, V.; Klein, R.M.; Berman, N.E. Differential expression of cytokines and chemokines during secondary neuron death following brain injury in old and young mice. Neurosci. Lett. 2004, 369, 28–32. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Stoica, B.A.; Sabirzhanov, B.; Burns, M.P.; Faden, A.I.; Loane, D.J. Traumatic Brain injury in aged animals increases lesion size and chronically alters microglial/macrophage classical and alternative activation states. Neurobiol. Aging 2013, 34, 1397–1411. [Google Scholar] [CrossRef] [PubMed]
- Morganti, J.M.; Riparip, L.K.; Chou, A.; Liu, S.; Gupta, N.; Rosi, S. Age Exacerbates the ccr2/5-mediated neuroinflammatory response to traumatic brain injury. J. Neuroinflamm. 2016, 13, 80. [Google Scholar] [CrossRef] [PubMed]
- Sandhir, R.; Onyszchuk, G.; Berman, N.E. Exacerbated glial response in the aged mouse hippocampus following controlled cortical impact injury. Exp. Neurol. 2008, 213, 372–380. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, C.L.; Niemi, E.C.; Wang, S.H.; Lee, C.C.; Bingham, D.; Zhang, J.; Cozen, M.L.; Charo, I.; Huang, E.J.; Liu, J.; et al. Ccr2 deficiency impairs macrophage infiltration and improves cognitive function after traumatic brain injury. J. Neurotrauma 2014, 31, 1677–1688. [Google Scholar] [CrossRef] [PubMed]
- Morganti, J.M.; Jopson, T.D.; Liu, S.; Riparip, L.K.; Guandique, C.K.; Gupta, N.; Ferguson, A.R.; Rosi, S. Ccr2 Antagonism alters brain macrophage polarization and ameliorates cognitive dysfunction induced by traumatic brain injury. J. Neurosci. 2015, 35, 748–760. [Google Scholar] [CrossRef] [PubMed]
- Semple, B.D.; Bye, N.; Rancan, M.; Ziebell, J.M.; Morganti-Kossmann, M.C. Role of Ccl2 (Mcp-1) in traumatic brain Injury (Tbi): Evidence from Severe tbi patients and Ccl2-/-Mice. J. Cereb. Blood Flow Metab. 2010, 30, 769–782. [Google Scholar] [CrossRef] [PubMed]
- Prinz, M.; Priller, J. Tickets to the Brain: Role of Ccr2 and Cx3cr1 in myeloid cell entry in the Cns. J. Neuroimmunol. 2010, 224, 80–84. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Zhang, L.; Wu, Q.; Wu, Q.; Wang, T. Chemokine Ccl2 Induces apoptosis in cortex following traumatic brain injury. J. Mol. Neurosci. 2013, 51, 1021–1029. [Google Scholar] [CrossRef] [PubMed]
- Shi, C.; Pamer, E.G. Monocyte Recruitment during infection and inflammation. Nat. Rev. Immunol. 2011, 11, 762–774. [Google Scholar] [CrossRef] [PubMed]
- Gyoneva, S.; Kim, D.; Katsumoto, A.; Kokiko-Cochran, O.N.; Lamb, B.T.; Ransohoff, R.M. Ccr2 Deletion Dissociates cavity size and tau pathology after mild traumatic brain injury. J. Neuroinflamm. 2015, 12, 228. [Google Scholar] [CrossRef] [PubMed]
- Geissmann, F.; Manz, M.G.; Jung, S.; Sieweke, M.H.; Merad, M.; Ley, K. Development of monocytes, macrophages, dendritic cells. Science 2010, 327, 656–661. [Google Scholar] [CrossRef] [PubMed]
- Villeda, S.A.; Plambeck, K.E.; Middeldorp, J.; Castellano, J.M.; Mosher, K.I.; Luo, J.; Smith, L.K.; Bieri, G.; Lin, K.; Berdnik, D.; et al. Young Blood reverses age-related impairments in cognitive function and synaptic plasticity in mice. Nat. Med. 2014, 20, 659–663. [Google Scholar] [CrossRef] [PubMed]
- Rosenzweig, E.S.; Barnes, C.A. Impact of aging on hippocampal Function: Plasticity, network dynamics, cognition. Prog. Neurobiol. 2003, 69, 143–179. [Google Scholar] [CrossRef]
- Kawata, K.; Liu, C.Y.; Merkel, S.F.; Ramirez, S.H.; Tierney, R.T.; Langford, D. Blood Biomarkers for Brain Injury: What Are We Measuring? Neurosci. Biobehav. Rev. 2016, 68, 460–473. [Google Scholar] [CrossRef] [PubMed]
- Agoston, D.V.; Shutes-David, A.; Peskind, E.R. Biofluid Biomarkers of traumatic brain injury. Brain Inj. 2017, 31, 1195–1203. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Liu, P.; Guo, F.; Zhang, Z.Y.; Zhang, Z. Oxidative Burst of circulating neutrophils following traumatic brain injury in human. PLoS ONE 2013, 8, e68963. [Google Scholar] [CrossRef]
- Hazeldine, J.; Lord, J.M.; Belli, A. Traumatic brain injury and peripheral immune Suppression: Primer and prospectus. Front. Neurol. 2015, 6, 235. [Google Scholar] [CrossRef] [PubMed]
- Alliot, F.; Godin, I.; Pessac, B. Microglia derive from progenitors, originating from the yolk sac, which proliferate in the brain. Brain Res. Dev. Brain Res. 1999, 117, 145–152. [Google Scholar] [CrossRef]
- Ginhoux, F.; Greter, M.; Leboeuf, M.; Nandi, S.; See, P.; Gokhan, S.; Mehler, M.F.; Conway, S.J.; Ng, L.G.; Stanley, E.R.; et al. Fate Mapping analysis reveals that adult microglia derive from primitive macrophages. Science 2010, 330, 841–845. [Google Scholar] [CrossRef] [PubMed]
- Saederup, N.; Cardona, A.E.; Croft, K.; Mizutani, M.; Cotleur, A.C.; Tsou, C.L.; Ransohoff, R.M.; Charo, I.F. Selective chemokine receptor usage by central nervous system myeloid cells in ccr2-red fluorescent protein knock-in mice. PLoS ONE 2010, 5, e13693. [Google Scholar] [CrossRef] [PubMed]
- Davies, L.C.; Rosas, M.; Jenkins, S.J.; Liao, C.T.; Scurr, M.J.; Brombacher, F.; Fraser, D.J.; Allen, J.E.; Jones, S.A.; Taylor, P.R. Distinct bone marrow-derived and tissue-resident macrophage lineages proliferate at key stages during inflammation. Nat. Commun. 2013, 4, 1886. [Google Scholar] [CrossRef] [PubMed]
- Ajami, B.; Bennett, J.L.; Krieger, C.; Tetzlaff, W.; Rossi, F.M. Local Self-Renewal Can Sustain Cns Microglia Maintenance and Function Throughout Adult Life. Nat. Neurosci. 2007, 10, 1538–1543. [Google Scholar] [CrossRef] [PubMed]
- Yona, S.; Kim, K.W.; Wolf, Y.; Mildner, A.; Varol, D.; Breker, M.; Strauss-Ayali, D.; Viukov, S.; Guilliams, M.; Misharin, A.; et al. Fate Mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity 2013, 38, 79–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loane, D.J.; Kumar, A. Microglia in the Tbi Brain: The good, the bad, the dysregulated. Exp. Neurol. 2016, 3, 316–327. [Google Scholar] [CrossRef] [PubMed]
- Jin, X.; Ishii, H.; Bai, Z.; Itokazu, T.; Yamashita, T. Temporal changes in cell marker expression and cellular infiltration in a controlled cortical impact model in adult male c57bl/6 mice. PLoS ONE 2012, 7, e41892. [Google Scholar] [CrossRef] [PubMed]
- Gordon, S. Alternative activation of macrophages. Nat. Rev. Immunol. 2003, 3, 23–35. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Alvarez-Croda, D.M.; Stoica, B.A.; Faden, A.I.; Loane, D.J. Microglial/Macrophage polarization dynamics following traumatic brain injury. J. Neurotrauma 2016, 33, 1732–1750. [Google Scholar] [CrossRef] [PubMed]
- Morganti, J.M.; Riparip, L.K.; Rosi, S. Call off the dog (Ma): M1/M2 polarization is concurrent following traumatic brain injury. PLoS ONE 2016, 11, e0148001. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.C.; Nakamura, M.C.; Hsieh, C.L. Brain trauma elicits non-canonical macrophage activation states. J. Neuroinflamm. 2016, 13, 117. [Google Scholar] [CrossRef] [PubMed]
- Jablonski, K.A.; Amici, S.A.; Webb, L.M.; de Dios Ruiz-Rosado, J.; Popovich, P.G.; Partida-Sanchez, S.; Guerau-de-Arellano, M. Novel markers to delineate murine M1 and M2 macrophages. PLoS ONE 2015, 10, e0145342. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.C.; Ruiz, C.R.; Lebson, L.; Selenica, M.L.; Rizer, J.; Hunt, J.B., Jr.; Rojiani, R.; Reid, P.; Kammath, S.; Nash, K.; et al. Aging Enhances classical activation but mitigates alternative activation in the central nervous system. Neurobiol. Aging 2013, 34, 1610–1620. [Google Scholar] [CrossRef] [PubMed]
- Gong, D.; Shi, W.; Yi, S.J.; Chen, H.; Groffen, J.; Heisterkamp, N. Tgfbeta Signaling Plays a Critical Role in Promoting Alternative Macrophage Activation. BMC Immunol. 2012, 13, 31. [Google Scholar] [CrossRef] [PubMed]
- Wattananit, S.; Tornero, D.; Graubardt, N.; Memanishvili, T.; Monni, E.; Tatarishvili, J.; Miskinyte, G.; Ge, R.; Ahlenius, H.; Lindvall, O.; et al. Monocyte-Derived macrophages contribute to spontaneous long-term functional recovery after stroke in mice. J. Neurosci. 2016, 36, 4182–4195. [Google Scholar] [CrossRef] [PubMed]
- Gliem, M.; Schwaninger, M.; Jander, S. Protective features of peripheral monocytes/macrophages in stroke. Biochim. Biophys. Acta 2016, 1862, 329–338. [Google Scholar] [CrossRef] [PubMed]
- Shechter, R.; London, A.; Varol, C.; Raposo, C.; Cusimano, M.; Yovel, G.; Rolls, A.; Mack, M.; Pluchino, S.; Martino, G.; et al. Infiltrating Blood-derived macrophages are vital cells playing an anti-inflammatory role in recovery from spinal cord injury in mice. PLoS Med. 2009, 6, e1000113. [Google Scholar] [CrossRef] [PubMed]
- Lighthall, J.W. Controlled cortical Impact: A new experimental brain injury model. J. Neurotrauma 1988, 5, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Alamed, J.; Wilcock, D.M.; Diamond, D.M.; Gordon, M.N.; Morgan, D. Two-day radial-arm water maze learning and memory task; robust resolution of amyloid-related memory deficits in transgenic mice. Nat. Protoc. 2006, 1, 1671–1679. [Google Scholar] [CrossRef] [PubMed]





© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chou, A.; Krukowski, K.; Morganti, J.M.; Riparip, L.-K.; Rosi, S. Persistent Infiltration and Impaired Response of Peripherally-Derived Monocytes after Traumatic Brain Injury in the Aged Brain. Int. J. Mol. Sci. 2018, 19, 1616. https://doi.org/10.3390/ijms19061616
Chou A, Krukowski K, Morganti JM, Riparip L-K, Rosi S. Persistent Infiltration and Impaired Response of Peripherally-Derived Monocytes after Traumatic Brain Injury in the Aged Brain. International Journal of Molecular Sciences. 2018; 19(6):1616. https://doi.org/10.3390/ijms19061616
Chicago/Turabian StyleChou, Austin, Karen Krukowski, Josh M. Morganti, Lara-Kirstie Riparip, and Susanna Rosi. 2018. "Persistent Infiltration and Impaired Response of Peripherally-Derived Monocytes after Traumatic Brain Injury in the Aged Brain" International Journal of Molecular Sciences 19, no. 6: 1616. https://doi.org/10.3390/ijms19061616