Bacterial Toxins and Targeted Brain Therapy: New Insights from Cytotoxic Necrotizing Factor 1 (CNF1)


Abstract
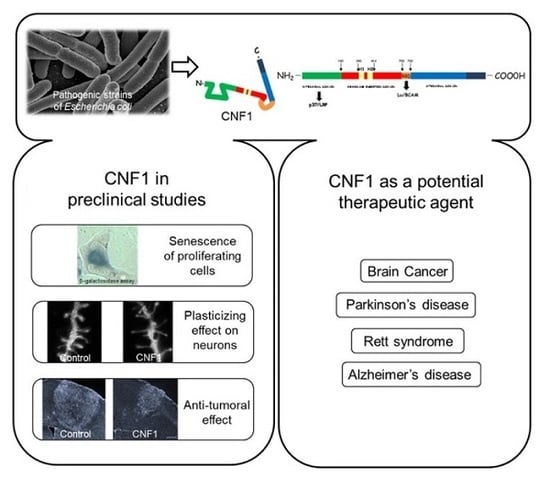
:
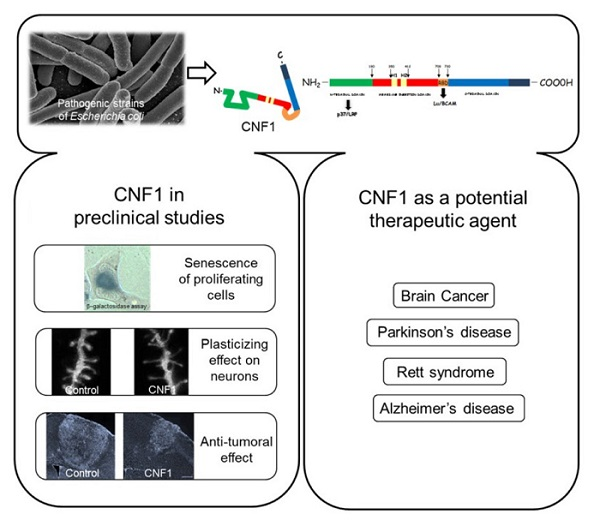
1. Introduction
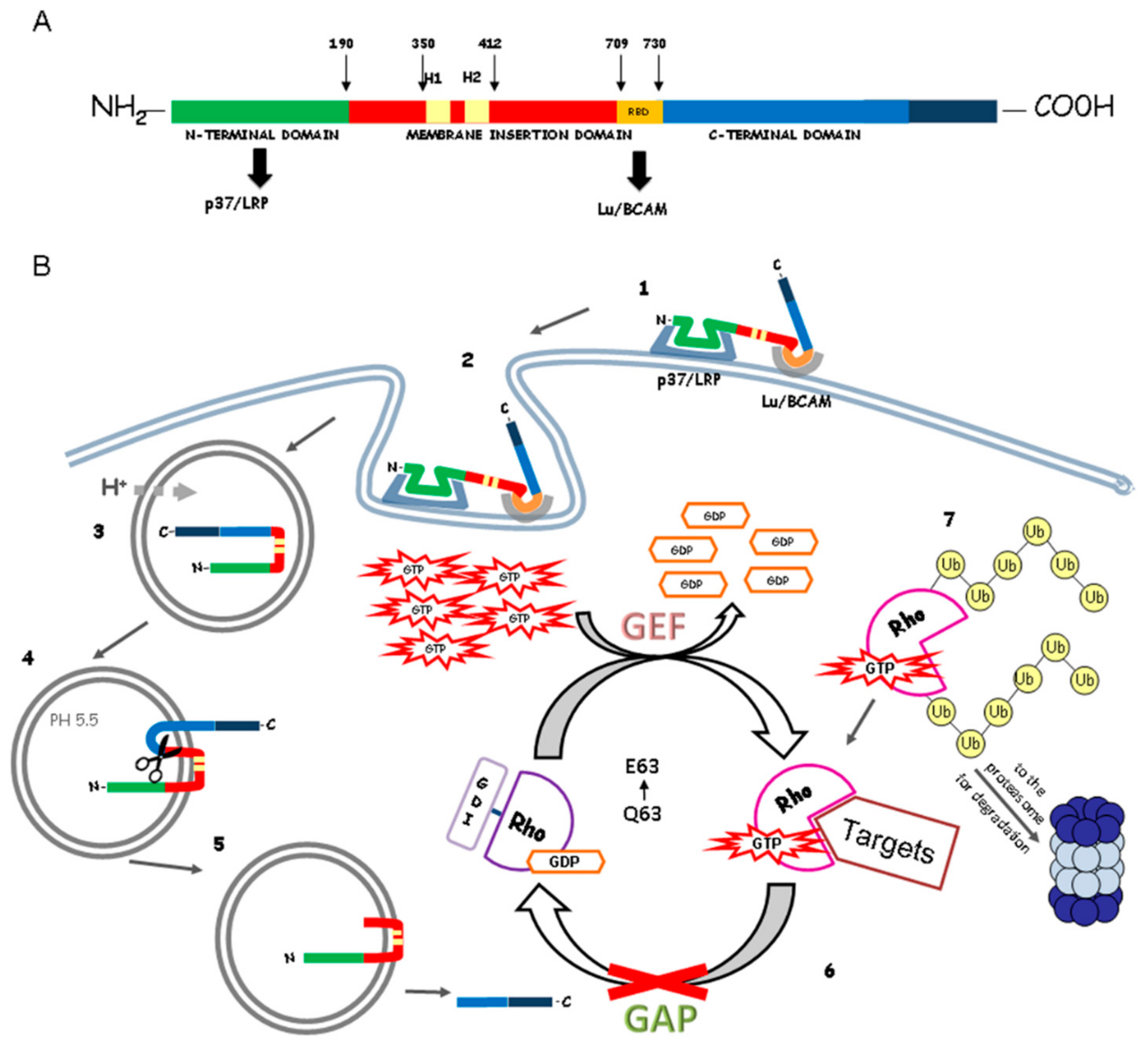
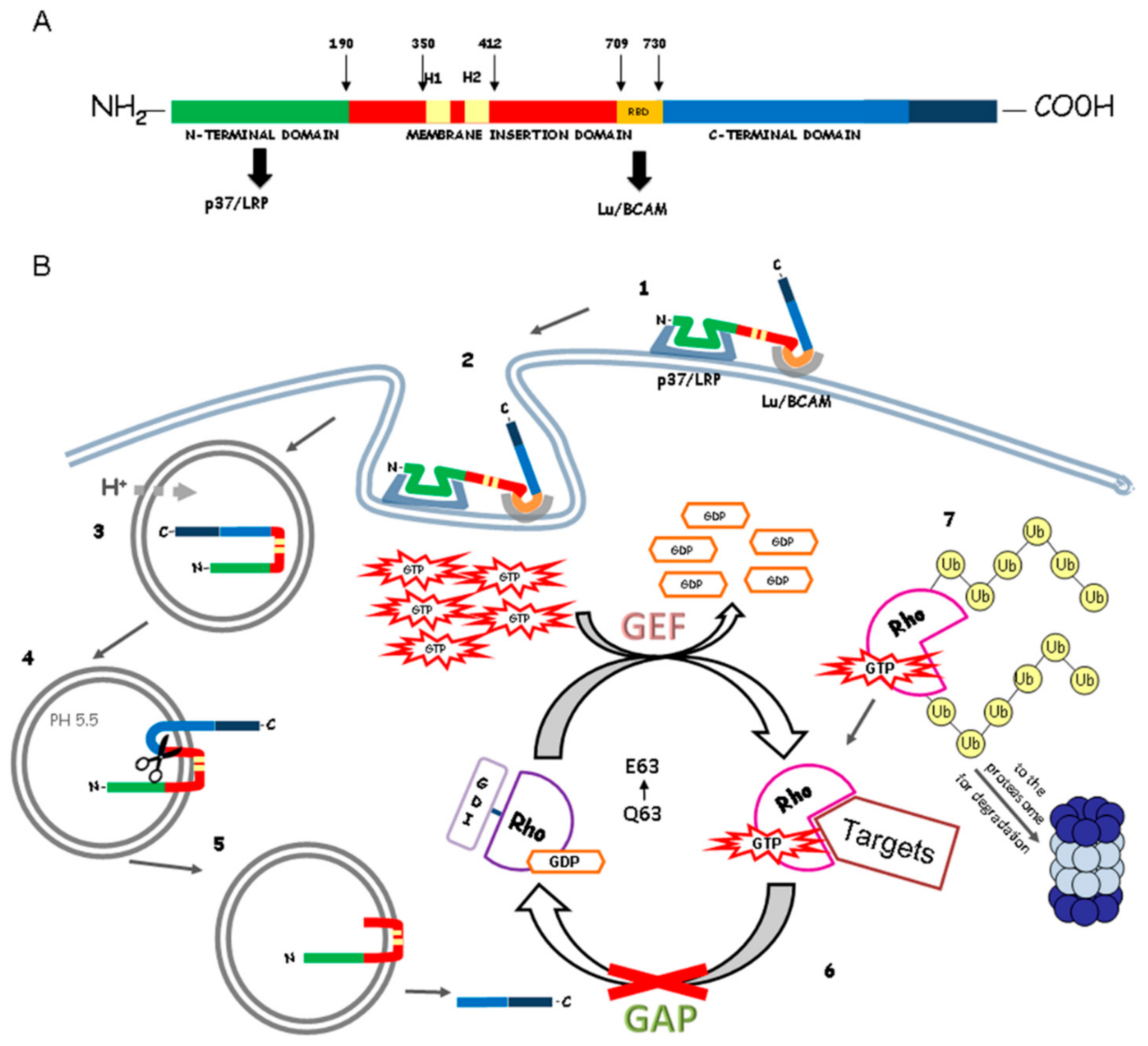
2. Cytotoxic Necrotizing Factor (CNF1) Structure and Mechanism of Action
3. Effects of CNF1 on Neurons
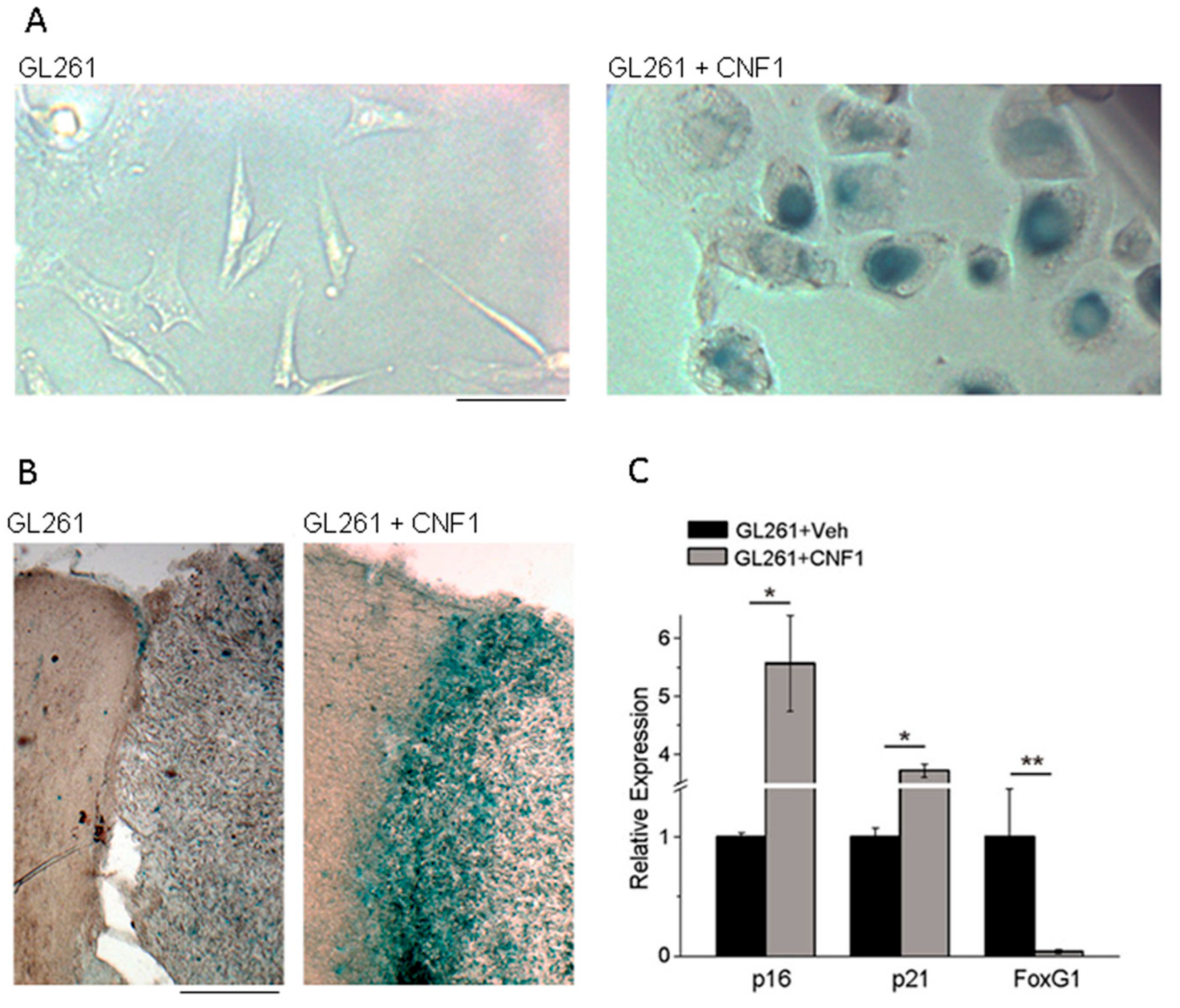
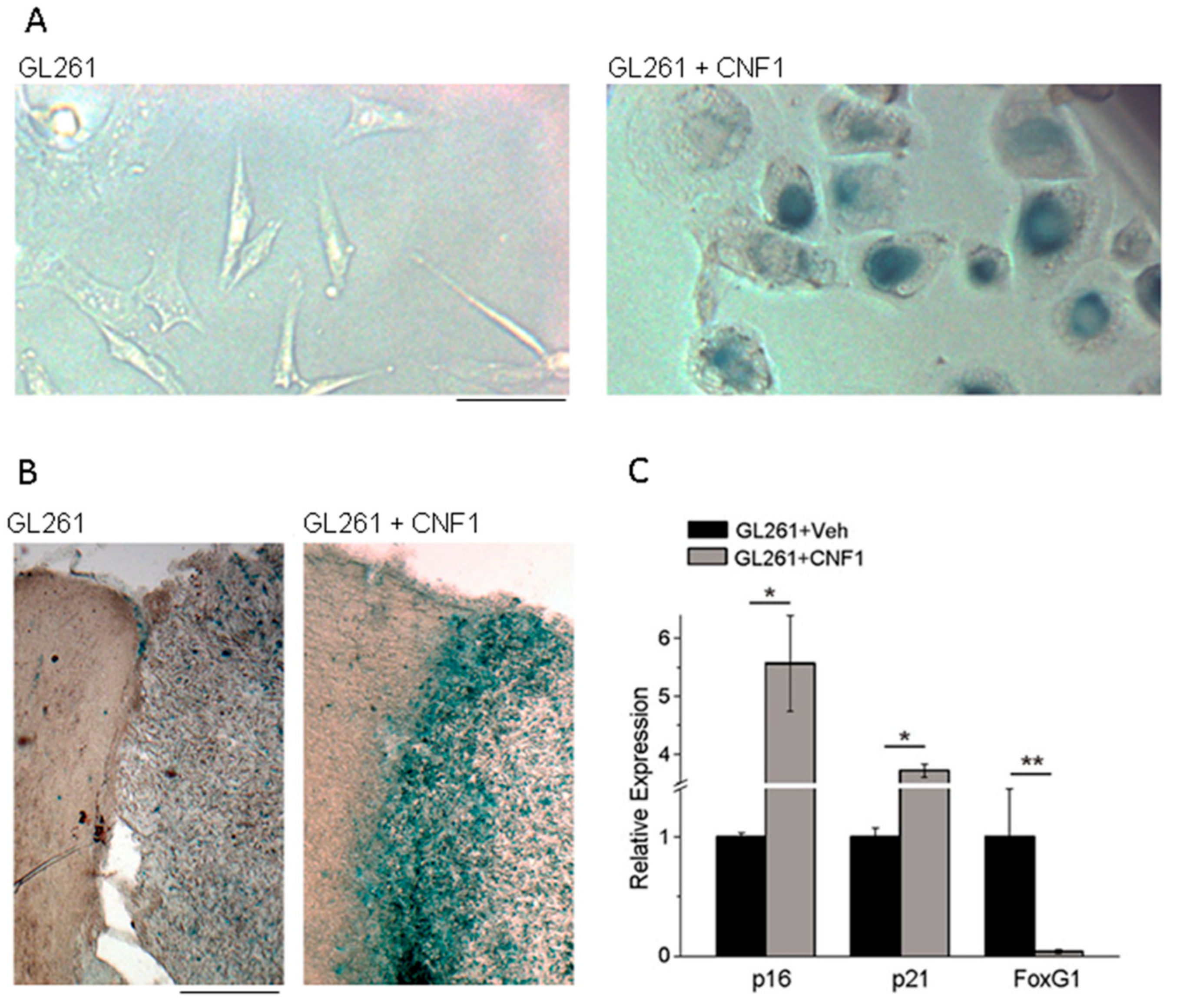
4. Effects of CNF1 on Cancer Cells
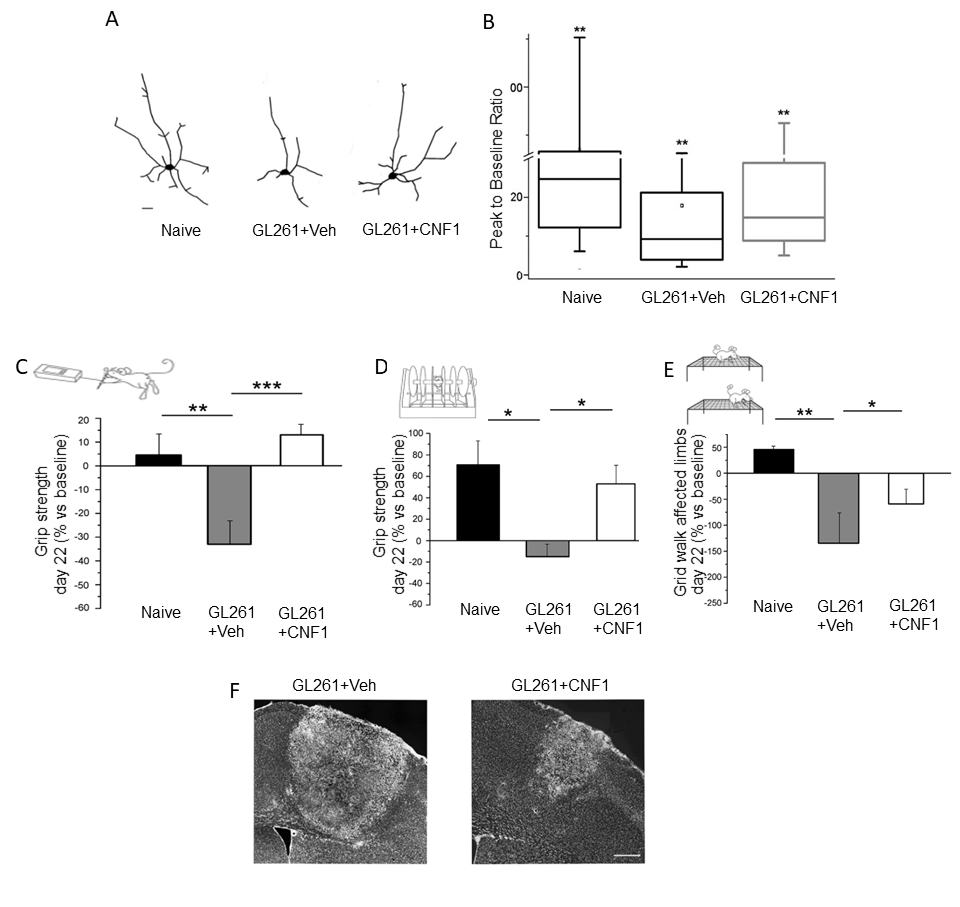
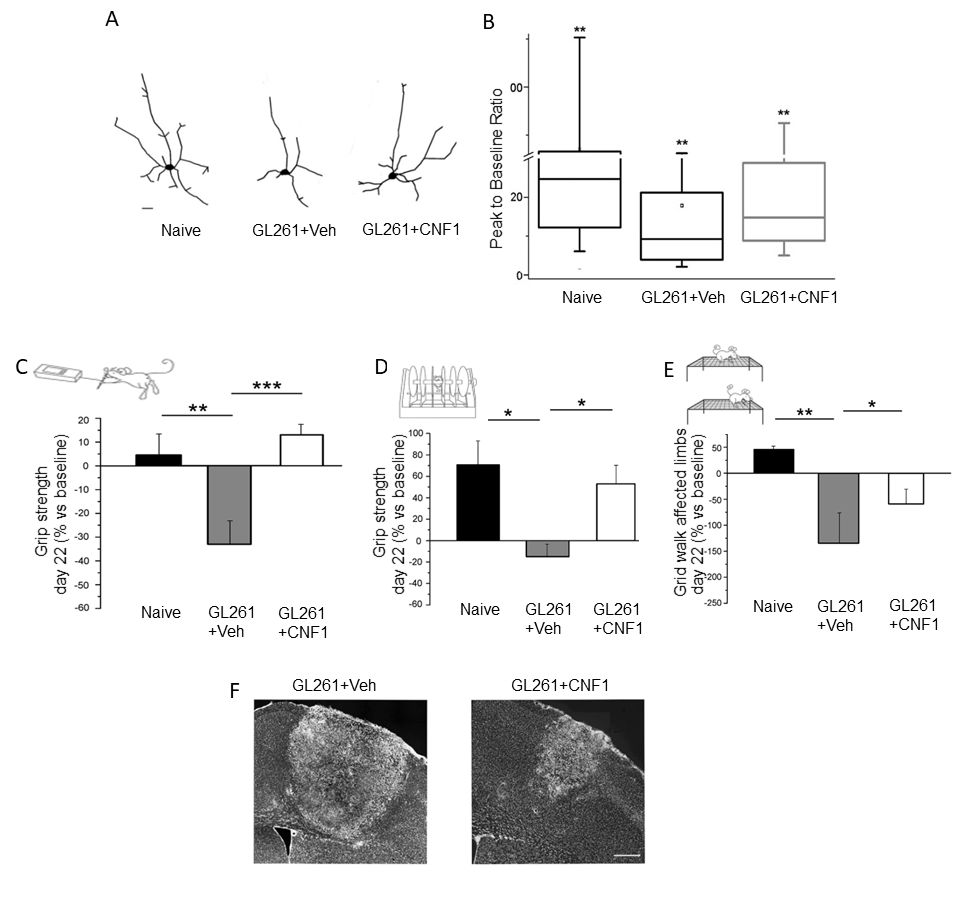
5. CNF1 Action in Glioma: Functional Sparing of Peritumoral Neurons
6. Concluding Remarks
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Popoff, M.R.; Poulain, B. Bacterial toxins and the nervous system: Neurotoxins and multipotential toxins interacting with neuronal cells. Toxins 2010, 2, 683–737. [Google Scholar] [CrossRef] [PubMed]
- Aktories, K.; Barbieri, J.T. Bacterial cytotoxins: Targeting eukaryotic switches. Nat. Rev. Microbiol. 2005, 3, 397–410. [Google Scholar] [CrossRef] [PubMed]
- Boquet, P. The cytotoxic necrotizing factor 1 (CNF1) from Escherichia coli. Toxicon 2001, 39, 1673–1680. [Google Scholar] [CrossRef]
- Middlebrook, J.L.; Dorland, R.B. Bacterial toxins: Cellular mechanisms of action. Microbiol. Rev. 1984, 48, 199–221. [Google Scholar] [PubMed]
- Pirazzini, M.; Rossetto, O.; Eleopra, R.; Montecucco, C. Botulinum Neurotoxins: Biology, Pharmacology, and Toxicology. Pharmacol. Rev. 2017, 69, 200–235. [Google Scholar] [CrossRef] [PubMed]
- Mazzocchio, R.; Caleo, M. More than at the neuromuscular synapse: Actions of botulinum neurotoxin A in the central nervous system. Neuroscientist 2015, 21, 44–61. [Google Scholar] [CrossRef] [PubMed]
- Pavone, F.; Luvisetto, S.; Marinelli, S.; Straface, E.; Fabbri, A.; Falzano, L.; Fiorentini, C.; Malorni, W. The Rac GTPase-activating bacterial protein toxin CNF1 induces analgesia up-regulating mu-opioid receptors. Pain 2009, 145, 219–229. [Google Scholar] [CrossRef] [PubMed]
- Ney, J.P.; Joseph, K.R. Neurologic uses of botulinum neurotoxin type A. Neuropsychiatr. Dis. Treat. 2007, 3, 785–798. [Google Scholar] [CrossRef] [PubMed]
- Nigam, P.K.; Nigam, A. Botulinum toxin. Indian J. Dermatol. 2010, 55, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Caleo, M.; Restani, L. Direct central nervous system effects of botulinum neurotoxin. Toxicon 2017, 147, 68–72. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.R.; O’Neill, B.P. Glioblastoma survival in the United States before and during the temozolomide era. J. Neuro-Oncol. 2012, 107, 359–364. [Google Scholar] [CrossRef] [PubMed]
- Weller, M.; Cloughesy, T.; Perry, J.R.; Wick, W. Standards of care for treatment of recurrent glioblastoma—Are we there yet? Neuro Oncol. 2013, 15, 4–27. [Google Scholar] [CrossRef] [PubMed]
- Zahaf, N.; Schmidt, G. Bacterial Toxins for Cancer Therapy. Toxins 2017, 9, 236. [Google Scholar] [CrossRef] [PubMed]
- Aktories, K. Bacterial protein toxins that modify host regulatory GTPases. Nat. Rev. Microbiol. 2011, 9, 487–498. [Google Scholar] [CrossRef] [PubMed]
- De Filippis, B.; Fabbri, A.; Simone, D.; Canese, R.; Ricceri, L.; Malchiodi-Albedi, F.; Laviola, G.; Fiorentini, C. Modulation of RhoGTPases improves the behavioral phenotype and reverses astrocytic deficits in a mouse model of rett syndrome. Neuropsychopharmacology 2012, 37, 1152–1163. [Google Scholar] [CrossRef] [PubMed]
- Travaglione, S.; Loizzo, S.; Ballan, G.; Fiorentini, C.; Fabbri, A. The E. coli CNF1 as a pioneering therapy for the central nervous system diseases. Toxins 2013, 6, 270–282. [Google Scholar] [CrossRef] [PubMed]
- Loizzo, S.; Rimondini, R.; Travaglione, S.; Fabbri, A.; Guidotti, M.; Ferri, A.; Campana, G.; Fiorentini, C. CNF1 Increases Brain Energy Level, Counteracts Neuroinflammatory Markers and Rescues Cognitive Deficits in a Murine Model of Alzheimer’s Disease. PLoS ONE 2013, 8, e65898. [Google Scholar] [CrossRef]
- Musilli, M.; Ciotti, M.T.; Pieri, M.; Martino, A.; Borrelli, S.; Dinallo, V.; Diana, G. Therapeutic effects of the Rho GTPase modulator CNF1 in a model of Parkinson’s disease. Neuropharmacology 2016, 109, 357–365. [Google Scholar] [CrossRef] [PubMed]
- Fabbri, A.; Travaglione, S.; Fiorentini, C. Escherichia coli cytotoxic necrotizing factor 1 (CNF1): Toxin biology, in vivo applications and therapeutic potential. Toxins 2010, 2, 283–296. [Google Scholar] [CrossRef] [PubMed]
- Caprioli, A.; Falbo, V.; Roda, L.G.; Ruggeri, F.M.; Zona, C. Partial purification and characterization of an Escherichia coli toxic factor that induces morphological cell alterations. Infect. Immun. 1983, 39, 1300–1306. [Google Scholar] [PubMed]
- Piteau, M.; Papatheodorou, P.; Schwan, C.; Schlosser, A.; Aktories, K.; Schmidt, G. Lu/BCAM Adhesion Glycoprotein Is a Receptor for Escherichia coli Cytotoxic Necrotizing Factor 1 (CNF1). PLoS Pathog. 2014, 10, e1003884. [Google Scholar] [CrossRef]
- Reppin, F.; Cochet, S.; El Nemer, W.; Fritz, G.; Schmidt, G. High Affinity Binding of Escherichia coli Cytotoxic Necrotizing Factor 1 (CNF1) to Lu/BCAM Adhesion Glycoprotein. Toxins 2017, 10, 3. [Google Scholar] [CrossRef] [PubMed]
- Contamin, S.; Galmiche, A.; Doye, A.; Flatau, G.; Benmerah, A.; Boquet, P. The p21 Rho-activating toxin cytotoxic necrotizing factor 1 is endocytosed by a clathrin-independent mechanism and enters the cytosol by an acidic-dependent membrane translocation step. Mol. Biol. Cell 2000, 11, 1775–1787. [Google Scholar] [CrossRef] [PubMed]
- Pei, S.; Doye, A.; Boquet, P. Mutation of specific acidic residues of the CNF1 T domain into lysine alters cell membrane translocation of the toxin. Mol. Microbiol. 2001, 41, 1237–1247. [Google Scholar] [CrossRef] [PubMed]
- Blumenthal, B.; Hoffmann, C.; Aktories, K.; Backert, S.; Schmidt, G. The cytotoxic necrotizing factors from Yersinia pseudotuberculosis and from Escherichia coli bind to different cellular receptors but take the same route to the cytosol. Infect. Immun. 2007, 75, 3344–3353. [Google Scholar] [CrossRef] [PubMed]
- Aktories, K.; Schmidt, G.; Just, I. Rho GTPases as targets of bacterial protein toxins. Biol. Chem. 2000, 381, 421–426. [Google Scholar] [CrossRef] [PubMed]
- Falzano, L.; Filippini, P.; Travaglione, S.; Miraglia, A.G.; Fabbri, A.; Fiorentini, C. Escherichia coli cytotoxic necrotizing factor 1 blocks cell cycle G 2/M transition in uroepithelial cells. Infect. Immun. 2006, 74, 3765–3772. [Google Scholar] [CrossRef] [PubMed]
- Flatau, G.; Lemichez, E.; Gauthier, M.; Chardin, P.; Paris, S.; Florentini, C.; Boquet, P. Toxin-induced activation of the G protein p21 Rho by deamidation of glutamine. Nature 1997, 387, 729–733. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, G.; Sehr, P.; Wilm, M.; Selzer, J.; Mann, M.; Aktories, K. Gin 63 of Rho is deamidated by Escherichia coli cytotoxic necrotizing factor-1. Nature 1997, 387, 725–729. [Google Scholar] [CrossRef] [PubMed]
- Lemonnier, M.; Landraud, L.; Lemichez, E. Rho GTPase-activating bacterial toxins: From bacterial virulence regulation to eukaryotic cell biology. FEMS Microbiol. Rev. 2007, 31, 515–534. [Google Scholar] [CrossRef] [PubMed]
- Boyer, L.; Travaglione, S.; Falzano, L.; Gauthier, N.C.; Popoff, M.R.; Lemichez, E.; Fiorentini, C.; Fabbri, A. Rac GTPase Instructs Nuclear Facteor-κB Activation by Conveying the SCF Complex and IkBa to the Ruffling Membranes. Mol. Biol. Cell 2004, 15, 1895–1903. [Google Scholar] [CrossRef] [PubMed]
- Falzano, L.; Quaranta, M.G.; Travaglione, S.; Filippini, P.; Fabbri, A.; Viora, M.; Donelli, G.; Fiorentini, C. Cytotoxic necrotizing factor 1 enhances reactive oxygen species-dependent transcription and secretion of proinflammatory cytokines in human uroepithelial cells. Infect. Immun. 2003, 71, 4178–4181. [Google Scholar] [CrossRef] [PubMed]
- Fiorentini, C.; Matarrese, P.; Straface, E.; Falzano, L.; Donelli, G.; Boquet, P.; Malorni, W. Rho-dependent cell spreading activated by E. coli cytotoxic necrotizing factor 1 hinders apoptosis in epithelial cells. Cell Death Differ. 1998, 5, 921–929. [Google Scholar] [CrossRef] [PubMed]
- Etienne-Manneville, S.; Hall, A. Rho GTPases in cell biology. Nature 2002, 420, 629–635. [Google Scholar] [CrossRef] [PubMed]
- Luo, L. RHO GTPASES in neuronal morphogenesis. Nat. Rev. Neurosci. 2000, 1, 173–180. [Google Scholar] [CrossRef] [PubMed]
- Lorenzetto, E.; Ettorre, M.; Pontelli, V.; Bolomini-Vittori, M.; Bolognin, S.; Zorzan, S.; Laudanna, C.; Buffelli, M. Rac1 Selective Activation Improves Retina Ganglion Cell Survival and Regeneration. PLoS ONE 2013, 8, e64350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiorentini, C.; Arancia, G.; Caprioli, A.; Falbo, V.; Ruggeri, F.M.; Donelli, G. Cytoskeletal changes induced in HEp-2 cells by the cytotoxic necrotizing factor of Escherichia coli. Toxicon 1988, 26, 1047–1056. [Google Scholar] [CrossRef]
- Fiorentini, C.; Fabbri, A.; Flatau, G.; Donelli, G.; Matarrese, P.; Lemichez, E.; Falzano, L.; Boquet, P. Escherichia coli cytotoxic necrotizing factor 1 (CNF1), a toxin that activates the Rho GTPase. J. Biol. Chem. 1997, 272, 19532–19537. [Google Scholar] [CrossRef] [PubMed]
- Diana, G.; Valentini, G.; Travaglione, S.; Falzano, L.; Pieri, M.; Zona, C.; Meschini, S.; Fabbri, A.; Fiorentini, C. Enhancement of learning and memory after activation of cerebral Rho GTPases. Proc. Natl. Acad. Sci. USA 2007, 104, 636–641. [Google Scholar] [CrossRef] [PubMed]
- Cerri, C.; Fabbri, A.; Vannini, E.; Spolidoro, M.; Costa, M.; Maffei, L.; Fiorentini, C.; Caleo, M. Activation of Rho GTPases triggers structural remodeling and functional plasticity in the adult rat visual cortex. J. Neurosci. 2011, 31, 15163–15172. [Google Scholar] [CrossRef] [PubMed]
- Martino, A.; Ettorre, M.; Musilli, M.; Lorenzetto, E.; Buffelli, M.; Diana, G. Rho GTPase-dependent plasticity of dendritic spines in the adult brain. Front. Cell. Neurosci. 2013, 7. [Google Scholar] [CrossRef] [PubMed]
- Malchiodi-Albedi, F.; Paradisi, S.; Di Nottia, M.; Simone, D.; Travaglione, S.; Falzano, L.; Guidotti, M.; Frank, C.; Cutarelli, A.; Fabbri, A.; et al. Cnf1 improves astrocytic ability to support neuronal growth and differentiation in vitro. PLoS ONE 2012, 7, e34115. [Google Scholar] [CrossRef] [PubMed]
- De Viti, S.; Martino, A.; Musilli, M.; Fiorentini, C.; Diana, G. The Rho GTPase activating CNF1 improves associative working memory for object-in-place. Behav. Brain Res. 2010, 212, 78–83. [Google Scholar] [CrossRef] [PubMed]
- De Filippis, B.; Valenti, D.; Chiodi, V.; Ferrante, A.; de Bari, L.; Fiorentini, C.; Domenici, M.R.; Ricceri, L.; Vacca, R.A.; Fabbri, A.; et al. Modulation of Rho GTPases rescues brain mitochondrial dysfunction, cognitive deficits and aberrant synaptic plasticity in female mice modeling Rett syndrome. Eur. Neuropsychopharmacol. 2015, 25, 889–901. [Google Scholar] [CrossRef] [PubMed]
- De Filippis, B.; Valenti, D.; de Bari, L.; de Rasmo, D.; Musto, M.; Fabbri, A.; Ricceri, L.; Fiorentini, C.; Laviola, G.; Vacca, R.A. Mitochondrial free radical overproduction due to respiratory chain impairment in the brain of a mouse model of Rett syndrome: Protective effect of CNF1. Free Radic. Biol. Med. 2015, 83, 167–177. [Google Scholar] [CrossRef] [PubMed]
- Travaglione, S.; Ballan, G.; Fortuna, A.; Ferri, A.; Guidotti, M.; Campana, G.; Fiorentini, C.; Loizzo, S. CNF1 enhances brain energy content and counteracts spontaneous epileptiform phenomena in aged DBA/2J Mice. PLoS ONE 2015, 10, e0140495. [Google Scholar] [CrossRef] [PubMed]
- Rosadi, F.; Fiorentini, C.; Fabbri, A. Bacterial protein toxins in human cancers. Pathog. Dis. 2016, 74. [Google Scholar] [CrossRef] [PubMed]
- Augspach, A.; List, J.H.; Wolf, P.; Bielek, H.; Schwan, C.; Elsässer-Beile, U.; Aktories, K.; Schmidt, G. Activation of RhoA,B,C by Yersinia Cytotoxic Necrotizing Factor (CNFy) induces apoptosis in LNCaP prostate cancer cells. Toxins 2013, 5, 2241–2257. [Google Scholar] [CrossRef] [PubMed]
- Miraglia, A.G.; Travaglione, S.; Meschini, S.; Falzano, L.; Matarrese, P.; Quaranta, M.G.; Viora, M.; Fiorentini, C.; Fabbri, A. Cytotoxic necrotizing factor 1 prevents apoptosis via the Akt/IκB kinase pathway: Role of nuclear factor-κB and Bcl-2. Mol. Biol. Cell 2007, 18, 2735–2744. [Google Scholar] [CrossRef] [PubMed]
- Sander, E.E.; Ten Klooster, J.P.; Van Delft, S.; Van Der Kammen, R.A.; Collard, J.G. Rac downregulates Rho activity: Reciprocal balance between both GTPases determines cellular morphology and migratory behavior. J. Cell Biol. 1999, 147, 1009–1021. [Google Scholar] [CrossRef] [PubMed]
- Mills, M.; Meysick, K.C.; O’Brien, A.D. Cytotoxic necrotizing factor type 1 of uropathogenic Escherichia coli kills cultured human uroepithelial 5637 cells by an apoptotic mechanism. Infect. Immun. 2000, 68, 5869–5880. [Google Scholar] [CrossRef] [PubMed]
- Horoszewicz, J.S.; Leong, S.S.; Kawinski, E.; Karr, J.P.; Rosenthal, H.; Chu, T.M.; Mirand, E.A.; Murphy, G.P. LNCaP model of human prostatic carcinoma. Cancer Res. 1983, 43, 1809–1818. [Google Scholar] [PubMed]
- Xiao, L.; Eto, M.; Kazanietz, M.G. ROCK mediates phorbol ester-induced apoptosis in prostate cancer cells via p21Cip1 up-regulation and JNK. J. Biol. Chem. 2009, 284, 29365–29375. [Google Scholar] [CrossRef] [PubMed]
- Burridge, K.; Wennerberg, K. Rho and Rac Take Center Stage. Cell 2004, 116, 167–179. [Google Scholar] [CrossRef]
- Jaffe, A.B.; Hall, A. Rho GTPases: Biochemistry and Biology. Annu. Rev. Cell Dev. Biol. 2005, 21, 247–269. [Google Scholar] [CrossRef] [PubMed]
- Fabbri, A.; Travaglione, S.; Ballan, G.; Loizzo, S.; Fiorentini, C. The cytotoxic necrotizing factor 1 from E. coli: A janus toxin playing with cancer regulators. Toxins 2013, 5, 1462–1474. [Google Scholar] [CrossRef] [PubMed]
- Doye, A.; Mettouchi, A.; Bossis, G.; Clément, R.; Buisson-Touati, C.; Flatau, G.; Gagnoux, L.; Piechaczyk, M.; Boquet, P.; Lemichez, E. CNF1 exploits the ubiquitin-proteasome machinery to restrict Rho GTPase activation for bacterial host cell invasion. Cell 2002, 111, 553–564. [Google Scholar] [CrossRef]
- Guo, Y.; Zhang, Z.; Wei, H.; Wang, J.; Lv, J.; Zhang, K.; Keller, E.T.; Yao, Z.; Wang, Q. Cytotoxic necrotizing factor 1 promotes prostate cancer progression through activating the Cdc42–PAK1 axis. J. Pathol. 2017, 243, 208–219. [Google Scholar] [CrossRef] [PubMed]
- Fiorentini, C.; Falzano, L.; Fabbri, A.; Stringaro, A.; Logozzi, M.; Travaglione, S.; Contamin, S.; Arancia, G.; Malorni, W.; Fais, S. Activation of Rho GTPases by Cytotoxic Necrotizing Factor 1 Induces Macropinocytosis and Scavenging Activity in Epithelial Cells. Mol. Biol. Cell 2001, 12, 2061–2073. [Google Scholar] [CrossRef] [PubMed]
- Knust, Z.; Blumenthal, B.; Aktories, K.; Schmidt, G. Cleavage of Escherichia coli cytotoxic necrotizing factor 1 is required for full biologic activity. Infect. Immun. 2009, 77, 1835–1841. [Google Scholar] [CrossRef] [PubMed]
- De Rycke, J.; Mazars, P.; Nougayrede, J.P.; Tasca, C.; Boury, M.; Herault, F.; Valette, A.; Oswald, E. Mitotic block and delayed lethality in HeLa epithelial cells exposed to Escherichia coli BM2-1 producing cytotoxic necrotizing factor type 1. Infect. Immun. 1996, 64, 1694–1705. [Google Scholar] [PubMed]
- Vannini, E.; Panighini, A.; Cerri, C.; Fabbri, A.; Lisi, S.; Pracucci, E.; Benedetto, N.; Vannozzi, R.; Fiorentini, C.; Caleo, M.; et al. The bacterial protein toxin, cytotoxic necrotizing factor 1 (CNF1) provides long-term survival in a murine glioma model. BMC Cancer 2014, 14. [Google Scholar] [CrossRef] [PubMed]
- Vannini, E.; Olimpico, F.; Middei, S.; Ammassari-Teule, M.; de Graaf, E.L.; McDonnell, L.; Schmidt, G.; Fabbri, A.; Fiorentini, C.; Baroncelli, L.; et al. Electrophysiology of glioma: A Rho GTPase-activating protein reduces tumor growth and spares neuron structure and function. Neuro Oncol. 2016, 18, 1634–1643. [Google Scholar] [CrossRef] [PubMed]
- Vannini, E.; Maltese, F.; Olimpico, F.; Fabbri, A.; Costa, M.; Caleo, M.; Baroncelli, L. Progression of motor deficits in glioma-bearing mice: Impact of CNF1 therapy at symptomatic stages. Oncotarget 2017, 8, 23539–23550. [Google Scholar] [CrossRef] [PubMed]
- Guadagni, V.; Cerri, C.; Piano, I.; Novelli, E.; Gargini, C.; Fiorentini, C.; Caleo, M.; Strettoi, E. The bacterial toxin CNF1 as a tool to induce retinal degeneration reminiscent of retinitis pigmentosa. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Vega, F.M.; Ridley, A.J. Rho GTPases in cancer cell biology. FEBS Lett. 2008, 582, 2093–2101. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, R.; Pedersen, E.D.; Wang, Z.; Brakebusch, C. Rho GTPase function in tumorigenesis. Biochim. Biophys. Acta 2009, 1796, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.M.; Hall, W.A. Targeted toxins in brain tumor therapy. Toxins 2010, 2, 2645–2662. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Toxin | Therapeutic Application |
|---|---|
| Botulinum neurotoxin (BoNT) from C. botulinum | Dystonia Muscle tone disorders Autonomic disorders Cosmetic use Pain therapy |
| Lethal toxin (LF) from B. anthracis | Potential treatment of cancer |
| Pertussis toxin (PTX) from B. pertussis | Potential use in control of HIV replication |
| Cytotoxic nectorizing factor 1 (CNF1) from E. coli | Potential use in learning and memory enhancement Potential treatment for neurodegenerative disorders Potential treatment of primary brain tumors |
| Immunotoxins | Cancer therapy |
| Chlorotoxins from Leiurus quinquestriatus scorpion venom | Potential treatment of primary brain tumors, currently used to deliver anti-cancer drugs specifically to cancer cells |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tantillo, E.; Colistra, A.; Vannini, E.; Cerri, C.; Pancrazi, L.; Baroncelli, L.; Costa, M.; Caleo, M. Bacterial Toxins and Targeted Brain Therapy: New Insights from Cytotoxic Necrotizing Factor 1 (CNF1). Int. J. Mol. Sci. 2018, 19, 1632. https://doi.org/10.3390/ijms19061632
Tantillo E, Colistra A, Vannini E, Cerri C, Pancrazi L, Baroncelli L, Costa M, Caleo M. Bacterial Toxins and Targeted Brain Therapy: New Insights from Cytotoxic Necrotizing Factor 1 (CNF1). International Journal of Molecular Sciences. 2018; 19(6):1632. https://doi.org/10.3390/ijms19061632
Chicago/Turabian StyleTantillo, Elena, Antonella Colistra, Eleonora Vannini, Chiara Cerri, Laura Pancrazi, Laura Baroncelli, Mario Costa, and Matteo Caleo. 2018. "Bacterial Toxins and Targeted Brain Therapy: New Insights from Cytotoxic Necrotizing Factor 1 (CNF1)" International Journal of Molecular Sciences 19, no. 6: 1632. https://doi.org/10.3390/ijms19061632