Exploring the Roles of CREBRF and TRIM2 in the Regulation of Angiogenesis by High-Density Lipoproteins

 and
and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
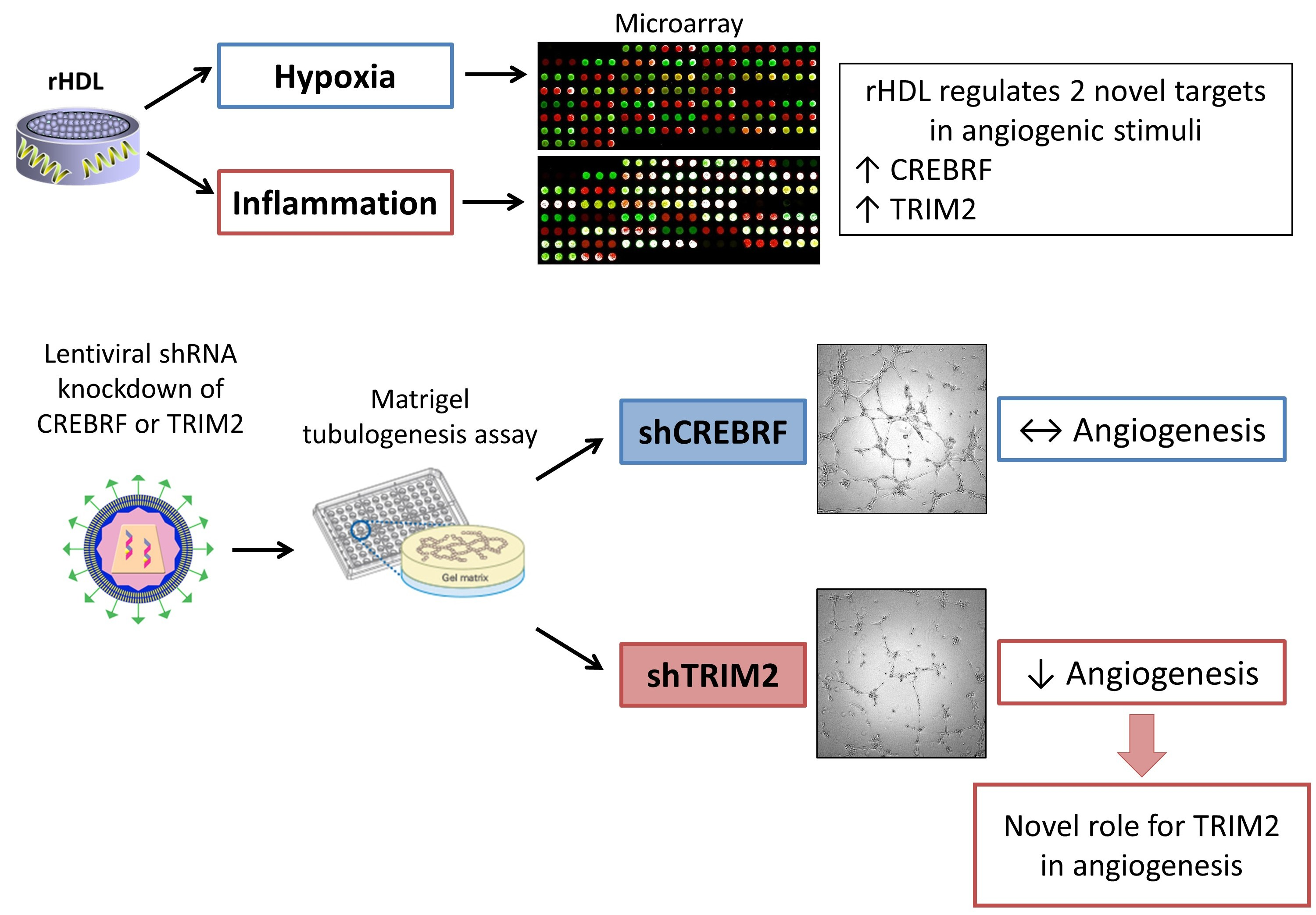
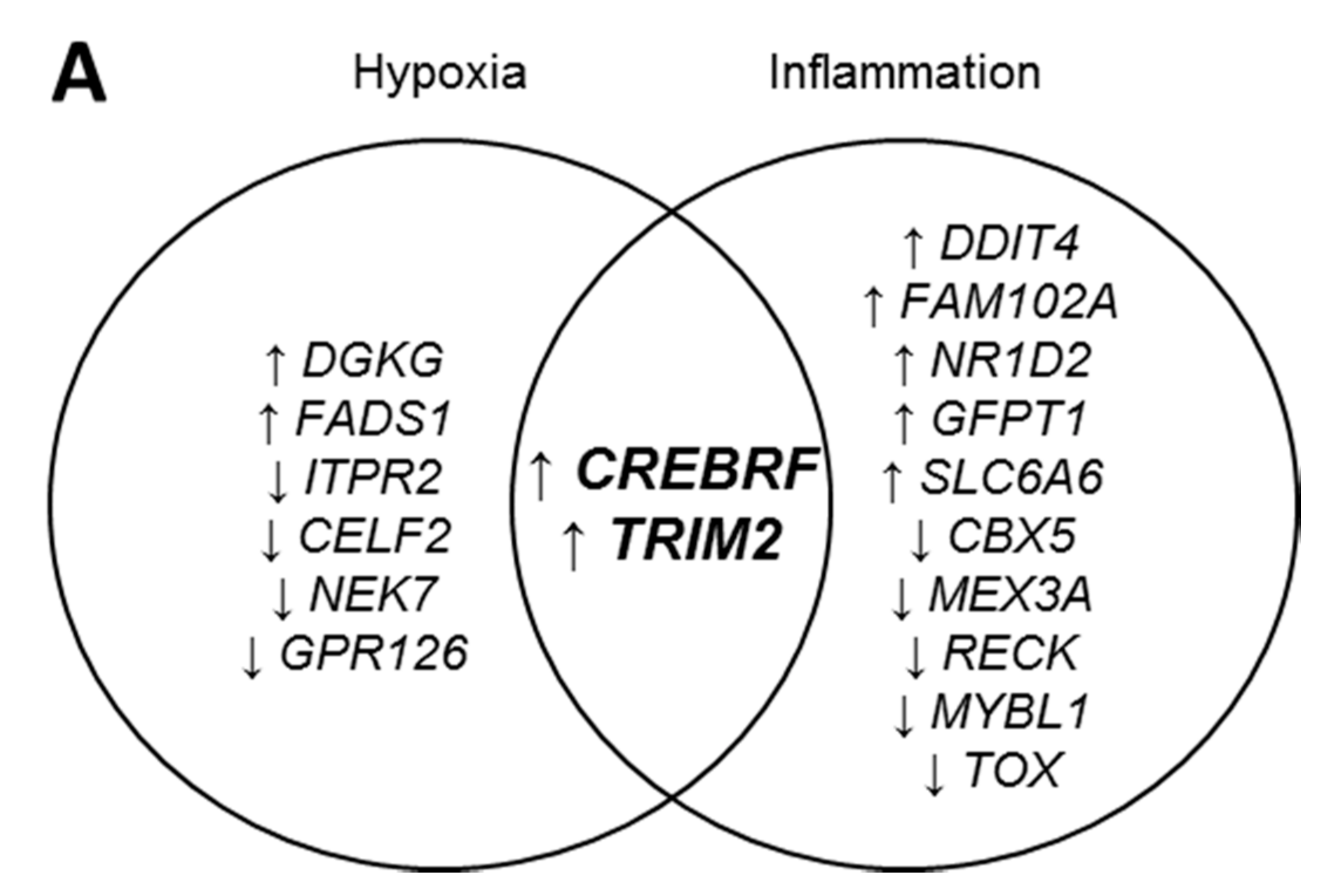
2.1. CREBRF and TRIM2 Are Novel Targets of rHDL in Response to Angiogenic Stimuli
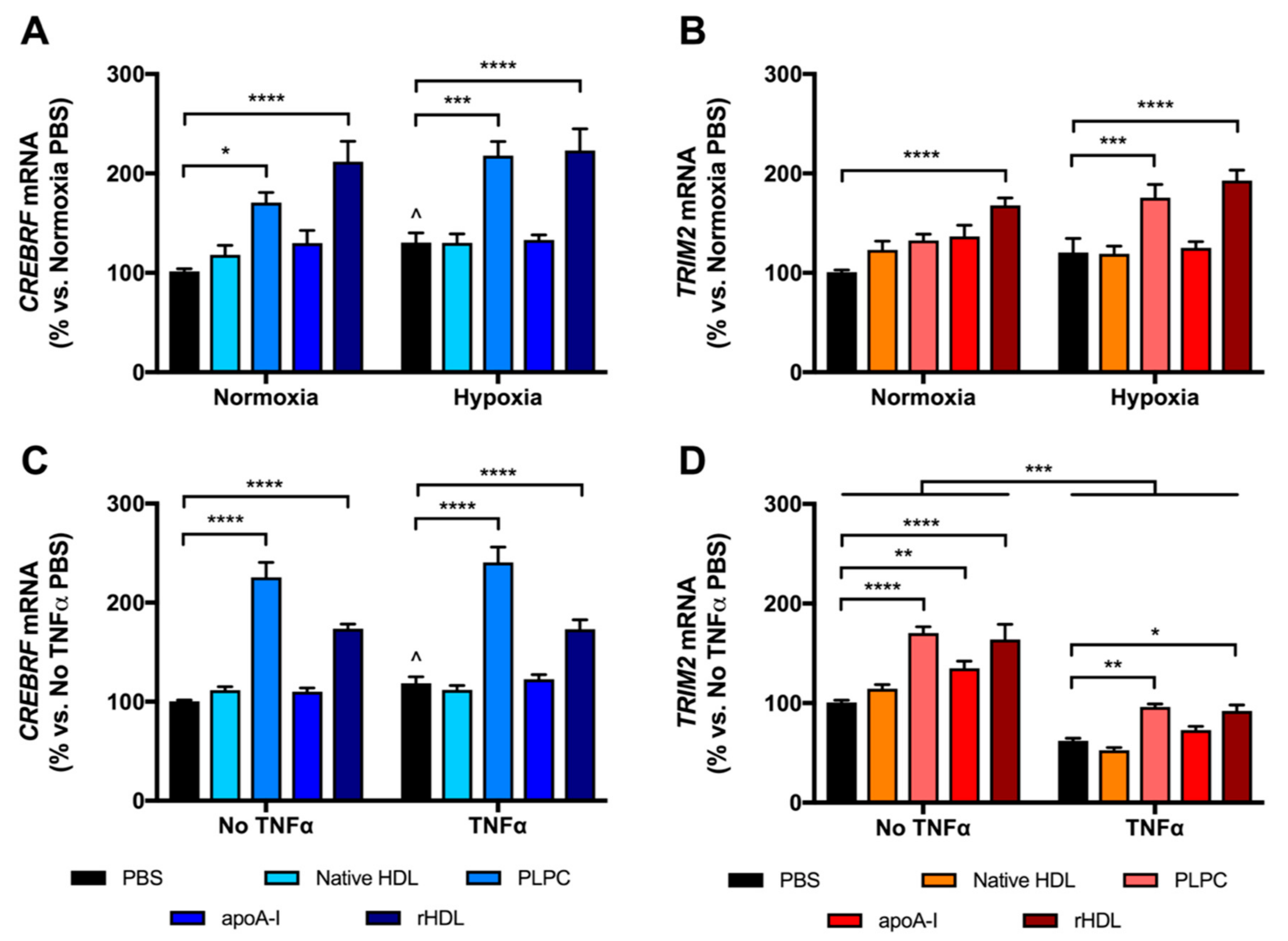
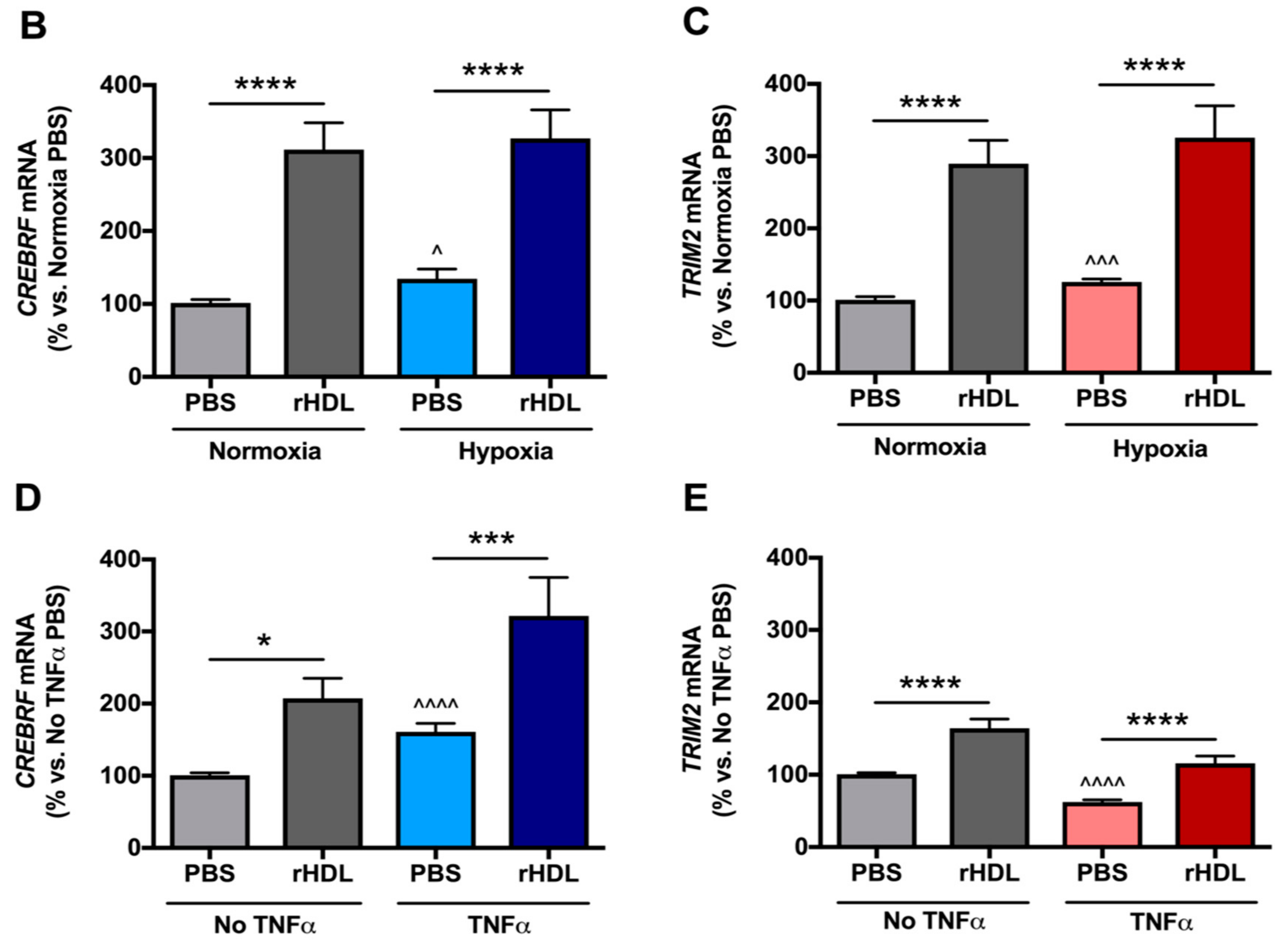
2.2. rHDL Modulates CREBRF and TRIM2 Expression in Human Coronary Artery Endothelial Cells (HCAECs) under Hypoxia and Inflammation
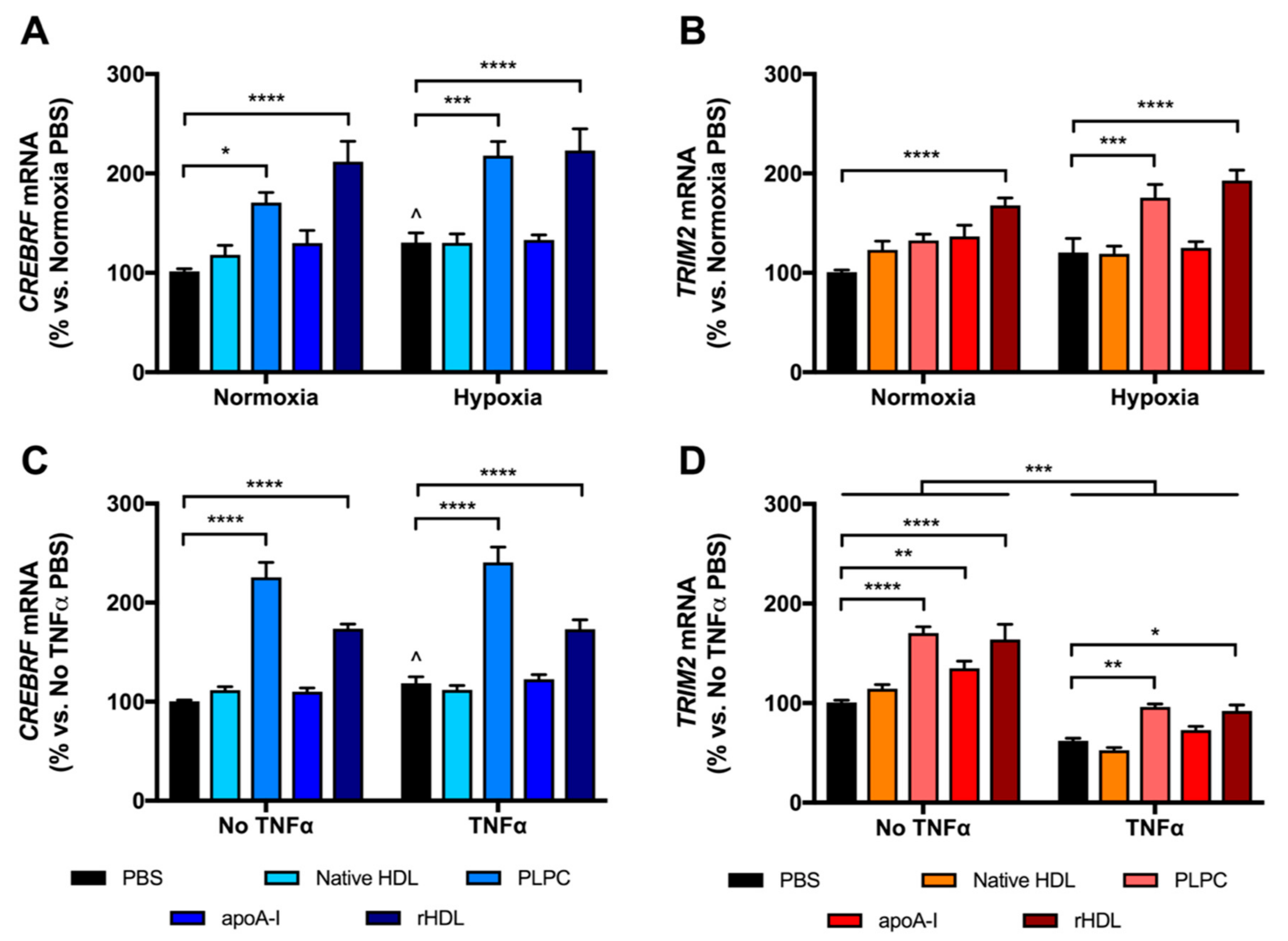
2.3. rHDL Upregulates CREBRF and TRIM2 mRNA Expression in HCAECs Predominantly through PLPC
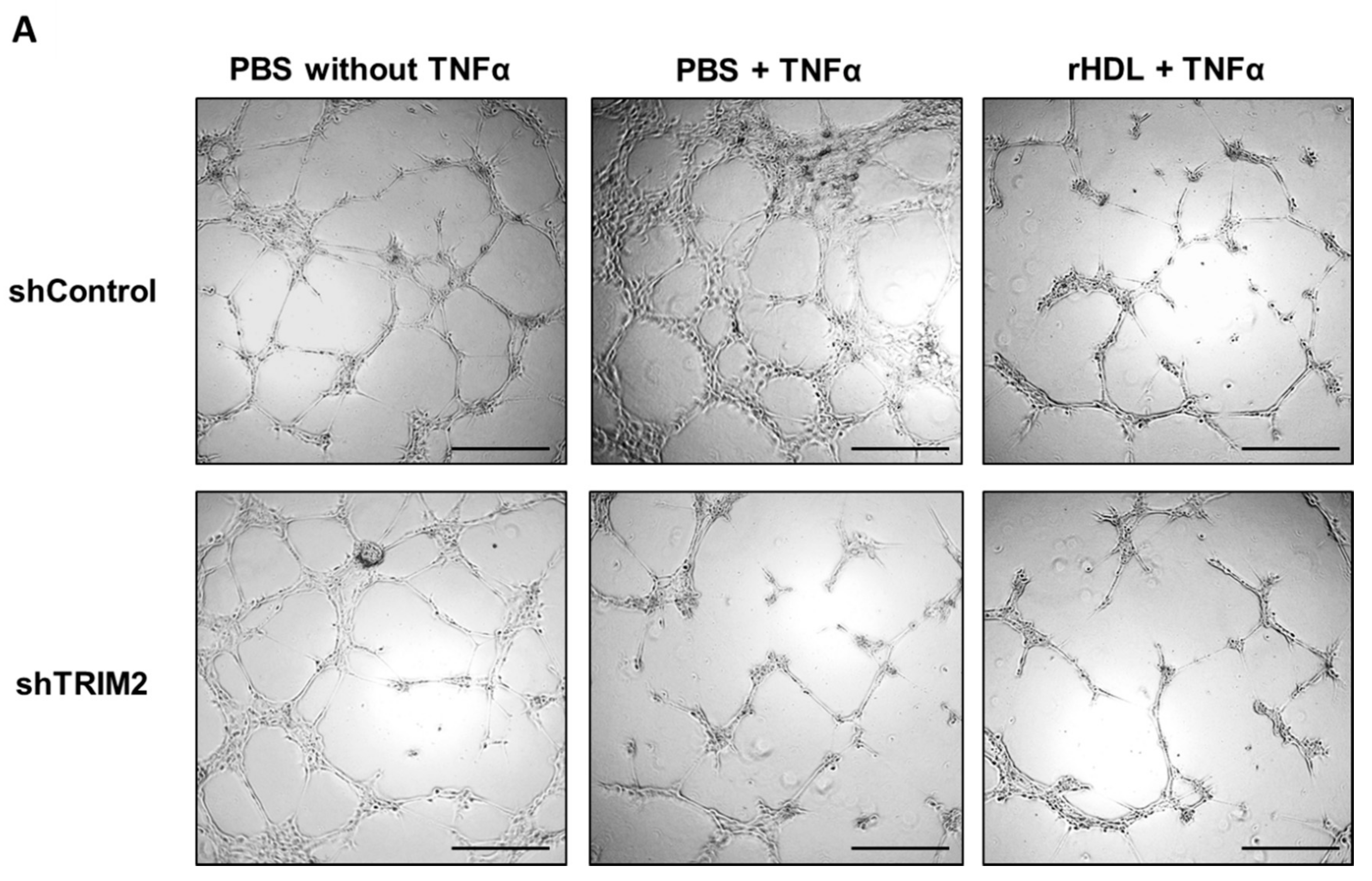
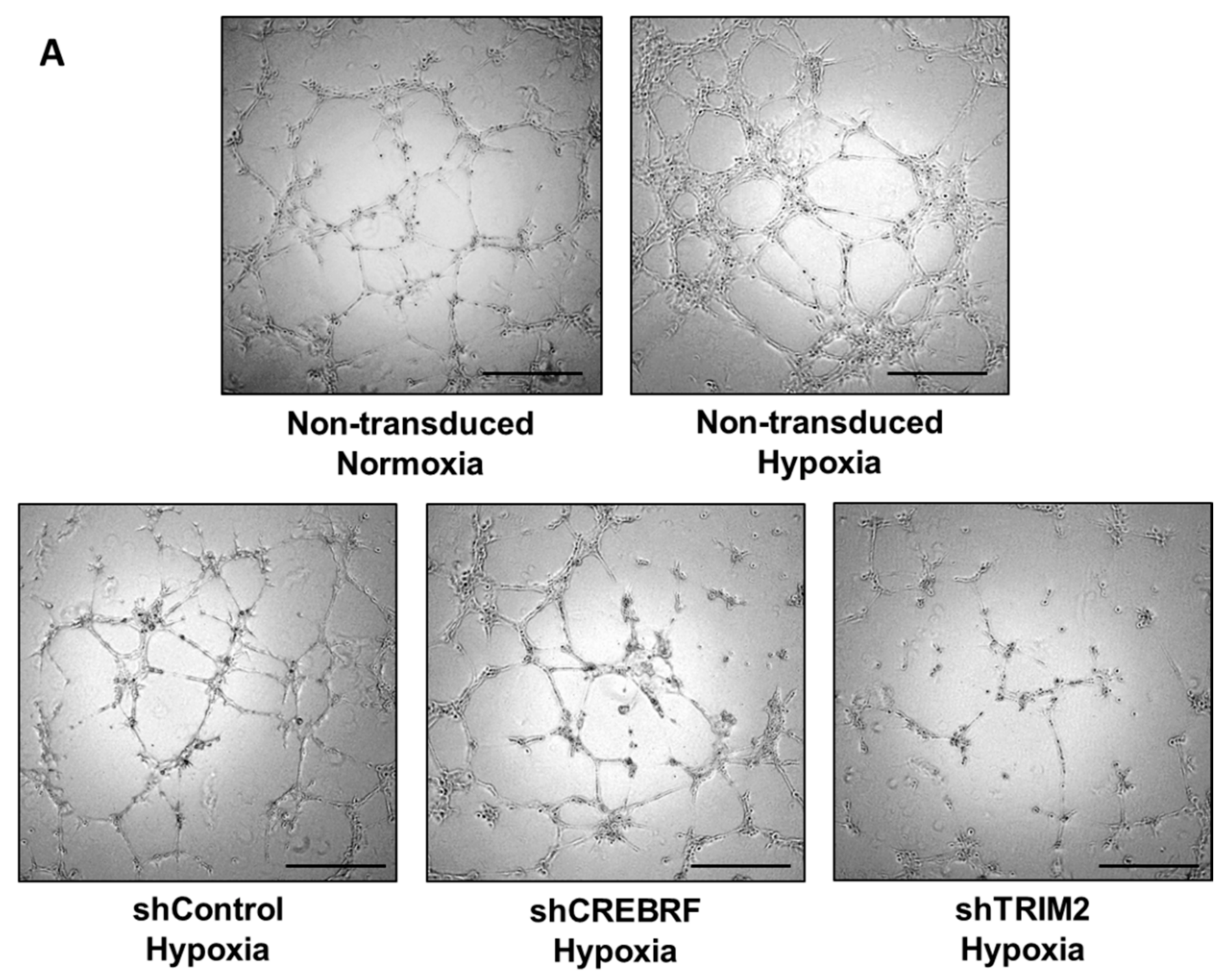
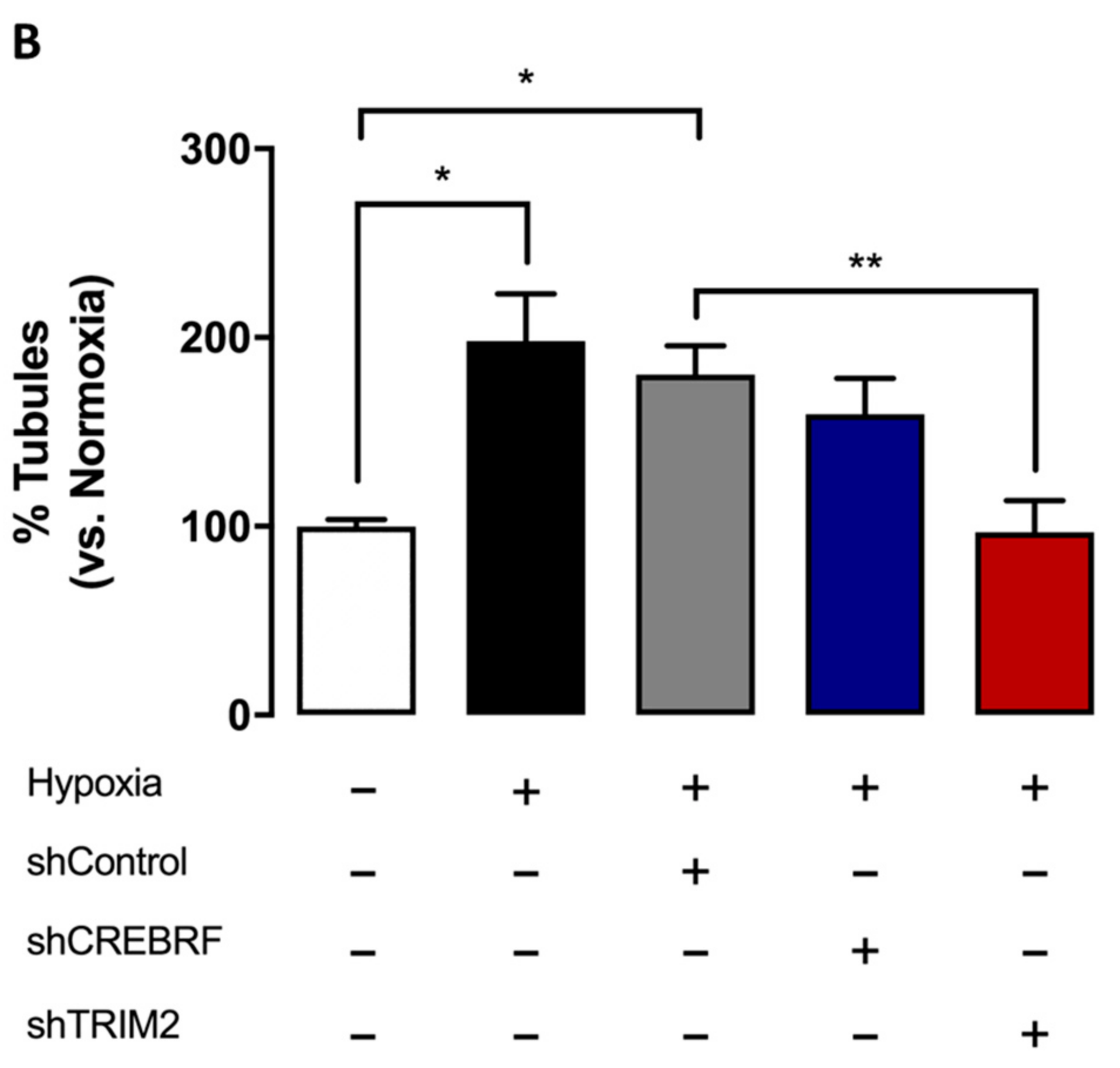
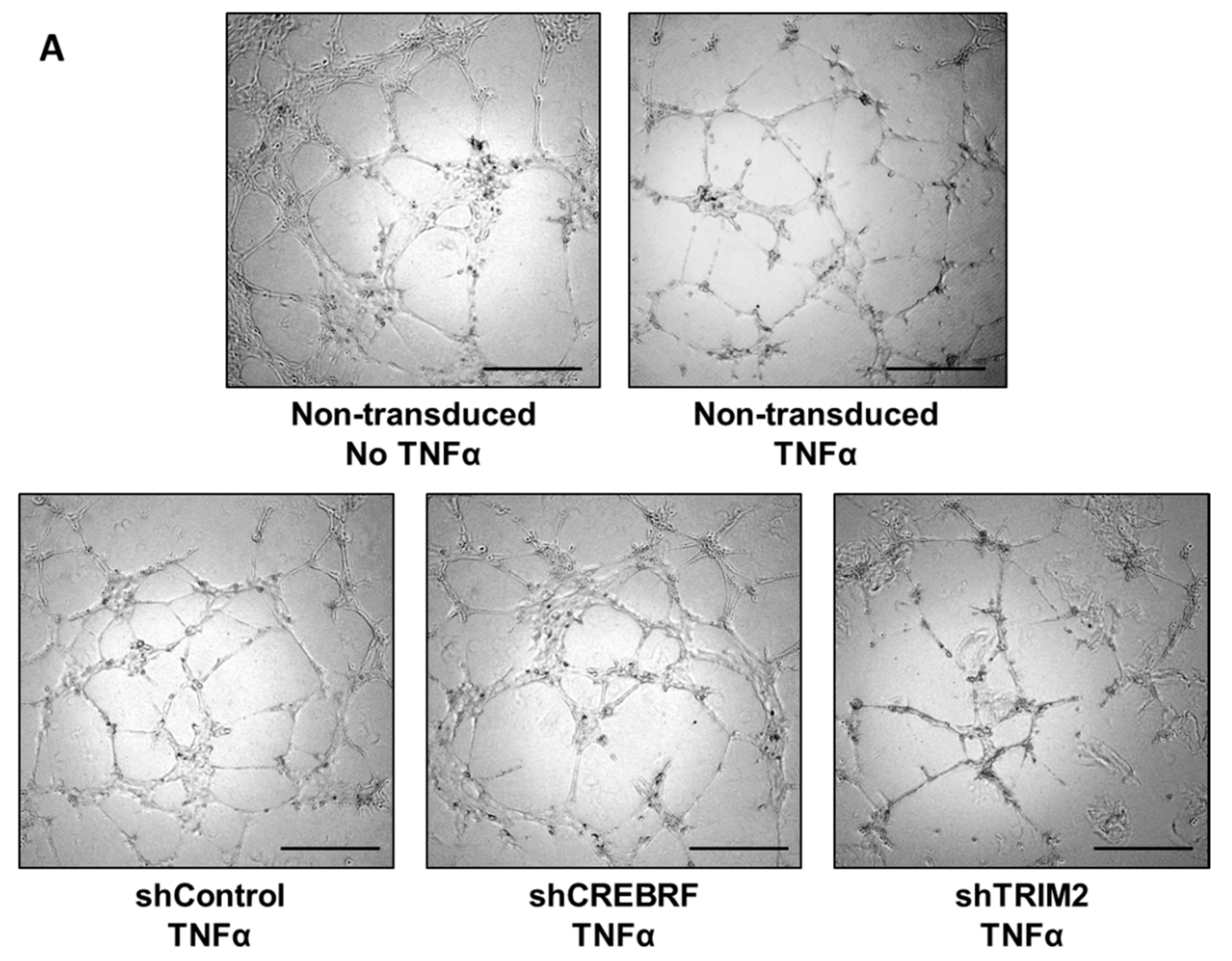
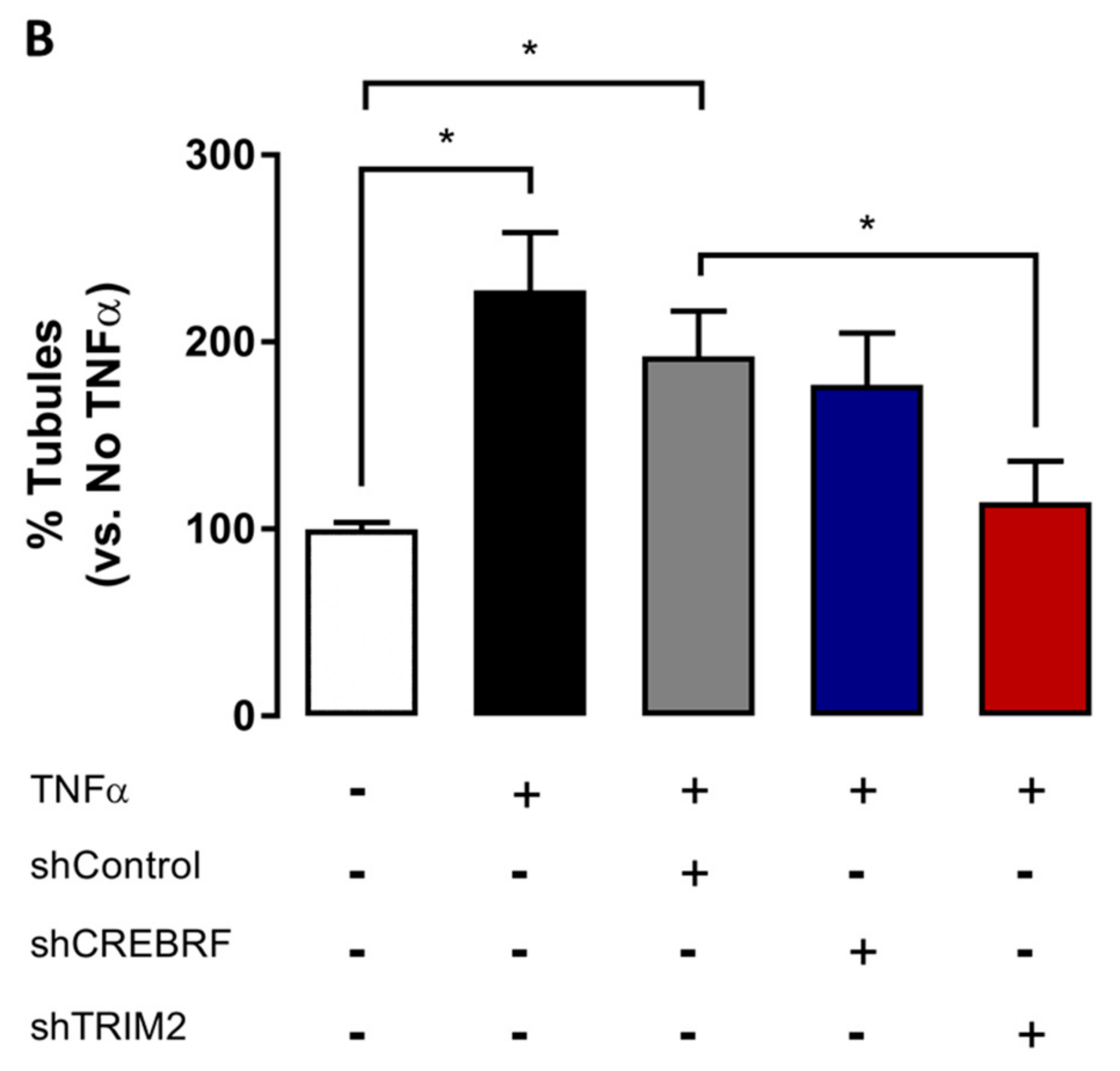
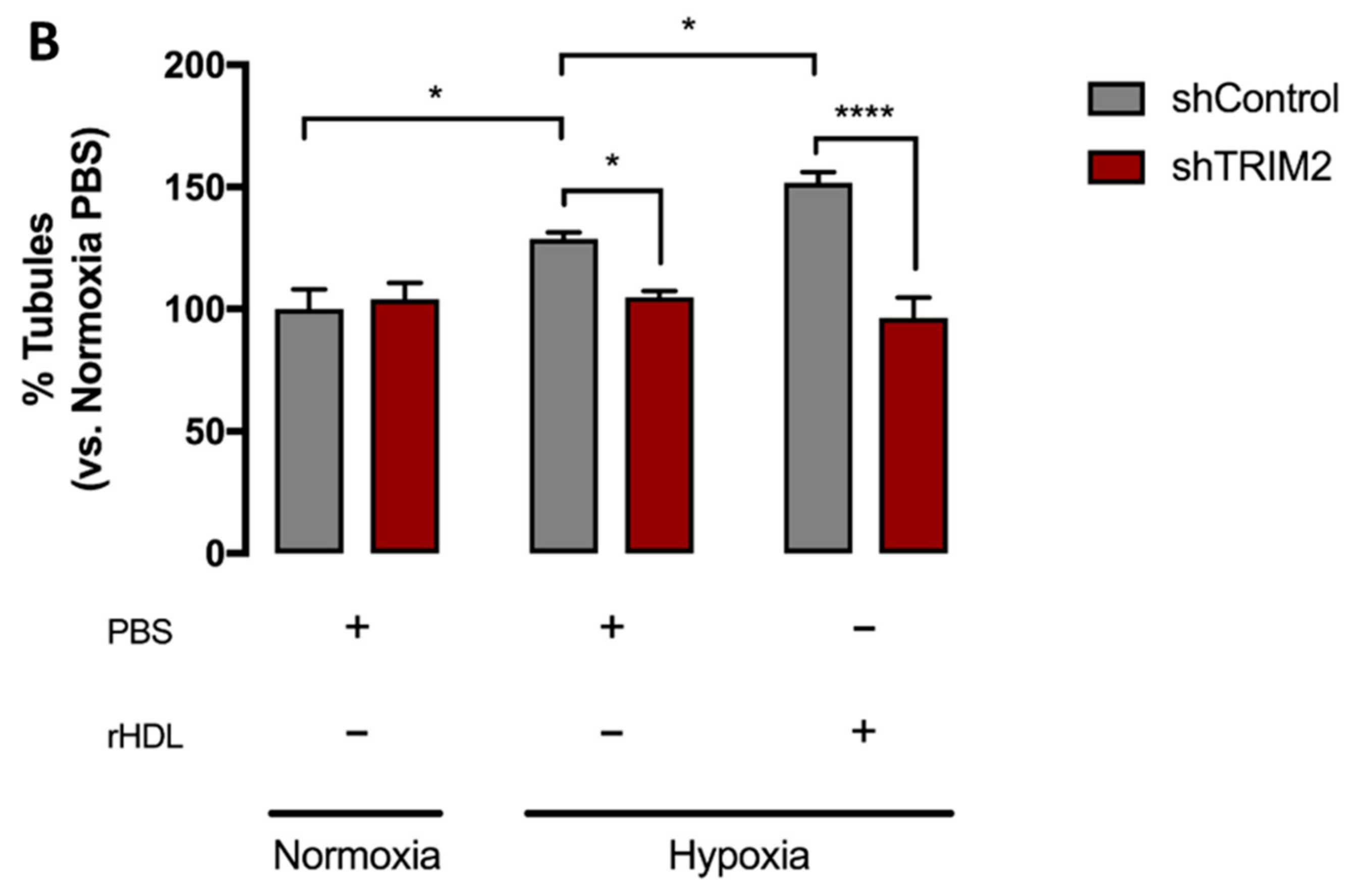
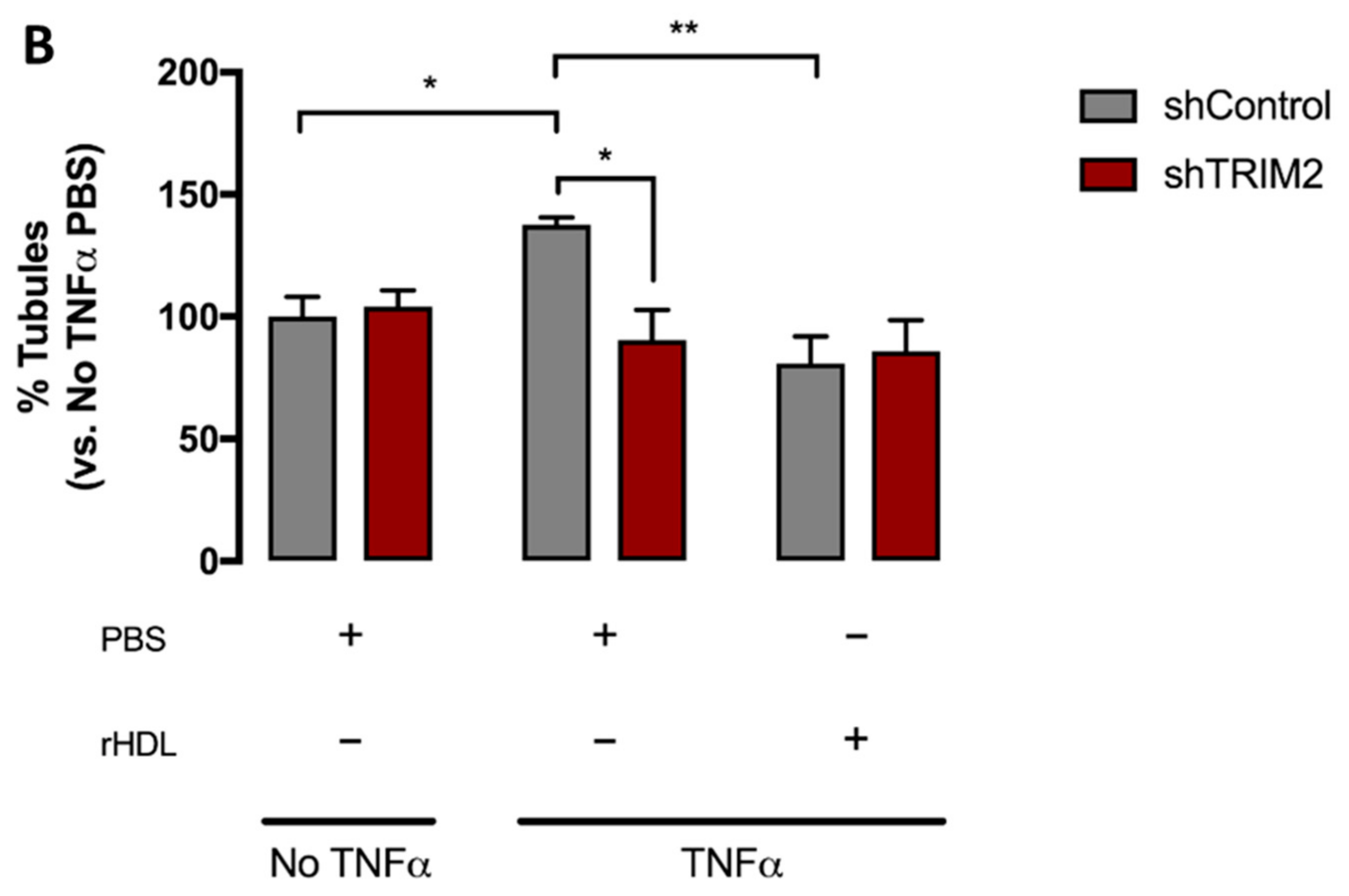
2.4. Tubulogenesis is Impaired by Lentiviral Knockdown of TRIM2 in Hypoxia and Inflammation
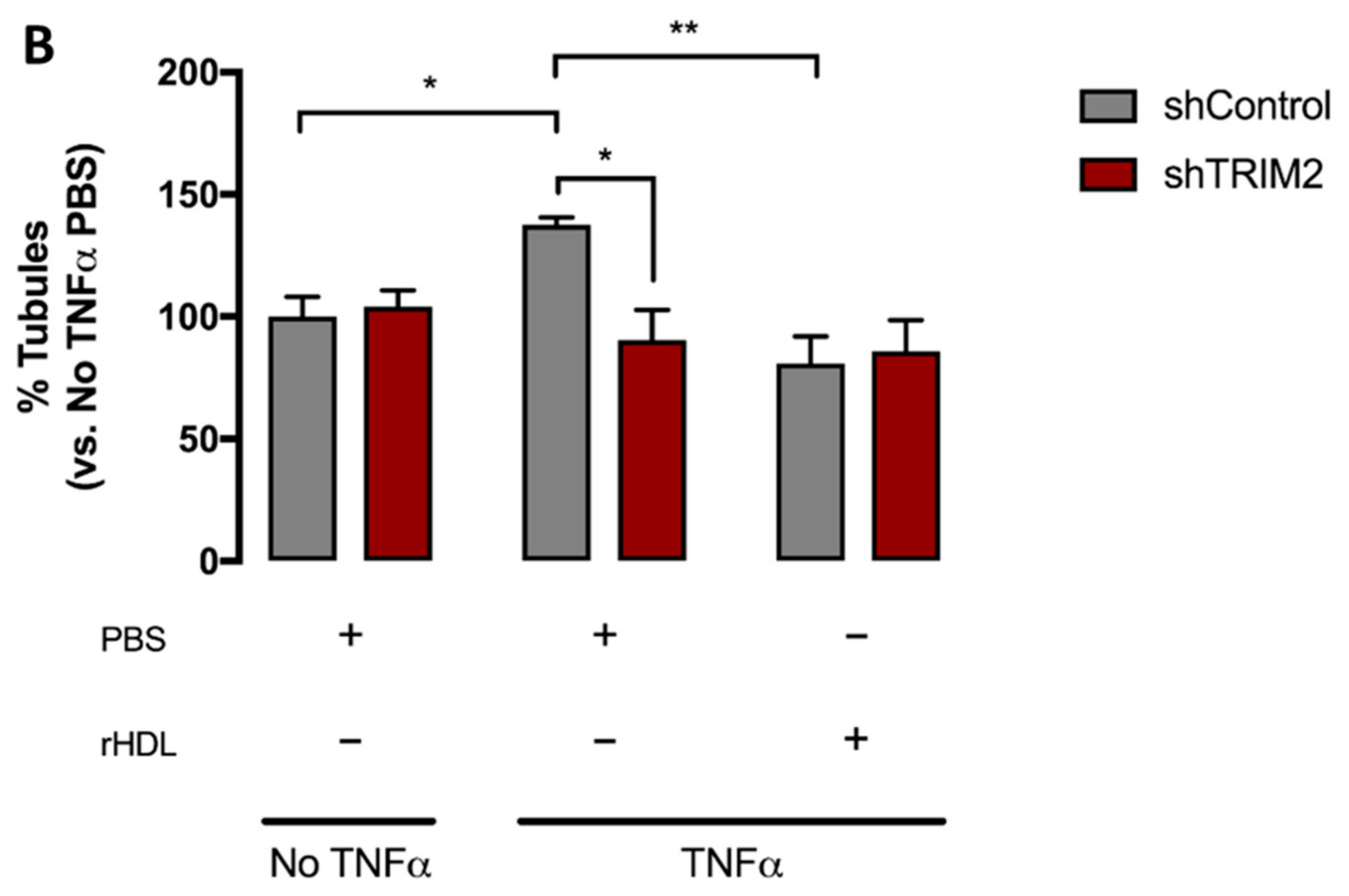
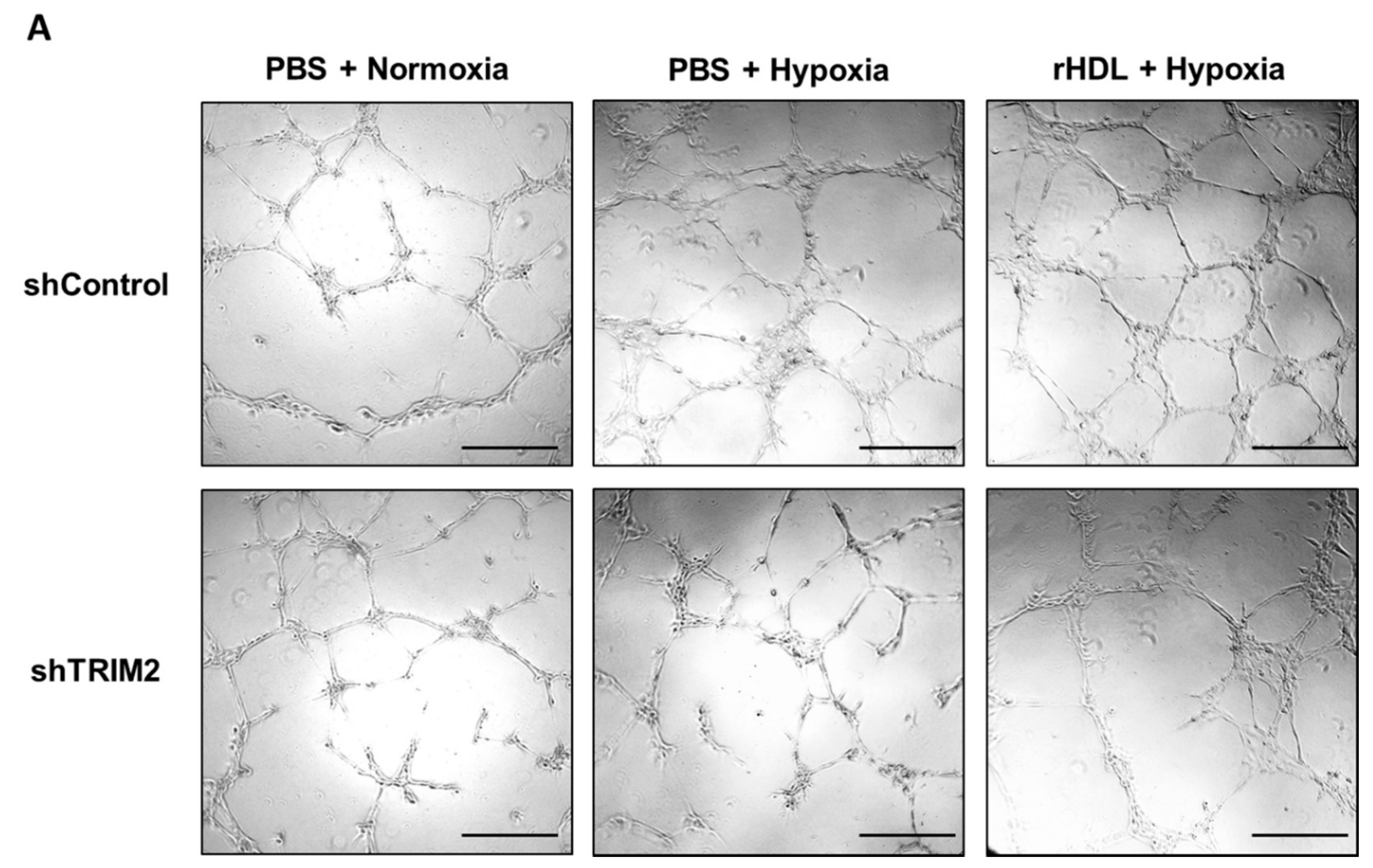
2.5. Lentiviral Knockdown of TRIM2 Attenuates the Pro-Angiogenic Action of rHDL in Hypoxia
3. Discussion
4. Materials and Methods
4.1. Preparation of Native HDL, Discoidal rHDL, ApoA-I, and Phospholipid Vesicles
4.2. Microarray
4.3. Cell Culture
4.4. mRNA Expression
4.5. Protein Expression
4.6. Lentiviral shRNA Knockdown of CREBRF and TRIM2—Matrigel Tubulogenesis Assay
4.7. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Semenza, G.L. Vascular responses to hypoxia and ischemia. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 648–652. [Google Scholar] [CrossRef] [PubMed]
- Costa, C.; Incio, J.; Soares, R. Angiogenesis and chronic inflammation: Cause or consequence? Angiogenesis 2007, 10, 149–166. [Google Scholar] [CrossRef] [PubMed]
- Carmeliet, P.; Jain, R.K. Angiogenesis in cancer and other diseases. Nature 2000, 407, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F. Cancer-related inflammation. Nature 2008, 454, 436–444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Global Burden of Disease Cancer Collaboration; Fitzmaurice, C.; Allen, C.; Barber, R.M.; Barregard, L.; Bhutta, Z.A.; Brenner, H.; Dicker, D.J.; Chimed-Orchir, O.; Dandona, R.; et al. Global, Regional, and National Cancer Incidence, Mortality, Years of Life Lost, Years Lived With Disability, and Disability-Adjusted Life-years for 32 Cancer Groups, 1990 to 2015: A Systematic Analysis for the Global Burden of Disease Study. JAMA Oncol. 2017, 3, 524–548. [Google Scholar] [CrossRef] [PubMed]
- Joseph, P.; Leong, D.; McKee, M.; Anand, S.S.; Schwalm, J.D.; Teo, K.; Mente, A.; Yusuf, S. Reducing the Global Burden of Cardiovascular Disease, Part 1: The Epidemiology and Risk Factors. Circ. Res. 2017, 121, 677–694. [Google Scholar] [CrossRef] [PubMed]
- Rye, K.A.; Barter, P.J. Cardioprotective functions of HDLs. J. Lipid Res. 2014, 55, 168–179. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.T.; Ng, M.K.; Bursill, C.A. The role of high-density lipoproteins in the regulation of angiogenesis. Cardiovasc. Res. 2015, 106, 184–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prosser, H.C.; Tan, J.T.; Dunn, L.L.; Patel, S.; Vanags, L.Z.; Bao, S.; Ng, M.K.; Bursill, C.A. Multifunctional regulation of angiogenesis by high-density lipoproteins. Cardiovasc. Res. 2014, 101, 145–154. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.T.; Prosser, H.C.; Vanags, L.Z.; Monger, S.A.; Ng, M.K.; Bursill, C.A. High-density lipoproteins augment hypoxia-induced angiogenesis via regulation of post-translational modulation of hypoxia-inducible factor 1alpha. FASEB J. 2014, 28, 206–217. [Google Scholar] [CrossRef] [PubMed]
- Sumi, M.; Sata, M.; Miura, S.; Rye, K.A.; Toya, N.; Kanaoka, Y.; Yanaga, K.; Ohki, T.; Saku, K.; Nagai, R. Reconstituted high-density lipoprotein stimulates differentiation of endothelial progenitor cells and enhances ischemia-induced angiogenesis. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 813–818. [Google Scholar] [CrossRef] [PubMed]
- Tsatralis, T.; Ridiandries, A.; Robertson, S.; Vanags, L.Z.; Lam, Y.T.; Tan, J.T.; Ng, M.K.; Bursill, C.A. Reconstituted high-density lipoproteins promote wound repair and blood flow recovery in response to ischemia in aged mice. Lipids Health Dis. 2016, 15, 150. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.T.; Prosser, H.C.; Dunn, L.L.; Vanags, L.Z.; Ridiandries, A.; Tsatralis, T.; Lecce, L.; Clayton, Z.E.; Yuen, S.C.; Robertson, S.; et al. High-Density Lipoproteins Rescue Diabetes-Impaired Angiogenesis via Scavenger Receptor Class B Type I. Diabetes 2016, 65, 3091–3103. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.J.; Shrestha, S.; Ong, K.L.; Johns, D.; Dunn, L.L.; Hou, L.; Barter, P.J.; Rye, K.A. Increasing HDL levels by inhibiting cholesteryl ester transfer protein activity in rabbits with hindlimb ischemia is associated with increased angiogenesis. Int. J. Cardiol. 2015, 199, 204–212. [Google Scholar] [CrossRef] [PubMed]
- Nicholls, S.J.; Dusting, G.J.; Cutri, B.; Bao, S.; Drummond, G.R.; Rye, K.A.; Barter, P.J. Reconstituted high-density lipoproteins inhibit the acute pro-oxidant and proinflammatory vascular changes induced by a periarterial collar in normocholesterolemic rabbits. Circulation 2005, 111, 1543–1550. [Google Scholar] [CrossRef] [PubMed]
- Song, G.J.; Kim, S.M.; Park, K.H.; Kim, J.; Choi, I.; Cho, K.H. SR-BI mediates high density lipoprotein (HDL)-induced anti-inflammatory effect in macrophages. Biochem. Biophys. Res. Commun. 2015, 457, 112–118. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Xi, D.; Zhao, J.; Luo, T.; Liu, J.; Lu, H.; Li, M.; Xiong, H.; Guo, Z. High-density Lipoprotein and Inflammation and Its Significance to Atherosclerosis. Am. J. Med. Sci. 2016, 352, 408–415. [Google Scholar] [CrossRef] [PubMed]
- Barter, P.J.; Nicholls, S.; Rye, K.A.; Anantharamaiah, G.M.; Navab, M.; Fogelman, A.M. Antiinflammatory properties of HDL. Circ. Res. 2004, 95, 764–772. [Google Scholar] [CrossRef] [PubMed]
- Balastik, M.; Ferraguti, F.; Pires-da Silva, A.; Lee, T.H.; Alvarez-Bolado, G.; Lu, K.P.; Gruss, P. Deficiency in ubiquitin ligase TRIM2 causes accumulation of neurofilament light chain and neurodegeneration. Proc. Natl. Acad. Sci. USA 2008, 105, 12016–12021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khazaei, M.R.; Bunk, E.C.; Hillje, A.L.; Jahn, H.M.; Riegler, E.M.; Knoblich, J.A.; Young, P.; Schwamborn, J.C. The E3-ubiquitin ligase TRIM2 regulates neuronal polarization. J. Neurochem. 2011, 117, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Ylikallio, E.; Poyhonen, R.; Zimon, M.; De Vriendt, E.; Hilander, T.; Paetau, A.; Jordanova, A.; Lonnqvist, T.; Tyynismaa, H. Deficiency of the E3 ubiquitin ligase TRIM2 in early-onset axonal neuropathy. Hum. Mol. Genet. 2013, 22, 2975–2983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pehlivan, D.; Coban Akdemir, Z.; Karaca, E.; Bayram, Y.; Jhangiani, S.; Yildiz, E.P.; Muzny, D.; Uluc, K.; Gibbs, R.A.; Baylor-Hopkins Center for Mendelian Genomics; et al. Exome sequencing reveals homozygous TRIM2 mutation in a patient with early onset CMT and bilateral vocal cord paralysis. Hum. Genet. 2015, 134, 671–673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thompson, S.; Pearson, A.N.; Ashley, M.D.; Jessick, V.; Murphy, B.M.; Gafken, P.; Henshall, D.C.; Morris, K.T.; Simon, R.P.; Meller, R. Identification of a novel Bcl-2-interacting mediator of cell death (Bim) E3 ligase, tripartite motif-containing protein 2 (TRIM2), and its role in rapid ischemic tolerance-induced neuroprotection. J. Biol. Chem. 2011, 286, 19331–19339. [Google Scholar] [CrossRef] [PubMed]
- Audas, T.E.; Li, Y.; Liang, G.; Lu, R. A novel protein, Luman/CREB3 recruitment factor, inhibits Luman activation of the unfolded protein response. Mol. Cell. Biol. 2008, 28, 3952–3966. [Google Scholar] [CrossRef] [PubMed]
- Binet, F.; Sapieha, P. ER Stress and Angiogenesis. Cell Metab. 2015, 22, 560–575. [Google Scholar] [CrossRef] [PubMed]
- Jain, B.P. An Overview of Unfolded Protein Response Signaling and Its Role in Cancer. Cancer Biother. Radiopharm. 2017, 32, 275–281. [Google Scholar] [CrossRef] [PubMed]
- Gargalovic, P.S.; Gharavi, N.M.; Clark, M.J.; Pagnon, J.; Yang, W.P.; He, A.; Truong, A.; Baruch-Oren, T.; Berliner, J.A.; Kirchgessner, T.G.; et al. The unfolded protein response is an important regulator of inflammatory genes in endothelial cells. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 2490–2496. [Google Scholar] [CrossRef] [PubMed]
- Lenna, S.; Han, R.; Trojanowska, M. Endoplasmic reticulum stress and endothelial dysfunction. IUBMB Life 2014, 66, 530–537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muller, C.; Salvayre, R.; Negre-Salvayre, A.; Vindis, C. HDLs inhibit endoplasmic reticulum stress and autophagic response induced by oxidized LDLs. Cell Death Differ. 2011, 18, 817–828. [Google Scholar] [CrossRef] [PubMed]
- Niculescu, L.S.; Sanda, G.M.; Sima, A.V. HDL inhibits endoplasmic reticulum stress by stimulating apoE and CETP secretion from lipid-loaded macrophages. Biochem. Biophys. Res. Commun. 2013, 434, 173–178. [Google Scholar] [CrossRef] [PubMed]
- Zamanian-Daryoush, M.; Lindner, D.; Tallant, T.C.; Wang, Z.; Buffa, J.; Klipfell, E.; Parker, Y.; Hatala, D.; Parsons-Wingerter, P.; Rayman, P.; et al. The cardioprotective protein apolipoprotein A1 promotes potent anti-tumorigenic effects. J. Biol. Chem. 2013, 288, 21237–21252. [Google Scholar] [CrossRef] [PubMed]
- Puranik, R.; Bao, S.; Nobecourt, E.; Nicholls, S.J.; Dusting, G.J.; Barter, P.J.; Celermajer, D.S.; Rye, K.A. Low dose apolipoprotein A-I rescues carotid arteries from inflammation in vivo. Atherosclerosis 2008, 196, 240–247. [Google Scholar] [CrossRef] [PubMed]
- Baker, P.W.; Rye, K.A.; Gamble, J.R.; Vadas, M.A.; Barter, P.J. Phospholipid composition of reconstituted high density lipoproteins influences their ability to inhibit endothelial cell adhesion molecule expression. J. Lipid Res. 2000, 41, 1261–1267. [Google Scholar] [PubMed]
- Schonrock, N.; Humphreys, D.T.; Preiss, T.; Gotz, J. Target gene repression mediated by miRNAs miR-181c and miR-9 both of which are down-regulated by amyloid-beta. J. Mol. Neurosci. 2012, 46, 324–335. [Google Scholar] [CrossRef] [PubMed]
- Kawano, M.; Tanaka, K.; Itonaga, I.; Iwasaki, T.; Tsumura, H. MicroRNA-181c prevents apoptosis by targeting of FAS receptor in Ewing’s sarcoma cells. Cancer Cell Int. 2018, 18, 37. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Liu, Z.; Peng, Y.; Yu, C. MicroRNA-181c inhibits glioblastoma cell invasion, migration and mesenchymal transition by targeting TGF-beta pathway. Biochem. Biophys. Res. Commun. 2016, 469, 1041–1048. [Google Scholar] [CrossRef] [PubMed]
- Cui, M.; Yue, L.; Fu, Y.; Yu, W.; Hou, X.; Zhang, X. Association of microRNA-181c expression with the progression and prognosis of human gastric carcinoma. Hepato-Gastroenterology 2013, 60, 961–964. [Google Scholar] [CrossRef] [PubMed]
- Fang, C.; Li, Q.; Min, G.; Liu, M.; Cui, J.; Sun, J.; Li, L. MicroRNA-181c Ameliorates Cognitive Impairment Induced by Chronic Cerebral Hypoperfusion in Rats. Mol. Neurobiol. 2017, 54, 8370–8385. [Google Scholar] [CrossRef] [PubMed]


© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wong, N.K.P.; Cheung, H.; Solly, E.L.; Vanags, L.Z.; Ritchie, W.; Nicholls, S.J.; Ng, M.K.C.; Bursill, C.A.; Tan, J.T.M. Exploring the Roles of CREBRF and TRIM2 in the Regulation of Angiogenesis by High-Density Lipoproteins. Int. J. Mol. Sci. 2018, 19, 1903. https://doi.org/10.3390/ijms19071903
Wong NKP, Cheung H, Solly EL, Vanags LZ, Ritchie W, Nicholls SJ, Ng MKC, Bursill CA, Tan JTM. Exploring the Roles of CREBRF and TRIM2 in the Regulation of Angiogenesis by High-Density Lipoproteins. International Journal of Molecular Sciences. 2018; 19(7):1903. https://doi.org/10.3390/ijms19071903
Chicago/Turabian StyleWong, Nathan K. P., Helena Cheung, Emma L. Solly, Laura Z. Vanags, William Ritchie, Stephen J. Nicholls, Martin K. C. Ng, Christina A. Bursill, and Joanne T. M. Tan. 2018. "Exploring the Roles of CREBRF and TRIM2 in the Regulation of Angiogenesis by High-Density Lipoproteins" International Journal of Molecular Sciences 19, no. 7: 1903. https://doi.org/10.3390/ijms19071903