Bosutinib Inhibits EGFR Activation in Head and Neck Cancer

 , , and
, , and
Abstract
:1. Introduction
2. Results
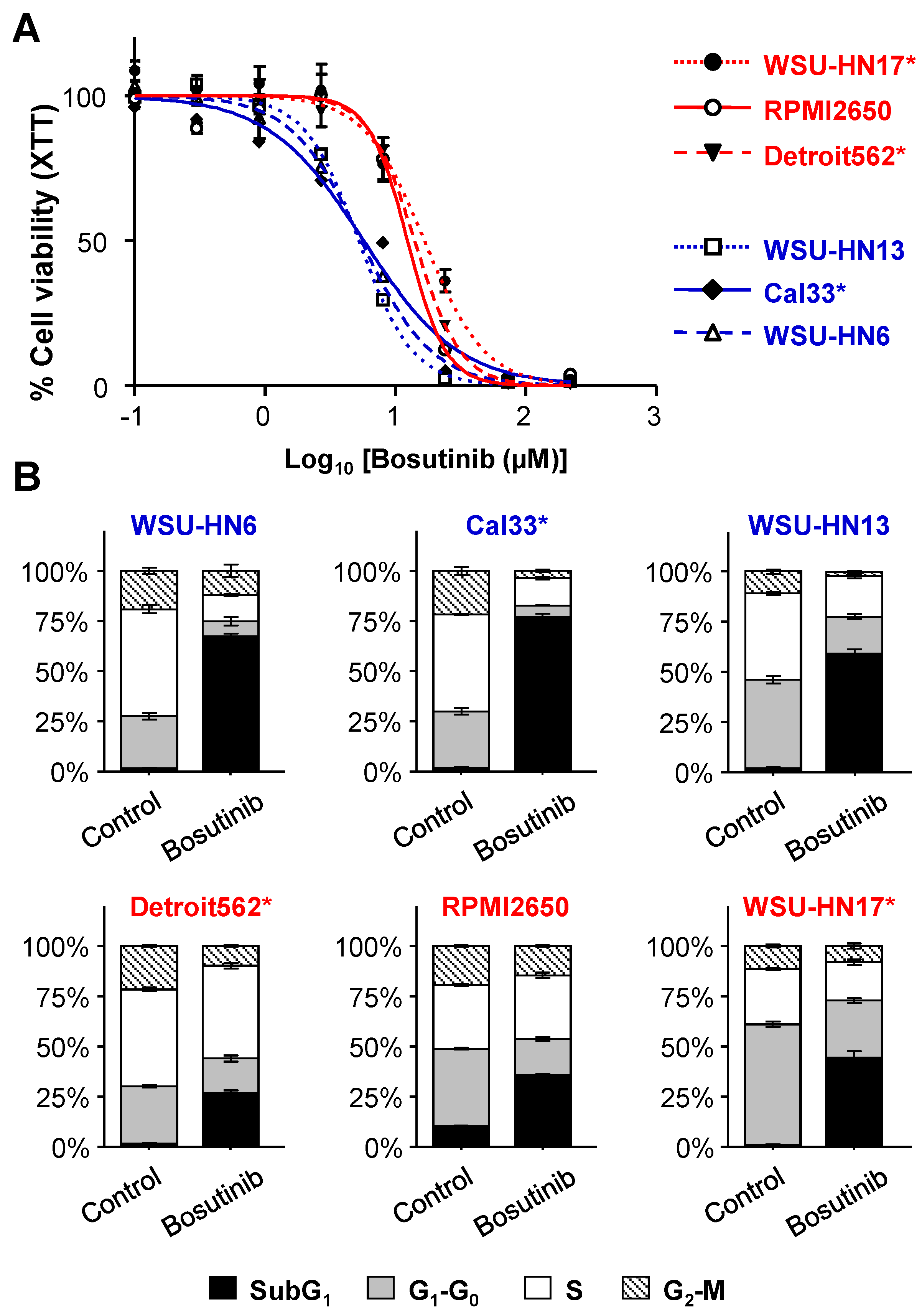
2.1. Sensitivity of HNSCC Cell Lines to Bosutinib
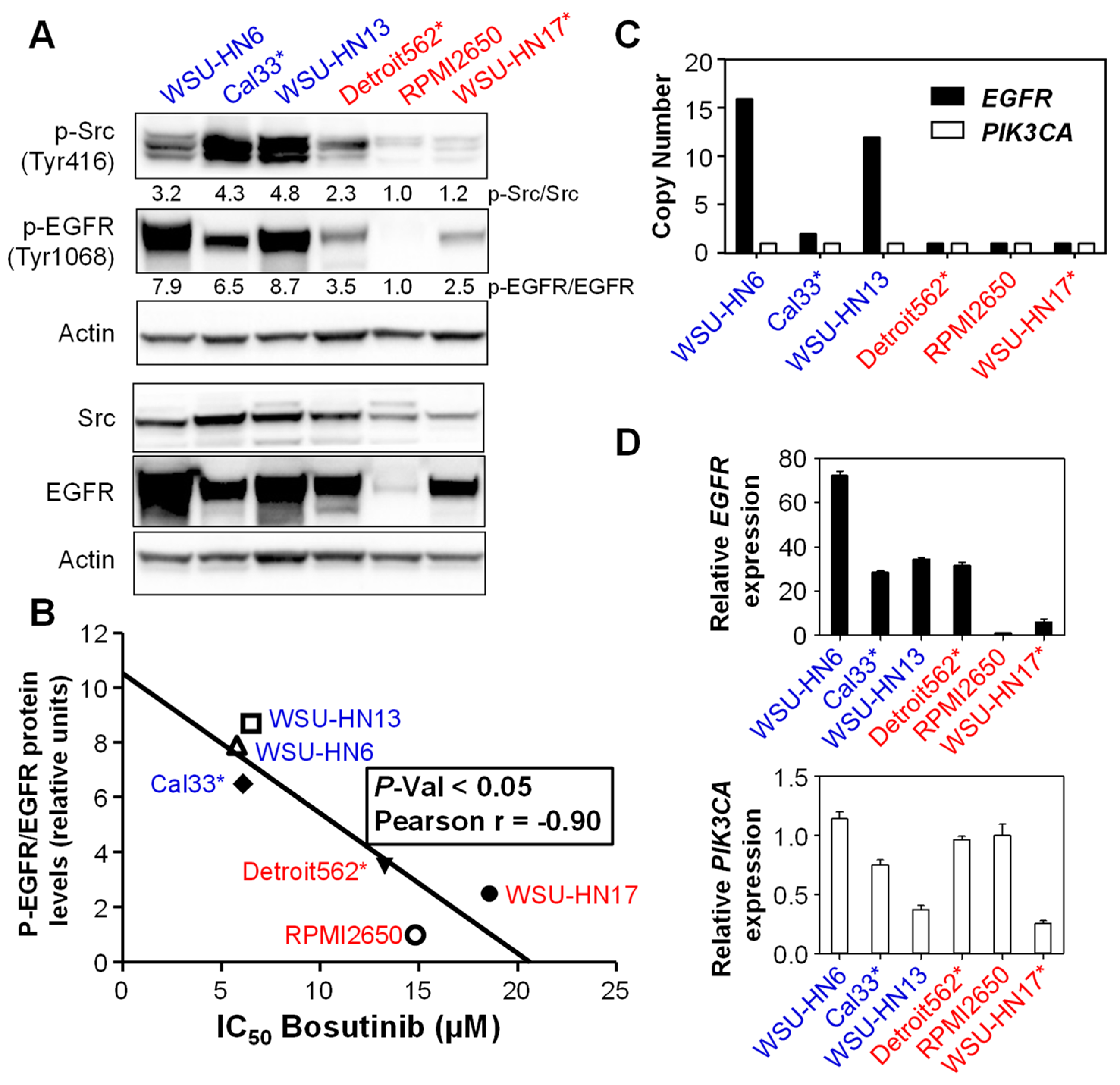
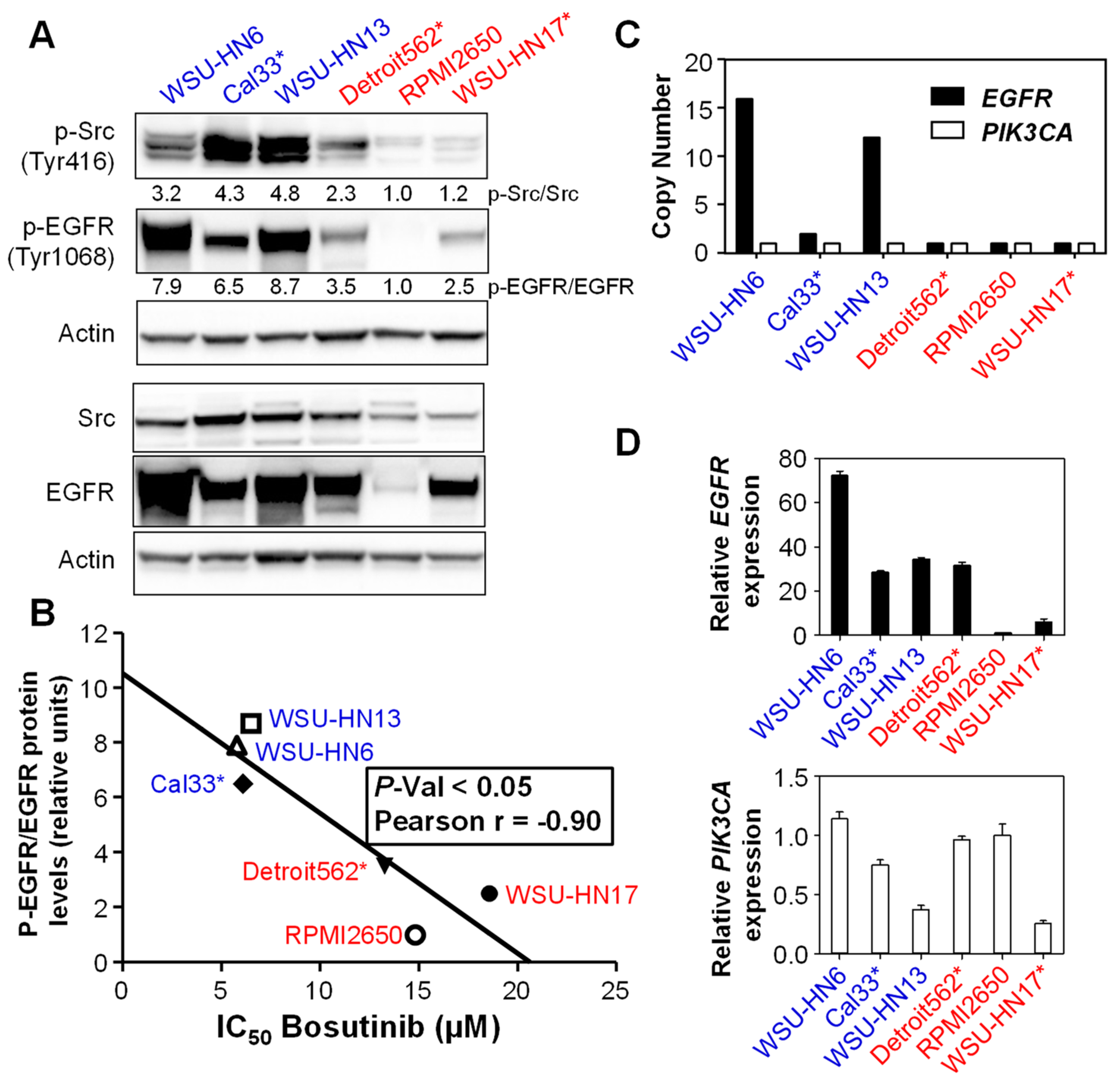
2.2. Sensitivity to Bosutinib Correlates with Phosphorylated EGFR Protein Levels
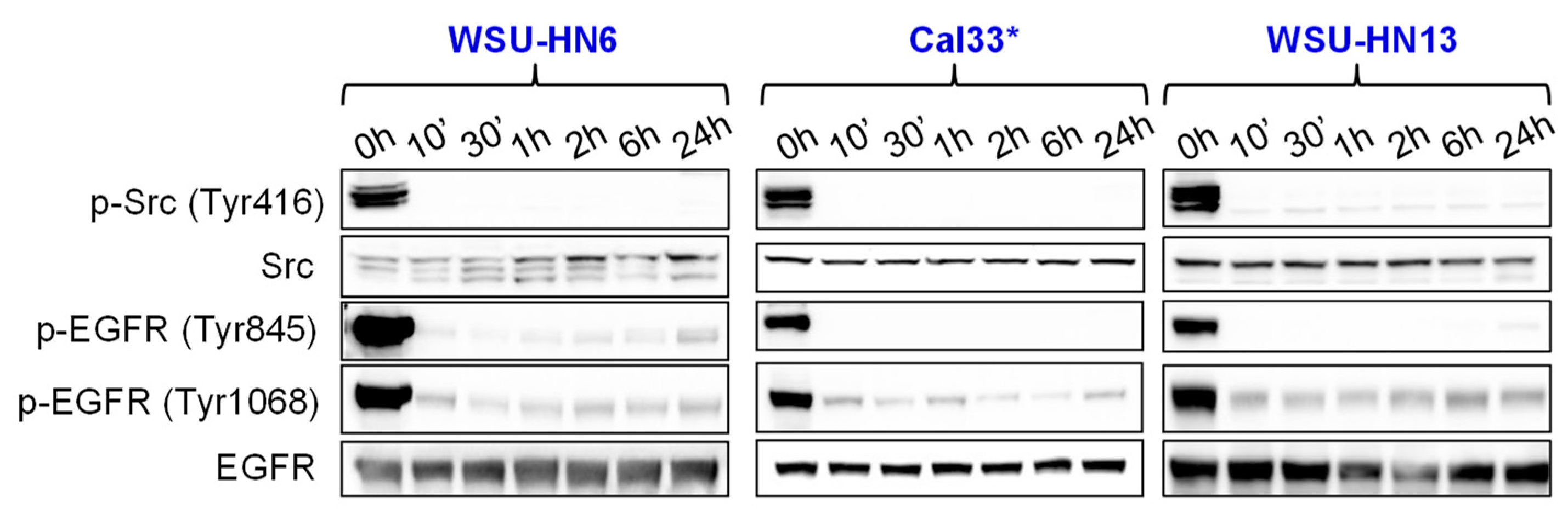
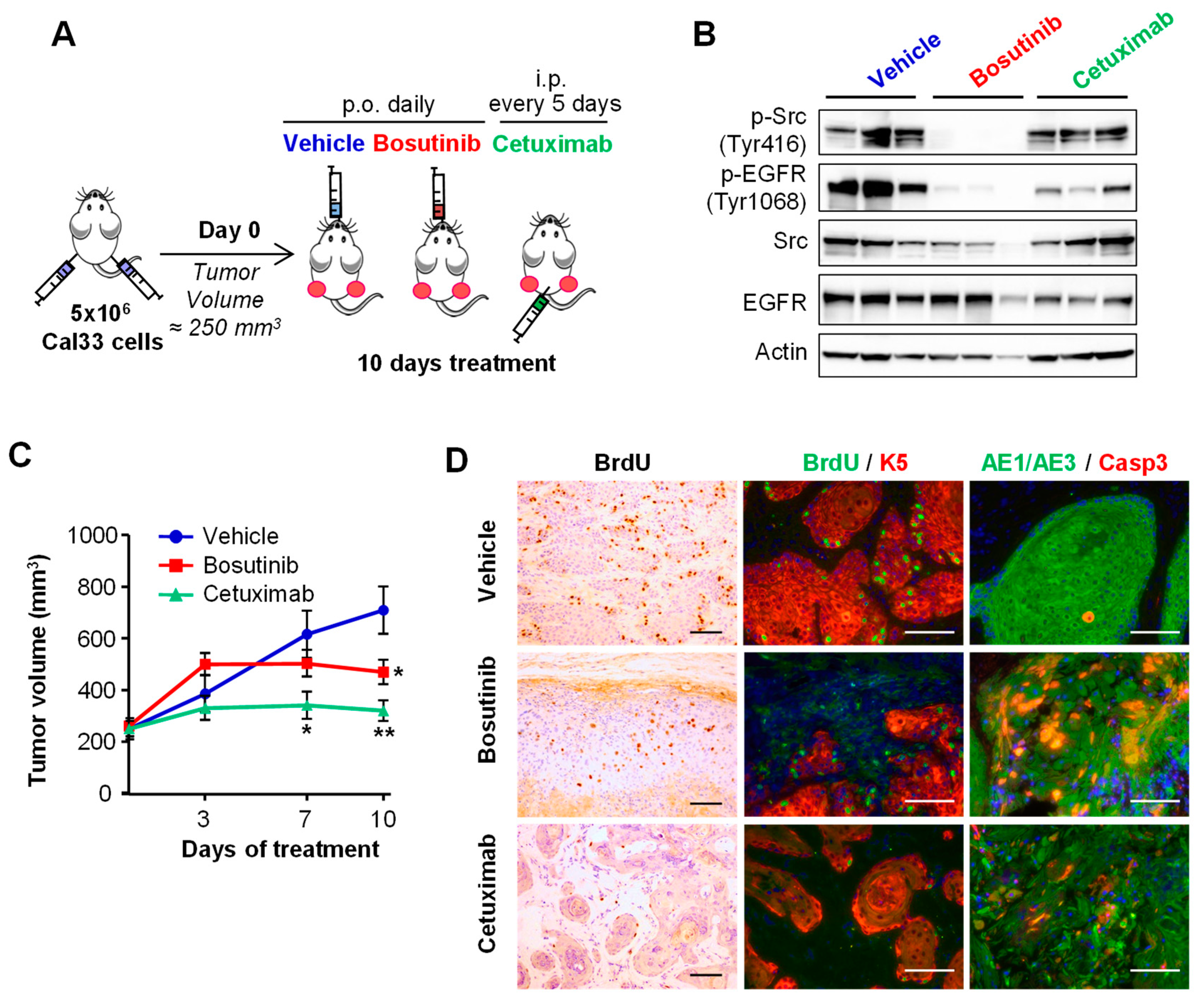
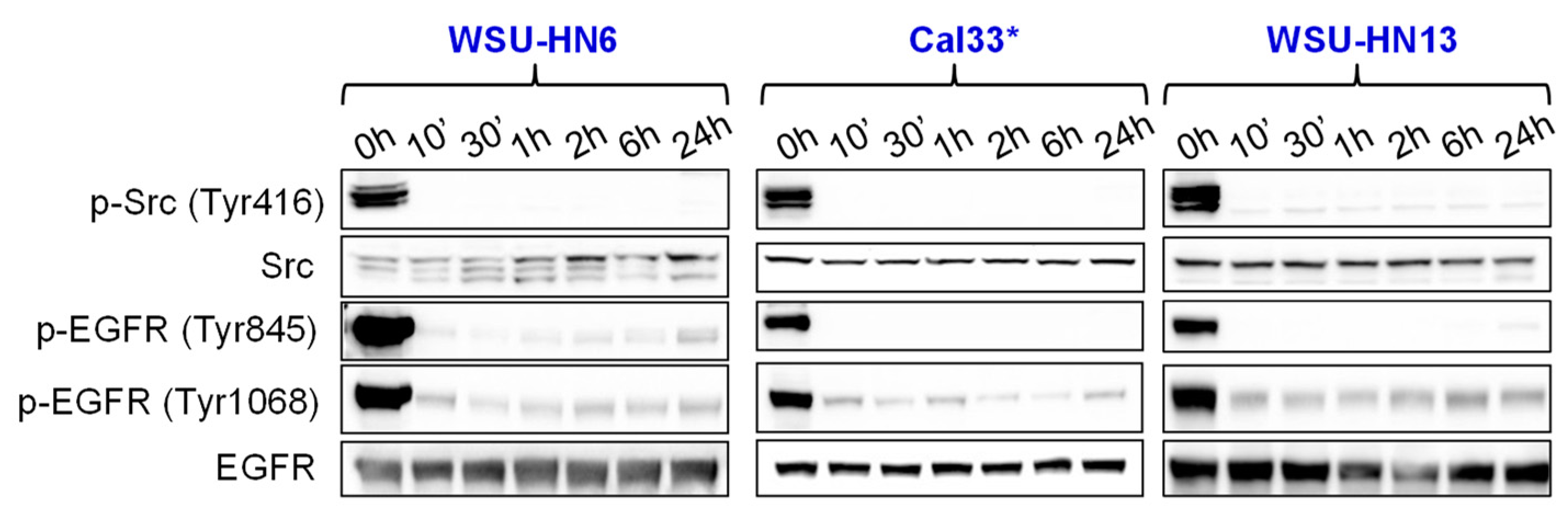
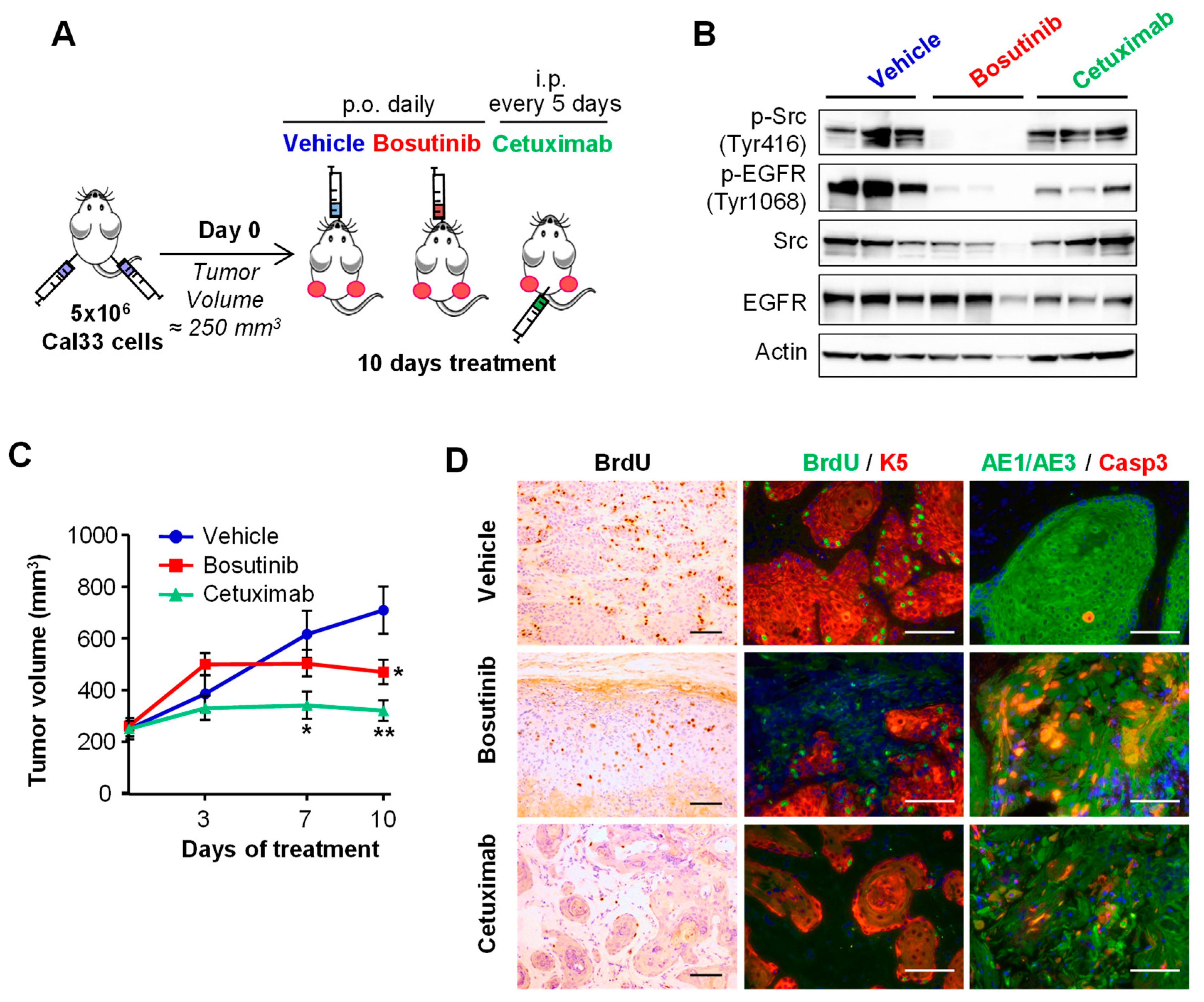
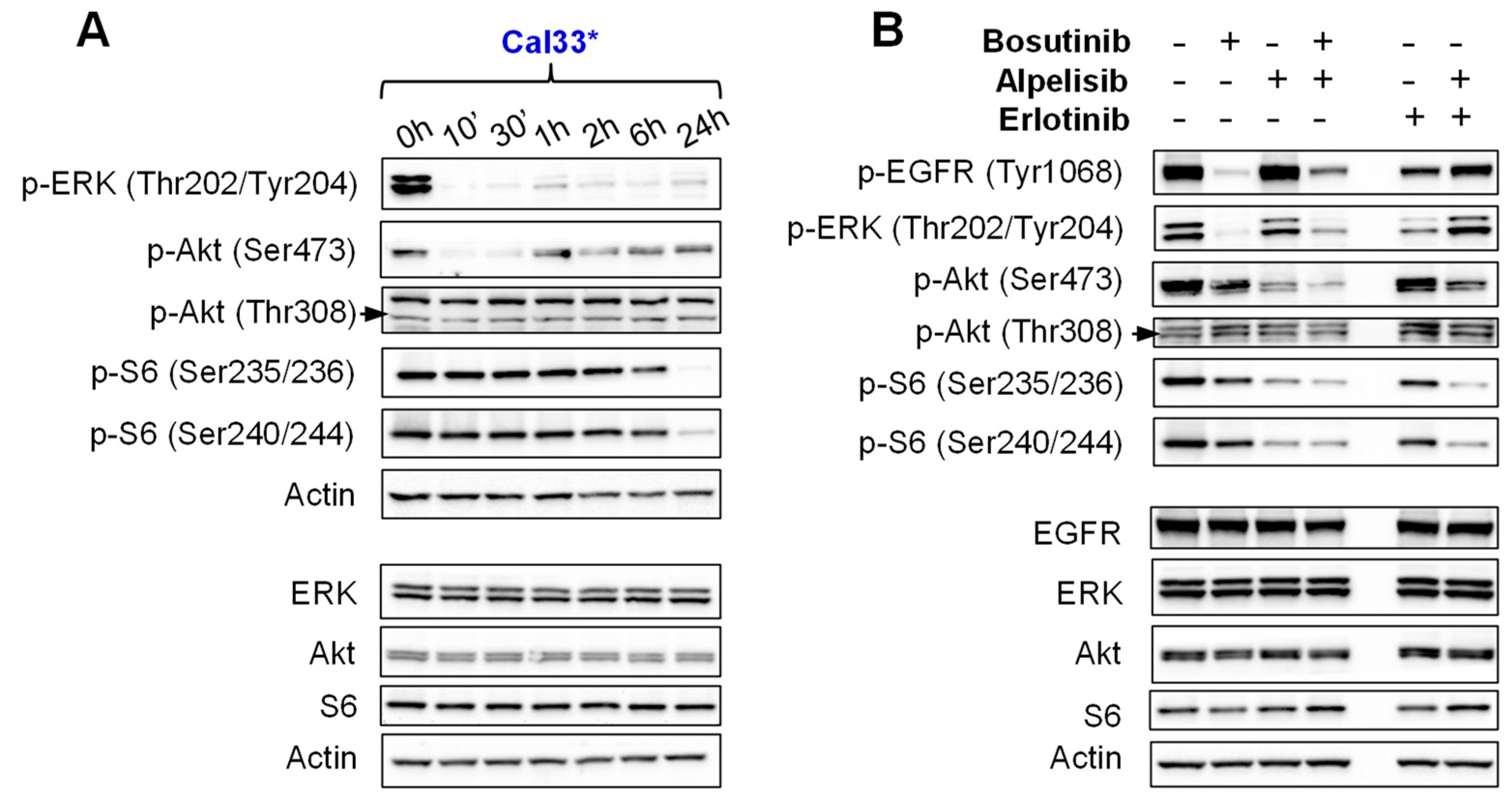
2.3. Bosutinib Effectively Inhibits Src and EGFR Phosphorylation in HNSCC Sensitive Cell Lines and in Xenografts
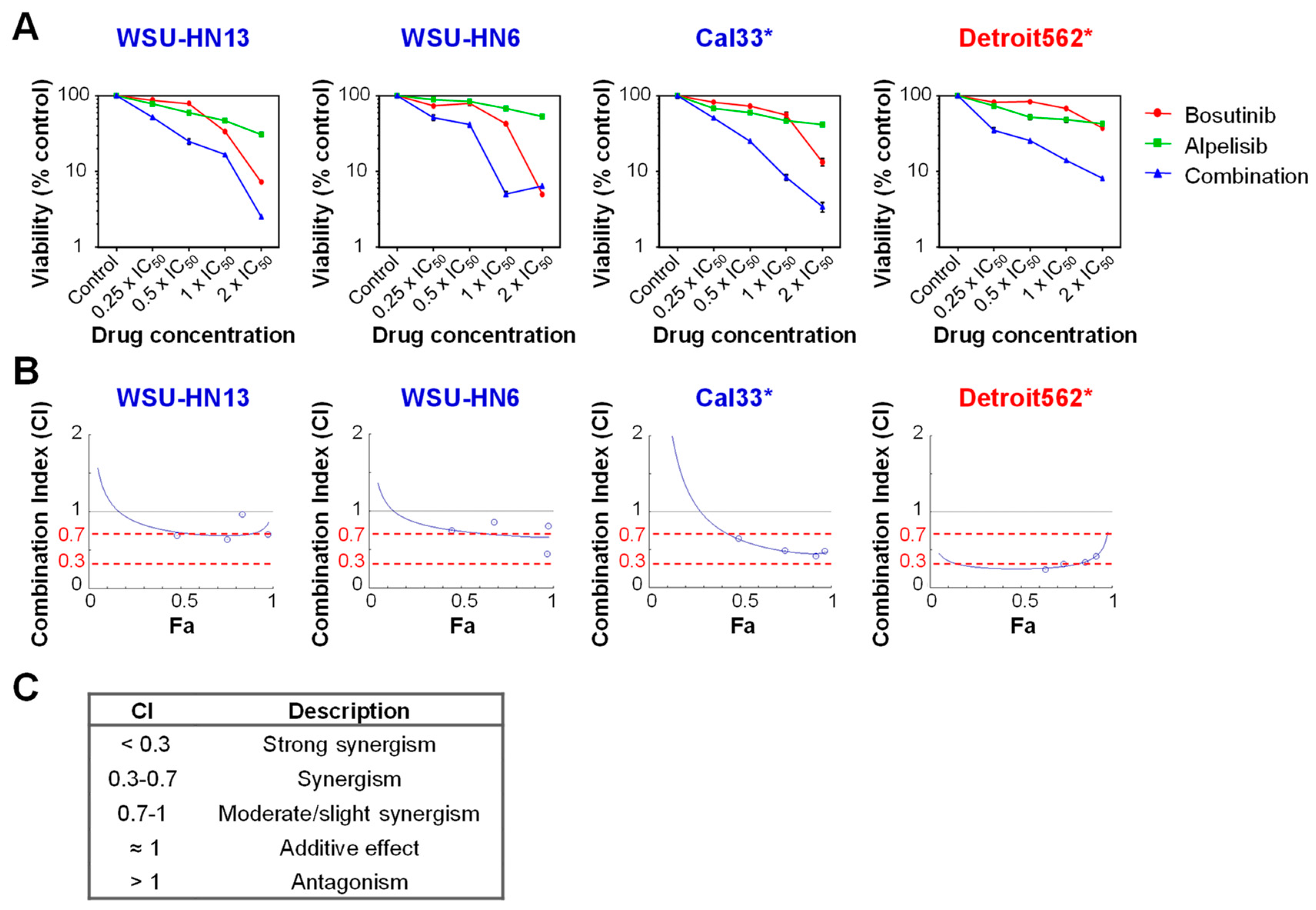
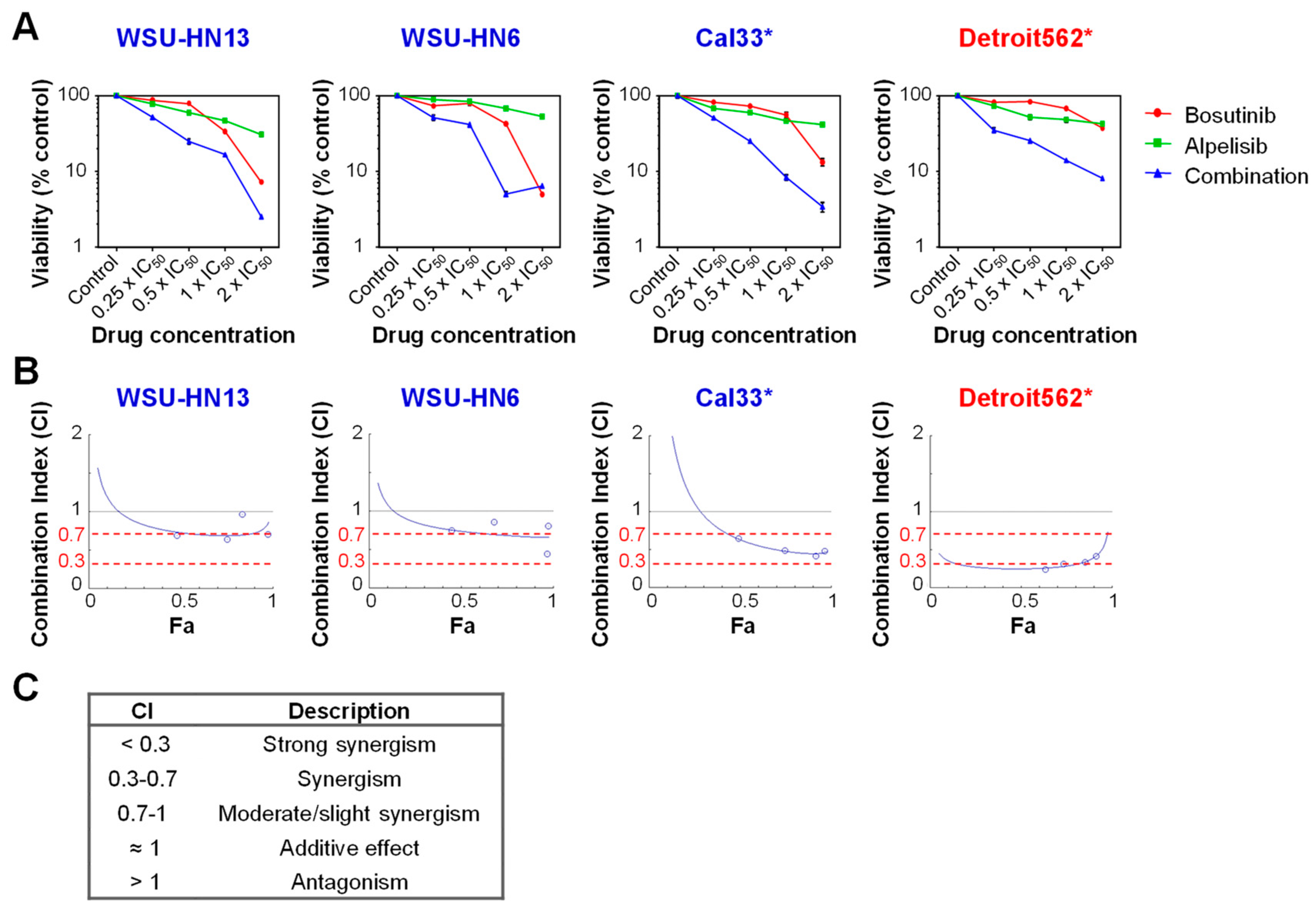
2.4. Bosutinib and PI3Kα Inhibitors Show a Synergistic Effect on Cell Viability in the Presence of Mutations in PIK3CA
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Inhibitors
4.2. Cell Viability Assays and Drug Combination Studies
4.3. Cell Cycle Analysis
4.4. Copy Number Variation (CNV) Analysis
4.5. mRNA Expression Analysis
4.6. Western Blotting
4.7. HNSCC Tumor-Derived Cell Line Xenograft Mouse Model
4.8. Immunohistochemistry and Immunofluorescence
4.9. Statistical Analysis
Supplementary Materials
Author Contributions
Founding
Acknowledgments
Conflicts of Interest
Abbreviations
| EGFR | Epidermal growth factor receptor |
| HNSCC | Head and neck squamous cell carcinoma |
| PIK3CA | Phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit α |
| PI3Kα | Phosphatidylinositol 3-kinase α |
| P.o. | Per Os (oral administration) |
| I.p. | Intraperitoneal |
References
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 2015, 136, E359–E386. [Google Scholar] [CrossRef] [PubMed]
- Leemans, C.R.; Braakhuis, B.J.; Brakenhoff, R.H. The molecular biology of head and neck cancer. Nat. Rev. Cancer 2011, 11, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Leon, X.; Hitt, R.; Constenla, M.; Rocca, A.; Stupp, R.; Kovacs, A.F.; Amellal, N.; Bessa, E.H.; Bourhis, J. A retrospective analysis of the outcome of patients with recurrent and/or metastatic squamous cell carcinoma of the head and neck refractory to a platinum-based chemotherapy. Clin. Oncol. 2005, 17, 418–424. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Network. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature 2015, 517, 576–582. [Google Scholar] [Green Version]
- Rabinowits, G.; Haddad, R.I. Overcoming resistance to EGFR inhibitor in head and neck cancer: A review of the literature. Oral. Oncol. 2012, 48, 1085–1089. [Google Scholar] [CrossRef] [PubMed]
- Vermorken, J.B.; Mesia, R.; Rivera, F.; Remenar, E.; Kawecki, A.; Rottey, S.; Erfan, J.; Zabolotnyy, D.; Kienzer, H.R.; Cupissol, D.; et al. Platinum-based chemotherapy plus cetuximab in head and neck cancer. N. Engl. J. Med. 2008, 359, 1116–1127. [Google Scholar] [CrossRef] [PubMed]
- Bonner, J.A.; Harari, P.M.; Giralt, J.; Azarnia, N.; Shin, D.M.; Cohen, R.B.; Jones, C.U.; Sur, R.; Raben, D.; Jassem, J.; et al. Radiotherapy plus cetuximab for squamous-cell carcinoma of the head and neck. N. Engl. J. Med. 2006, 354, 567–578. [Google Scholar] [CrossRef] [PubMed]
- Vlacich, G.; Coffey, R.J. Resistance to EGFR-targeted therapy: A family affair. Cancer Cell 2011, 20, 423–425. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, M.S.; Stojanov, P.; Mermel, C.H.; Robinson, J.T.; Garraway, L.A.; Golub, T.R.; Meyerson, M.; Gabriel, S.B.; Lander, E.S.; Getz, G. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature 2014, 505, 495–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García-Escudero, R.; Segrelles, C.; Dueñas, M.; Pombo, M.; Ballestín, C.; Alonso-Riaño, M.; Nenclares, P.; Álvarez-Rodríguez, R.; Sánchez-Aniceto, G.; Ruíz-Alonso, A.; López-Cedrún, J.L. Overexpression of PIK3CA in head and neck squamous cell carcinoma is associated with poor outcome and activation of the YAP pathway. Oral. Oncol. 2018, 79, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Garnett, M.J.; Edelman, E.J.; Heidorn, S.J.; Greenman, C.D.; Dastur, A.; Lau, K.W.; Greninger, P.; Thompson, I.R.; Luo, X.; Soares, J.; et al. Systematic identification of genomic markers of drug sensitivity in cancer cells. Nature 2012, 483, 570–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nichols, A.C.; Black, M.; Yoo, J.; Pinto, N.; Fernandes, A.; Haibe-Kains, B.; Boutros, P.C.; Barrett, J.W. Exploiting high-throughput cell line drug screening studies to identify candidate therapeutic agents in head and neck cancer. BMC Pharmacol. Toxicol. 2014, 15, 66. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.K.B.; Tiong, K.H.; Chang, J.K.; Liew, C.S.; Rahman, Z.A.A.; Tan, A.C.; Khang, T.F.; Cheong, S.C. DeSigN: Connecting gene expression with therapeutics for drug repurposing and development. BMC Genom. 2017, 18, 934. [Google Scholar] [CrossRef] [PubMed]
- Rusconi, F.; Piazza, R.; Vagge, E.; Gambacorti-Passerini, C. Bosutinib: A review of preclinical and clinical studies in chronic myelogenous leukemia. Expert Opin. Pharm. 2014, 15, 701–710. [Google Scholar] [CrossRef] [PubMed]
- Fung, C.; Grandis, J.R. Emerging drugs to treat squamous cell carcinomas of the head and neck. Expert Opin. Emerg. Drugs 2010, 15, 355–373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Formisano, L.; D’Amato, V.; Servetto, A.; Brillante, S.; Raimondo, L.; Di Mauro, C.; Marciano, R.; Orsini, R.C.; Cosconati, S.; Randazzo, A.; et al. Src inhibitors act through different mechanisms in Non-Small Cell Lung Cancer models depending on EGFR and RAS mutational status. Oncotarget 2015, 6, 26090–26103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elkabets, M.; Pazarentzos, E.; Juric, D.; Sheng, Q.; Pelossof, R.A.; Brook, S.; Benzaken, A.O.; Rodon, J.; Morse, N.; Yan, J.J.; et al. AXL mediates resistance to PI3Kα inhibition by activating the EGFR/PKC/mTOR axis in head and neck and esophageal squamous cell carcinomas. Cancer Cell 2015, 27, 533–546. [Google Scholar] [CrossRef] [PubMed]
- Fritsch, C.; Huang, A.; Chatenay-Rivauday, C.; Schnell, C.; Reddy, A.; Liu, M.; Kauffmann, A.; Guthy, D.; Erdmann, D.; De Pover, A.; et al. Characterization of the novel and specific PI3Kα inhibitor NVP-BYL719 and development of the patient stratification strategy for clinical trials. Mol. Cancer Ther. 2014, 13, 1117–1129. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.H.; Ma, B.B.; Cheong, H.T.; Hui, C.W.; Hui, E.P.; Chan, A.T. Preclinical evaluation of PI3K inhibitor BYL719 as a single agent and its synergism in combination with cisplatin or MEK inhibitor in nasopharyngeal carcinoma (NPC). Am. J. Cancer Res. 2015, 5, 1496–1506. [Google Scholar] [PubMed]
- Nam, A.R.; Kim, J.W.; Park, J.E.; Bang, J.H.; Jin, M.H.; Lee, K.H.; Kim, T.Y.; Han, S.W.; Im, S.A.; Kim, T.Y.; et al. Src as a Therapeutic Target in Biliary Tract Cancer. Mol. Cancer Ther. 2016, 15, 1515–1524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vojvodic, M.; Hansford, L.; Morozova, O.; Blakely, K.; Taylor, P.; Fathers, K.; Moffat, J.; Marra, M.; Smith, K.; Moran, M.; et al. A phosphoproteomics approach to identify candidate kinase inhibitor pathway targets in lymphoma-like primary cell lines. Curr. Drug Discov. Technol. 2013, 10, 283–304. [Google Scholar] [CrossRef] [PubMed]
- U.S. Food & Drug Administration. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2012/203341lbl.pdf (accessed on 19 June 2018).
- Stransky, N.; Egloff, A.M.; Tward, A.D.; Kostic, A.D.; Cibulskis, K.; Sivachenko, A.; Kryukov, G.V.; Lawrence, M.S.; Sougnez, C.; McKenna, A.; et al. The mutational landscape of head and neck squamous cell carcinoma. Science 2011, 333, 1157–1160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pickering, C.R.; Zhang, J.; Yoo, S.Y.; Bengtsson, L.; Moorthy, S.; Neskey, D.M.; Zhao, M.; Alves, M.V.O.; Chang, K.; Drummond, J.; et al. Integrative genomic characterization of oral squamous cell carcinoma identifies frequent somatic drivers. Cancer Discov. 2013, 3, 770–781. [Google Scholar] [CrossRef] [PubMed]
- Lui, V.W.; Hedberg, M.L.; Li, H.; Vangara, B.S.; Pendleton, K.; Zeng, Y.; Lu, Y.; Zhang, Q.; Du, Y.; Gilbert, B.R.; et al. Frequent mutation of the PI3K pathway in head and neck cancer defines predictive biomarkers. Cancer Discov. 2013, 3, 761–769. [Google Scholar] [CrossRef] [PubMed]
- Sato, K. Cellular functions regulated by phosphorylation of EGFR on Tyr845. Int. J. Mol. Sci. 2013, 14, 10761–10790. [Google Scholar] [CrossRef] [PubMed]
- Tice, D.A.; Biscardi, J.S.; Nickles, A.L.; Parsons, S.J. Mechanism of biological synergy between cellular Src and epidermal growth factor receptor. Proc. Natl. Acad. Sci. USA 1999, 96, 1415–1420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, D.; Abba, M.C.; Molinolo, A.A.; Vitale-Cross, L.; Wang, Z.; Zaida, M.; Delic, N.C.; Samuels, Y.; Lyons, J.G.; Gutkind, J.S. The head and neck cancer cell oncogenome: A platform for the development of precision molecular therapies. Oncotarget 2014, 5, 8906–8923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chou, T.C.; Talalay, P. Quantitative analysis of dose-effect relationships: The combined effects of multiple drugs or enzyme inhibitors. Adv. Enzyme. Regul. 1984, 22, 27–55. [Google Scholar] [CrossRef]
- Tomayko, M.M.; Reynolds, C.P. Determination of subcutaneous tumor size in athymic (nude) mice. Cancer Chemother. Pharmacol. 1989, 24, 148–154. [Google Scholar] [CrossRef] [PubMed]
- Golas, J.M.; Lucas, J.; Etienne, C.; Golas, J.; Discafani, C.; Sridharan, L.; Boghaert, E.; Arndt, K.; Ye, F.; Boschelli, D.H.; et al. SKI-606, a Src/Abl inhibitor with in vivo activity in colon tumor xenograft models. Cancer Res. 2005, 65, 5358–5364. [Google Scholar] [CrossRef] [PubMed]
- Chou, T.C. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol. Rev. 2006, 58, 621–681. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | Origin | EGFR | PIK3CA |
|---|---|---|---|
| Cal33 | Tongue SCC | Wt | Mut (H1047R) |
| Detroit562 | Pharynx/pleural effusion metastasis | Wt | Mut (H1047R) |
| RPMI2650 | Nasal septum metastasis | Wt | Wt |
| WSU-HN6 | Base of tongue SCC | Wt | Wt |
| WSU-HN13 | Tongue SCC | Wt | Wt |
| WSU-HN17 | Lymph node | Wt | Mut (E542K) |
| Cell Line | Bosutinib | Alpelisib IC50 | Combination Index IC50 | % of SubG1 Cells at IC75 of Bosutinib | ||
|---|---|---|---|---|---|---|
| IC25 | IC50 | IC75 | ||||
| WSU-HN17 * | 14.42 ± 3.43 † | 18.56 ± 2.49 ‡ | 25.02 ± 2.09 ‡ | - | - | 44.25 ± 3.09 ‡ |
| RPMI2650 | 10.89 ± 5.50 | 14.81 ± 4.31 ‡ | 23.82 ± 3.03 ‡ | - | - | 35.53 ± 0.63 ‡ |
| Detroit562 * | 8.27 ± 0.92 | 13.25 ± 0.52 ‡ | 21.52 ± 1.27 ‡ | 36.84 ± 6.79 | 0.25 | 26.76 ± 1.05 ‡ |
| WSU-HN13 | 5.07 ± 1.12 | 6.49 ± 0.68 | 8.57 ± 0.18 | 35.41 ± 6.84 | 0.73 | 59.14 ± 2.11 † |
| Cal33 * | 2.98 ± 0.10 | 6.09 ± 0.11 | 12.76 ± 0.09 | 12.67 ± 2.39 | 0.63 | 77.17 ±1.31 ‡ |
| WSU-HN6 | 2.94 ± 0.16 | 5.78 ± 0.29 | 11.35 ± 0.55 | 35.60 ± 4.60 | 0.74 | 67.17 ± 1.51 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Segrelles, C.; Contreras, D.; Navarro, E.M.; Gutiérrez-Muñoz, C.; García-Escudero, R.; Paramio, J.M.; Lorz, C. Bosutinib Inhibits EGFR Activation in Head and Neck Cancer. Int. J. Mol. Sci. 2018, 19, 1824. https://doi.org/10.3390/ijms19071824
Segrelles C, Contreras D, Navarro EM, Gutiérrez-Muñoz C, García-Escudero R, Paramio JM, Lorz C. Bosutinib Inhibits EGFR Activation in Head and Neck Cancer. International Journal of Molecular Sciences. 2018; 19(7):1824. https://doi.org/10.3390/ijms19071824
Chicago/Turabian StyleSegrelles, Carmen, David Contreras, Elena M. Navarro, Carmen Gutiérrez-Muñoz, Ramón García-Escudero, Jesús M. Paramio, and Corina Lorz. 2018. "Bosutinib Inhibits EGFR Activation in Head and Neck Cancer" International Journal of Molecular Sciences 19, no. 7: 1824. https://doi.org/10.3390/ijms19071824