Anti-Correlation between the Dynamics of the Active Site Loop and C-Terminal Tail in Relation to the Homodimer Asymmetry of the Mouse Erythroid 5-Aminolevulinate Synthase

 , and
, and 
Abstract
:1. Introduction
2. Results
2.1. Potential for Formation of a β-Strand in the ALAS2 C-Tail as Revealed Using Bioinformatics
2.2. Higher Backbone RMSD in the ALAS2 C-Tail Than in Other Protein Regions
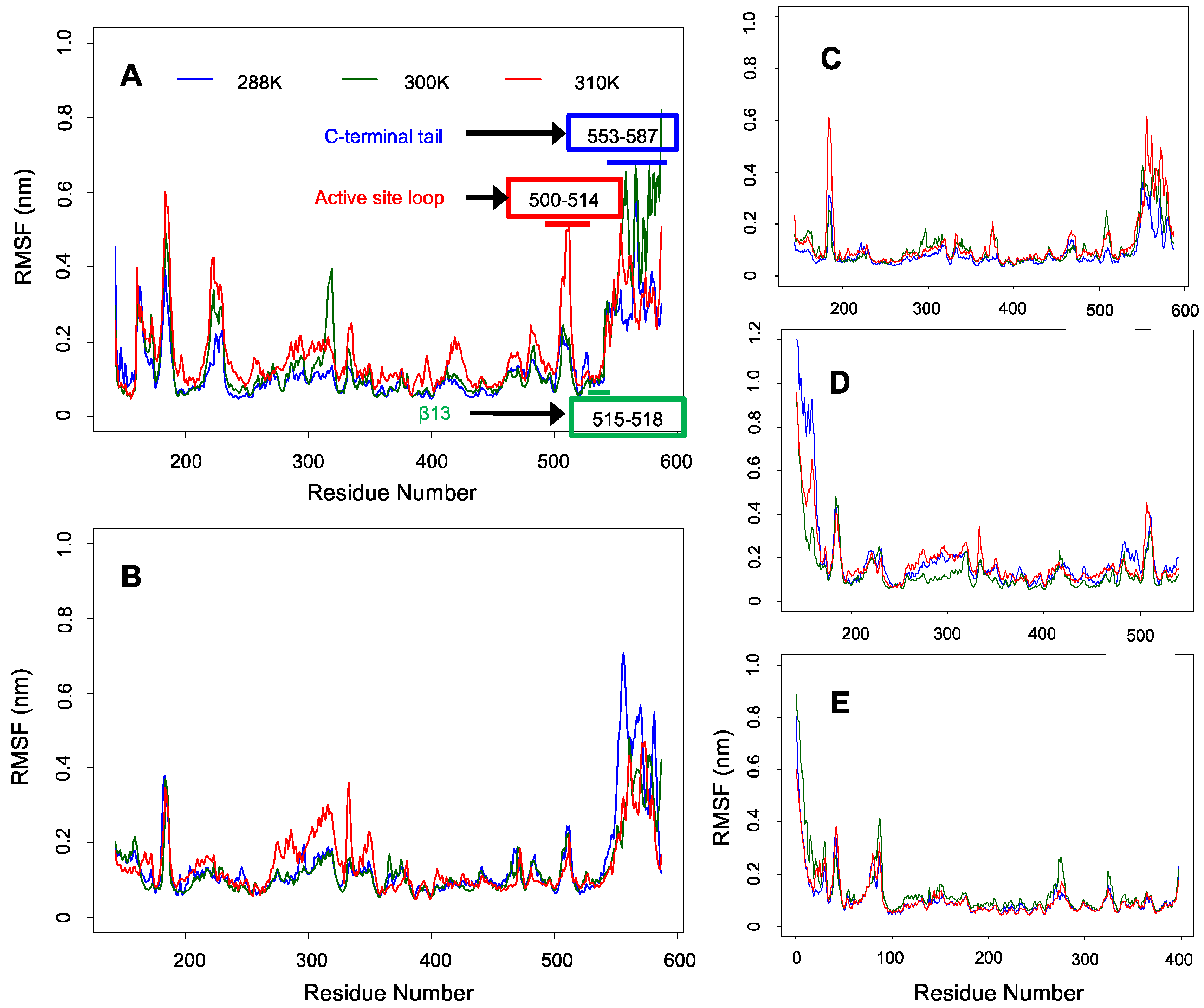
2.3. Fewer Structural Fluctuations in the Conserved Region Than in the C-Terminal Tail of ALAS2 as Deduced from the Backbone RMSF Analysis
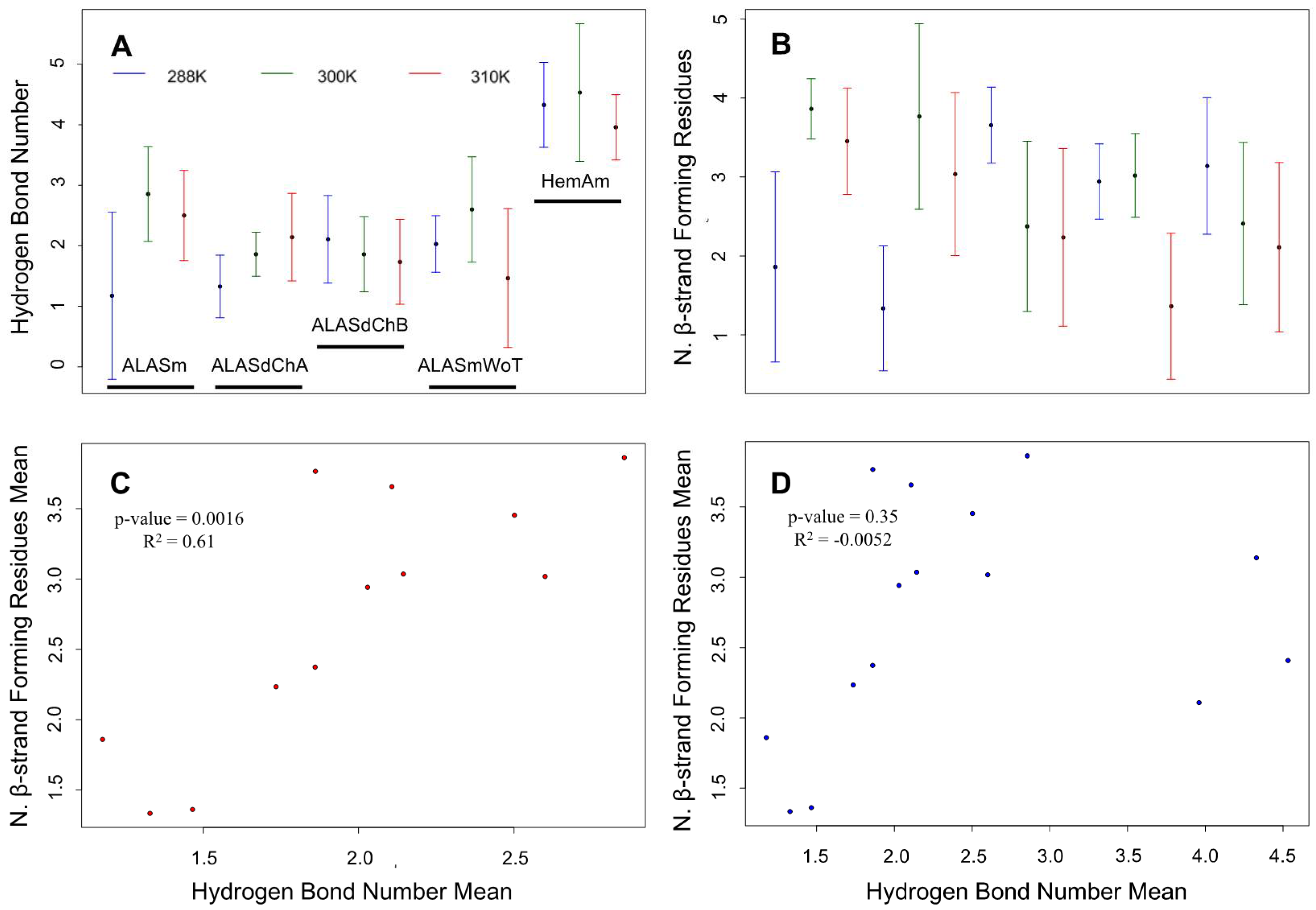
2.4. Confirmation of the Correlation between the Number of Hydrogen Bonds and the β-Strand-Forming Residues in the ALAS2 Active Site Loop as Revealed by MD Simulations
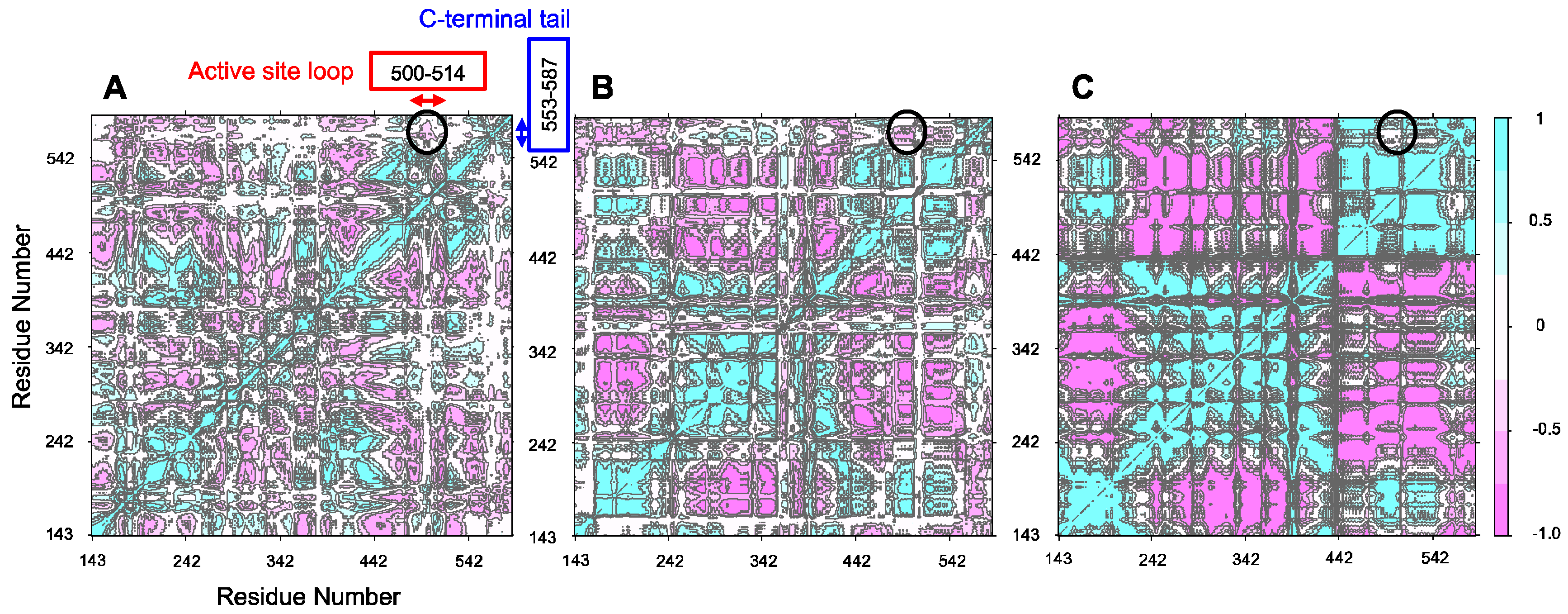
2.5. Anti-Correlation between the Active Site Loop and the C-Terminal Tail of Murine ALAS2
3. Discussion
4. Experimental Section
4.1. Sequence Analysis and Intrinsic Disorder Prediction
4.2. Molecular Dynamics (MD) Simulation Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Fratz, E.J.; Stojanovski, B.M.; Ferreira, G.C. Toward Heme: 5-Aminolevulinate Synthase and Initiation of Porphyrin Synthesis. In Handbook of Porphyrin Science, Heme Biochemistry; Ferreira, G.C., Kadish, K.M., Smith, K.M., Guilard, R., Eds.; World Scientific Publishing Co. Pte. Ltd.: Singapore, 2014; Volume 26, pp. 3–68. [Google Scholar]
- Hunter, G.A.; Ferreira, G.C. Molecular enzymology of 5-aminolevulinate synthase, the gatekeeper of heme biosynthesis. Biochim. Biophys. Acta 2011, 1814, 1467–1473. [Google Scholar] [CrossRef] [PubMed]
- Astner, I.; Schulze, J.O.; van den Heuvel, J.; Jahn, D.; Schubert, W.D.; Heinz, D.W. Crystal structure of 5-aminolevulinate synthase, the first enzyme of heme biosynthesis, and its link to XLSA in humans. EMBO J. 2005, 24, 3166–3177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hunter, G.A.; Ferreira, G.C. 5-aminolevulinate synthase: Catalysis of the first step of heme biosynthesis. Cell. Mol. Biol. 2009, 5, 102–110. [Google Scholar]
- Dailey, H.A.; Dailey, T.A.; Gerdes, S.; Jahn, D.; Jahn, M.; O’Brian, M.R.; Warren, M.J. Prokaryotic Heme Biosynthesis: Multiple Pathways to a Common Essential Product. Microbiol. Mol. Biol. Rev. 2017, 81, e00048-16. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Kaganjo, J.; Zhang, W.; Zeilstra-Ryalls, J. Investigating the bifunctionality of cyclizing and “classical” 5-aminolevulinate synthases. Protein Sci. 2018, 27, 402–410. [Google Scholar] [CrossRef] [PubMed]
- Bottomley, S.S.; Fleming, M.D. Sideroblastic anemia: Diagnosis and management. Hematol. Oncol. Clin. N. Am. 2014, 28, 653–670. [Google Scholar] [CrossRef] [PubMed]
- Bottomley, S.S.; Fleming, M.D. Sideroblastic Anemias: Molecular Basis, Pathophysiology, and Clinical Aspects. In Handbook of Porphyrin Science; World Scientific Publishing Company: Singapore, 2013; Volume 29, pp. 43–87. [Google Scholar]
- Fratz, E.J.; Clayton, J.; Hunter, G.A.; Ducamp, S.; Breydo, L.; Uversky, V.N.; Deybach, J.C.; Gouya, L.; Puy, H.; Ferreira, G.C. Human Erythroid 5-Aminolevulinate Synthase Mutations Associated with X-Linked Protoporphyria Disrupt the Conformational Equilibrium and Enhance Product Release. Biochemistry 2015, 54, 5617–5631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puy, H.; Gouya, L.; Deybach, J.C. Porphyrias. Lancet 2010, 375, 924–937. [Google Scholar] [CrossRef]
- Whatley, S.D.; Ducamp, S.; Gouya, L.; Grandchamp, B.; Beaumont, C.; Badminton, M.N.; Elder, G.H.; Holme, S.A.; Anstey, A.V.; Parker, M.; et al. C-terminal deletions in the ALAS2 gene lead to gain of function and cause X-linked dominant protoporphyria without anemia or iron overload. Am. J. Hum. Genet. 2008, 83, 408–414. [Google Scholar] [CrossRef] [PubMed]
- Fratz-Berilla, E.J.; Breydo, L.; Gouya, L.; Puy, H.; Uversky, V.N.; Ferreira, G.C. Isoniazid inhibits human erythroid 5-aminolevulinate synthase: Molecular mechanism and tolerance study with four x-linked protoporphyria patients. Biochim. Biophys. Acta 2017, 1863, 428–439. [Google Scholar] [CrossRef] [PubMed]
- Manceau, H.; Gouya, L.; Puy, H. Acute hepatic and erythropoietic porphyrias: From ALA synthases 1 and 2 to new molecular bases and treatments. Curr. Opin. Hemat. 2017, 24, 198–207. [Google Scholar] [CrossRef] [PubMed]
- Bishop, D.F.; Tchaikovskii, V.; Nazarenko, I.; Desnick, R.J. Molecular expression and characterization of erythroid-specific 5-aminolevulinate synthase gain-of-function mutations causing X-linked protoporphyria. Mol. Med. 2013, 19, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Ducamp, S.; Schneider-Yin, X.; de Rooij, F.; Clayton, J.; Fratz, E.J.; Rudd, A.; Ostapowicz, G.; Varigos, G.; Lefebvre, T.; Deybach, J.-C.; et al. Molecular and functional analysis of the C-terminal region of human erythroid-specific 5-aminolevulinic synthase associated with X-linked dominant protoporphyria (XLDPP). Hum. Mol. Genet. 2013, 22, 1280–1288. [Google Scholar] [CrossRef] [PubMed]
- Balwani, M.; Doheny, D.; Bishop, D.F.; Nazarenko, I.; Yasuda, M.; Dailey, H.A.; Anderson, K.E.; Bissell, D.M.; Bloomer, J.; Bonkovsky, H.L.; et al. Loss-of-Function Ferrochelatase and Gain-of-Function Erythroid-Specific 5-Aminolevulinate Synthase Mutations Causing Erythropoietic Protoporphyria and X-Linked Protoporphyria in North American Patients Reveal Novel Mutations and a High Prevalence of X-Linked Protoporphyria. Mol. Med. 2013, 19, 26–35. [Google Scholar] [PubMed]
- Hunter, G.A.; Ferreira, G.C. Pre-steady-state reaction of 5-aminolevulinate synthase. Evidence for a rate-determining product release. J. Biol. Chem. 1999, 274, 12222–12228. [Google Scholar] [CrossRef] [PubMed]
- Hunter, G.A.; Zhang, J.; Ferreira, G.C. Transient kinetic studies support refinements to the chemical and kinetic mechanisms of aminolevulinate synthase. J. Biol. Chem. 2007, 282, 23025–23035. [Google Scholar] [CrossRef] [PubMed]
- Stojanovski, B.M.; Hunter, G.A.; Jahn, M.; Jahn, D.; Ferreira, G.C. Unstable Reaction Intermediates and Hysteresis during the Catalytic Cycle of 5-Aminolevulinate Synthase: Implications from Using Pseudo and Alternate Substrates and a Promiscuous Enzyme Variant. J. Biol. Chem. 2014, 289, 22915–22925. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Ferreira, G.C. Transient state kinetic investigation of 5-aminolevulinate synthase reaction mechanism. J. Biol. Chem. 2002, 277, 44660–44669. [Google Scholar] [CrossRef] [PubMed]
- Lendrihas, T.; Hunter, G.A.; Ferreira, G.C. Targeting the active site gate to yield hyperactive variants of 5-aminolevulinate synthase. J. Biol. Chem. 2010, 285, 13704–13711. [Google Scholar] [CrossRef] [PubMed]
- Goodfellow, B.J.; Dias, J.S.; Ferreira, G.C.; Henklein, P.; Wray, V.; Macedo, A.L. The solution structure and heme binding of the presequence of murine 5-aminolevulinate synthase. FEBS Lett. 2001, 505, 325–331. [Google Scholar] [CrossRef] [Green Version]
- Dunker, A.K.; Obradovic, Z.; Romero, P.; Garner, E.C.; Brown, C.J. Intrinsic protein disorder in complete genomes. Genome Inform. 2000, 11, 161–171. [Google Scholar]
- Ward, J.J.; Sodhi, J.S.; McGuffin, L.J.; Buxton, B.F.; Jones, D.T. Prediction and functional analysis of native disorder in proteins from the three kingdoms of life. J. Mol. Biol. 2004, 337, 635–645. [Google Scholar] [CrossRef] [PubMed]
- Tokuriki, N.; Oldfield, C.J.; Uversky, V.N.; Berezovsky, I.N.; Tawfik, D.S. Do viral proteins possess unique biophysical features? Trends Biochem. Sci. 2009, 34, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Xue, B.; Williams, R.W.; Oldfield, C.J.; Dunker, A.K.; Uversky, V.N. Archaic chaos: Intrinsically disordered proteins in Archaea. BMC Syst. Biol. 2010, 4 (Suppl. 1), S1. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. The mysterious unfoldome: Structureless, underappreciated, yet vital part of any given proteome. J. Biomed. Biotechnol. 2010, 2010, 568068. [Google Scholar] [CrossRef] [PubMed]
- Xue, B.; Dunker, A.K.; Uversky, V.N. Orderly order in protein intrinsic disorder distribution: Disorder in thirty five hundred proteomes from viruses and the three domains of life. J. Biomol. Struct. Dyn. 2012, 30, 137–149. [Google Scholar] [CrossRef] [PubMed]
- Peng, Z.; Yan, J.; Fan, X.; Mizianty, M.J.; Xue, B.; Wang, K.; Hu, G.; Uversky, V.N.; Kurgan, L. Exceptionally abundant exceptions: Comprehensive characterization of intrinsic disorder in all domains of life. Cell. Mol. Life Sci. 2015, 72, 137–151. [Google Scholar] [CrossRef] [PubMed]
- DeForte, S.; Uversky, V.N. Order, Disorder, and Everything in between. Molecules 2016, 21, 1090. [Google Scholar] [CrossRef] [PubMed]
- Wright, P.E.; Dyson, H.J. Intrinsically unstructured proteins: Re-assessing the protein structure-function paradigm. J. Mol. Biol. 1999, 293, 321–331. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N.; Gillespie, J.R.; Fink, A.L. Why are “natively unfolded” proteins unstructured under physiologic conditions? Proteins 2000, 41, 415–427. [Google Scholar] [CrossRef]
- Dunker, A.K.; Lawson, J.D.; Brown, C.J.; Williams, R.M.; Romero, P.; Oh, J.S.; Oldfield, C.J.; Campen, A.M.; Ratliff, C.M.; Hipps, K.W.; et al. Intrinsically disordered protein. J. Mol. Graph. Model. 2001, 19, 26–59. [Google Scholar] [CrossRef] [Green Version]
- Dunker, A.K.; Obradovic, Z. The protein trinity—Linking function and disorder. Nat. Biotechnol. 2001, 19, 805–806. [Google Scholar] [CrossRef] [PubMed]
- Tompa, P. Intrinsically unstructured proteins. Trends Biochem. Sci. 2002, 27, 527–533. [Google Scholar] [CrossRef]
- Uversky, V.N. What does it mean to be natively unfolded? Eur. J. Biochem. 2002, 269, 2–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uversky, V.N. Natively unfolded proteins: A point where biology waits for physics. Protein Sci. 2002, 11, 739–756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dyson, H.J.; Wright, P.E. Intrinsically unstructured proteins and their functions. Nat. Rev. Mol. Cell Biol. 2005, 6, 197–208. [Google Scholar] [CrossRef] [PubMed]
- Xue, B.; Dunbrack, R.L.; Williams, R.W.; Dunker, A.K.; Uversky, V.N. PONDR-FIT: A meta-predictor of intrinsically disordered amino acids. Biochim. Biophys. Acta 2010, 1804, 996–1010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunker, A.K.; Silman, I.; Uversky, V.N.; Sussman, J.L. Function and structure of inherently disordered proteins. Curr. Opin. Struct. Biol. 2008, 18, 756–764. [Google Scholar] [CrossRef] [PubMed]
- Dunker, A.K.; Oldfield, C.J.; Meng, J.; Romero, P.; Yang, J.Y.; Chen, J.W.; Vacic, V.; Obradovic, Z.; Uversky, V.N. The unfoldomics decade: An update on intrinsically disordered proteins. BMC Genom. 2008, 9 (Suppl. 2), S1. [Google Scholar] [CrossRef] [PubMed]
- Dunker, A.K.; Bondos, S.E.; Huang, F.; Oldfield, C.J. Intrinsically disordered proteins and multicellular organisms. Semin. Cell Dev. Biol. 2015, 37, 44–55. [Google Scholar] [CrossRef] [PubMed]
- DeForte, S.; Uversky, V.N. Not an exception to the rule: The functional significance of intrinsically disordered protein regions in enzymes. Mol. Biosyst. 2017, 13, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Marsh, J.A.; Teichmann, S.A. Parallel dynamics and evolution: Protein conformational fluctuations and assembly reflect evolutionary changes in sequence and structure. Bioessays 2014, 36, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Xue, B.; Brown, C.J.; Dunker, A.K.; Uversky, V.N. Intrinsically disordered regions of p53 family are highly diversified in evolution. Biochim. Biophys. Acta 2013, 1834, 725–738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, C.J.; Takayama, S.; Campen, A.M.; Vise, P.; Marshall, T.W.; Oldfield, C.J.; Williams, C.J.; Dunker, A.K. Evolutionary rate heterogeneity in proteins with long disordered regions. J. Mol. Evol. 2002, 55, 104–110. [Google Scholar] [CrossRef] [PubMed]
- Na, I.; DeForte, S.; Stojanovski, B.M.; Ferreira, G.C.; Uversky, V.N. Molecular dynamics analysis of the structural and dynamic properties of the functionally enhanced hepta-variant of mouse 5-aminolevulinate synthase. J. Biomol. Struct. Dyn. 2018, 36, 152–165. [Google Scholar] [CrossRef] [PubMed]
- Sivaraman, J.; Myers, R.S.; Boju, L.; Sulea, T.; Cygler, M.; Jo Davisson, V.; Schrag, J.D. Crystal structure of Methanobacterium thermoautotrophicum phosphoribosyl-AMP cyclohydrolase HisI. Biochemistry 2005, 44, 10071–10080. [Google Scholar] [CrossRef] [PubMed]
- Lei, H.; Wu, C.; Liu, H.; Duan, Y. Folding free-energy landscape of villin headpiece subdomain from molecular dynamics simulations. Proc. Natl. Acad. Sci. USA 2007, 104, 4925–4930. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.H.; Kim, K.H.; Jun, G.; Greenfield, N.J.; Dominguez, R.; Volkmann, N.; Hitchcock-DeGregori, S.E.; Cohen, C. Deciphering the design of the tropomyosin molecule. Proc. Natl. Acad. Sci. USA 2001, 98, 8496–8501. [Google Scholar] [CrossRef] [PubMed]
- Madrazo, J.; Brown, J.H.; Litvinovich, S.; Dominguez, R.; Yakovlev, S.; Medved, L.; Cohen, C. Crystal structure of the central region of bovine fibrinogen (E5 fragment) at 1.4-A resolution. Proc. Natl. Acad. Sci. USA 2001, 98, 11967–11972. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.H. Breaking symmetry in protein dimers: Designs and functions. Protein Sci. 2006, 15, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caspar, D.L.; Klug, A. Physical principles in the construction of regular viruses. Cold Spring Harb. Symp. Quant. Biol. 1962, 27, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.E.; Speir, J.A. Quasi-equivalent viruses: A paradigm for protein assemblies. J. Mol. Biol. 1997, 269, 665–675. [Google Scholar] [CrossRef] [PubMed]
- Goodsell, D.S.; Olson, A.J. Structural symmetry and protein function. Annu. Rev. Biophys. Biomol. Struct. 2000, 29, 105–153. [Google Scholar] [CrossRef] [PubMed]
- Hilser, V.J.; Thompson, E.B. Intrinsic disorder as a mechanism to optimize allosteric coupling in proteins. Proc. Natl. Acad. Sci. USA 2007, 104, 8311–8315. [Google Scholar] [CrossRef] [PubMed]
- Hilser, V.J.; Wrabl, J.O.; Motlagh, H.N. Structural and energetic basis of allostery. Annu. Rev. Biophys. 2012, 41, 585–609. [Google Scholar] [CrossRef] [PubMed]
- Motlagh, H.N.; Li, J.; Thompson, E.B.; Hilser, V.J. Interplay between allostery and intrinsic disorder in an ensemble. Biochem. Soc. Trans. 2012, 40, 975–980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferreon, A.C.; Ferreon, J.C.; Wright, P.E.; Deniz, A.A. Modulation of allostery by protein intrinsic disorder. Nature 2013, 498, 390–394. [Google Scholar] [CrossRef] [PubMed]
- Motlagh, H.N.; Wrabl, J.O.; Li, J.; Hilser, V.J. The ensemble nature of allostery. Nature 2014, 508, 331–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, T.H.; Mehrabi, P.; Ren, Z.; Sljoka, A.; Ing, C.; Bezginov, A.; Ye, L.; Pomes, R.; Prosser, R.S.; Pai, E.F. The role of dimer asymmetry and protomer dynamics in enzyme catalysis. Science 2017, 355, eaag2355. [Google Scholar] [CrossRef] [PubMed]
- Levitzki, A.; Stallcup, W.B.; Koshland, D.E., Jr. Half-of-the-sites reactivity and the conformational states of cytidine triphosphate synthetase. Biochemistry 1971, 10, 3371–3378. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.; Giap, C.; Lazo, J.S.; Prochownik, E.V. Low molecular weight inhibitors of Myc-Max interaction and function. Oncogene 2003, 22, 6151–6159. [Google Scholar] [CrossRef] [PubMed]
- Follis, A.V.; Hammoudeh, D.I.; Wang, H.; Prochownik, E.V.; Metallo, S.J. Structural rationale for the coupled binding and unfolding of the c-Myc oncoprotein by small molecules. Chem. Biol. 2008, 15, 1149–1155. [Google Scholar] [CrossRef] [PubMed]
- Michel, J.; Cuchillo, R. The impact of small molecule binding on the energy landscape of the intrinsically disordered protein C-myc. PLoS ONE 2012, 7, e41070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Estacio, S.G.; Krobath, H.; Vila-Vicosa, D.; Machuqueiro, M.; Shakhnovich, E.I.; Faisca, P.F. A simulated intermediate state for folding and aggregation provides insights into DeltaN6 beta2-microglobulin amyloidogenic behavior. PLoS Comput. Biol. 2014, 10, e1003606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loureiro, R.J.S.; Vila-Vicosa, D.; Machuqueiro, M.; Shakhnovich, E.I.; Faisca, P.F.N. A tale of two tails: The importance of unstructured termini in the aggregation pathway of beta2-microglobulin. Proteins 2017, 85, 2045–2057. [Google Scholar] [CrossRef] [PubMed]
- Krobath, H.; Estacio, S.G.; Faisca, P.F.; Shakhnovich, E.I. Identification of a conserved aggregation-prone intermediate state in the folding pathways of Spc-SH3 amyloidogenic variants. J. Mol. Biol. 2012, 422, 705–722. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. The most important thing is the tail: Multitudinous functionalities of intrinsically disordered protein termini. FEBS Lett. 2013, 587, 1891–1901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, B.L.; Kardon, J.R.; Sauer, R.T.; Baker, T.A. Structure of the Mitochondrial Aminolevulinic Acid Synthase, a Key Heme Biosynthetic Enzyme. Structure 2018, 26, 580–589. [Google Scholar] [CrossRef] [PubMed]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef] [PubMed]
- The UniProt Consortium. UniProt: The universal protein knowledgebase. Nucleic Acids Res. 2017, 45, D158–D169. [Google Scholar]
- Henikoff, S.; Henikoff, J.G. Amino acid substitution matrices from protein blocks. Proc. Natl. Acad. Sci. USA 1992, 89, 10915–10919. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Romero, P.; Rani, M.; Dunker, A.K.; Obradovic, Z. Predicting Protein Disorder for N-, C-, and Internal Regions. Genome Inform. 1999, 10, 30–40. [Google Scholar]
- Obradovic, Z.; Peng, K.; Vucetic, S.; Radivojac, P.; Dunker, A.K. Exploiting heterogeneous sequence properties improves prediction of protein disorder. Proteins 2005, 61 (Suppl. 7), 176–182. [Google Scholar] [CrossRef] [PubMed]
- Peng, K.; Vucetic, S.; Radivojac, P.; Brown, C.J.; Dunker, A.K.; Obradovic, Z. Optimizing long intrinsic disorder predictors with protein evolutionary information. J. Bioinform. Comput. Biol. 2005, 3, 35–60. [Google Scholar] [CrossRef] [PubMed]
- Romero, P.; Obradovic, Z.; Li, X.; Garner, E.C.; Brown, C.J.; Dunker, A.K. Sequence complexity of disordered protein. Proteins 2001, 42, 38–48. [Google Scholar] [CrossRef]
- Zhang, Y. I-TASSER server for protein 3D structure prediction. BMC Bioinform. 2008, 9, 40. [Google Scholar] [CrossRef] [PubMed]
- Dolinsky, T.J.; Nielsen, J.E.; McCammon, J.A.; Baker, N.A. PDB2PQR: An automated pipeline for the setup of Poisson-Boltzmann electrostatics calculations. Nucleic Acids Res. 2004, 32, W665–W667. [Google Scholar] [CrossRef] [PubMed]
- Vanommeslaeghe, K.; Hatcher, E.; Acharya, C.; Kundu, S.; Zhong, S.; Shim, J.; Darian, E.; Guvench, O.; Lopes, P.; Vorobyov, I.; et al. CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem. 2010, 31, 671–690. [Google Scholar] [CrossRef] [PubMed]
- Best, R.B.; Zhu, X.; Shim, J.; Lopes, P.E.; Mittal, J.; Feig, M.; Mackerell, A.D., Jr. Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone phi, psi and side-chain chi(1) and chi(2) dihedral angles. J. Chem. Theory Comput. 2012, 8, 3257–3273. [Google Scholar] [CrossRef] [PubMed]
- Berendsen, H.J.C.; Vanderspoel, D.; Vandrunen, R. Gromacs—A Message-Passing Parallel Molecular-Dynamics Implementation. Comput. Phys. Commun. 1995, 91, 43–56. [Google Scholar] [CrossRef]
- Lee, P.; Ye, Z.; Van Dyke, K.; Kirk, R.G. X-ray microanalysis of Plasmodium falciparum and infected red blood cells: Effects of qinghaosu and chloroquine on potassium, sodium, and phosphorus composition. Am. J. Trop. Med. Hyg. 1988, 39, 157–165. [Google Scholar] [CrossRef] [PubMed]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef] [Green Version]
- Darden, T.; York, D.; Pedersen, L. Particle Mesh Ewald—An N.Log(N) Method for Ewald Sums in Large Systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A Smooth Particle Mesh Ewald Method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef]
- Pall, S.; Hess, B. A flexible algorithm for calculating pair interactions on SIMD architectures. Comput. Phys. Commun. 2013, 184, 2641–2650. [Google Scholar] [CrossRef] [Green Version]
- Nose, S.; Klein, M.L. Constant Pressure Molecular-Dynamics for Molecular-Systems. Mol. Phys. 1983, 50, 1055–1076. [Google Scholar] [CrossRef]
- Parrinello, M.; Rahman, A. Polymorphic Transitions in Single-Crystals—A New Molecular-Dynamics Method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- R-Core-Team. R: A Language and Environment for Statistical Computing. 2015. Availabe online: http://www.R-project.org/ (accessed on 27 June 2018).
- Grant, B.J.; Rodrigues, A.P.C.; ElSawy, K.M.; McCammon, J.A.; Caves, L.S.D. Bio3d: An R package for the comparative analysis of protein structures. Bioinformatics 2006, 22, 2695–2696. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Homodimer System | Intercept (α) | Slope (β) | p-Value |
|---|---|---|---|
| 288 K | 0.17 | −0.40 | 0.04 |
| 300 K | 0.42 | −0.50 | 0.04 |
| 310 K | −0.04 | −1.10 | 0.01 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Na, I.; Catena, D.; Kong, M.J.; Ferreira, G.C.; Uversky, V.N. Anti-Correlation between the Dynamics of the Active Site Loop and C-Terminal Tail in Relation to the Homodimer Asymmetry of the Mouse Erythroid 5-Aminolevulinate Synthase. Int. J. Mol. Sci. 2018, 19, 1899. https://doi.org/10.3390/ijms19071899
Na I, Catena D, Kong MJ, Ferreira GC, Uversky VN. Anti-Correlation between the Dynamics of the Active Site Loop and C-Terminal Tail in Relation to the Homodimer Asymmetry of the Mouse Erythroid 5-Aminolevulinate Synthase. International Journal of Molecular Sciences. 2018; 19(7):1899. https://doi.org/10.3390/ijms19071899
Chicago/Turabian StyleNa, Insung, Dominique Catena, Min J. Kong, Gloria C. Ferreira, and Vladimir N. Uversky. 2018. "Anti-Correlation between the Dynamics of the Active Site Loop and C-Terminal Tail in Relation to the Homodimer Asymmetry of the Mouse Erythroid 5-Aminolevulinate Synthase" International Journal of Molecular Sciences 19, no. 7: 1899. https://doi.org/10.3390/ijms19071899