Importance of Full-Collapse Vesicle Exocytosis for Synaptic Fatigue-Resistance at Rat Fast and Slow Muscle Neuromuscular Junctions

Abstract
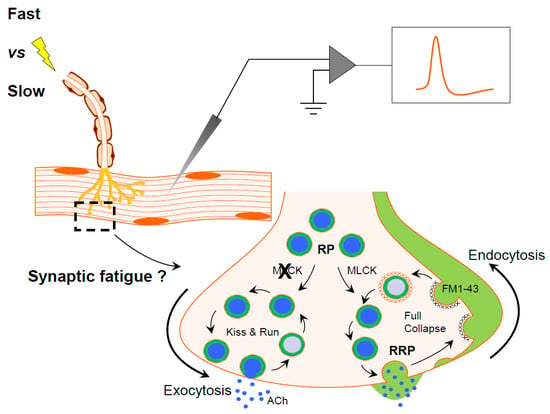
:
1. Introduction
2. Results
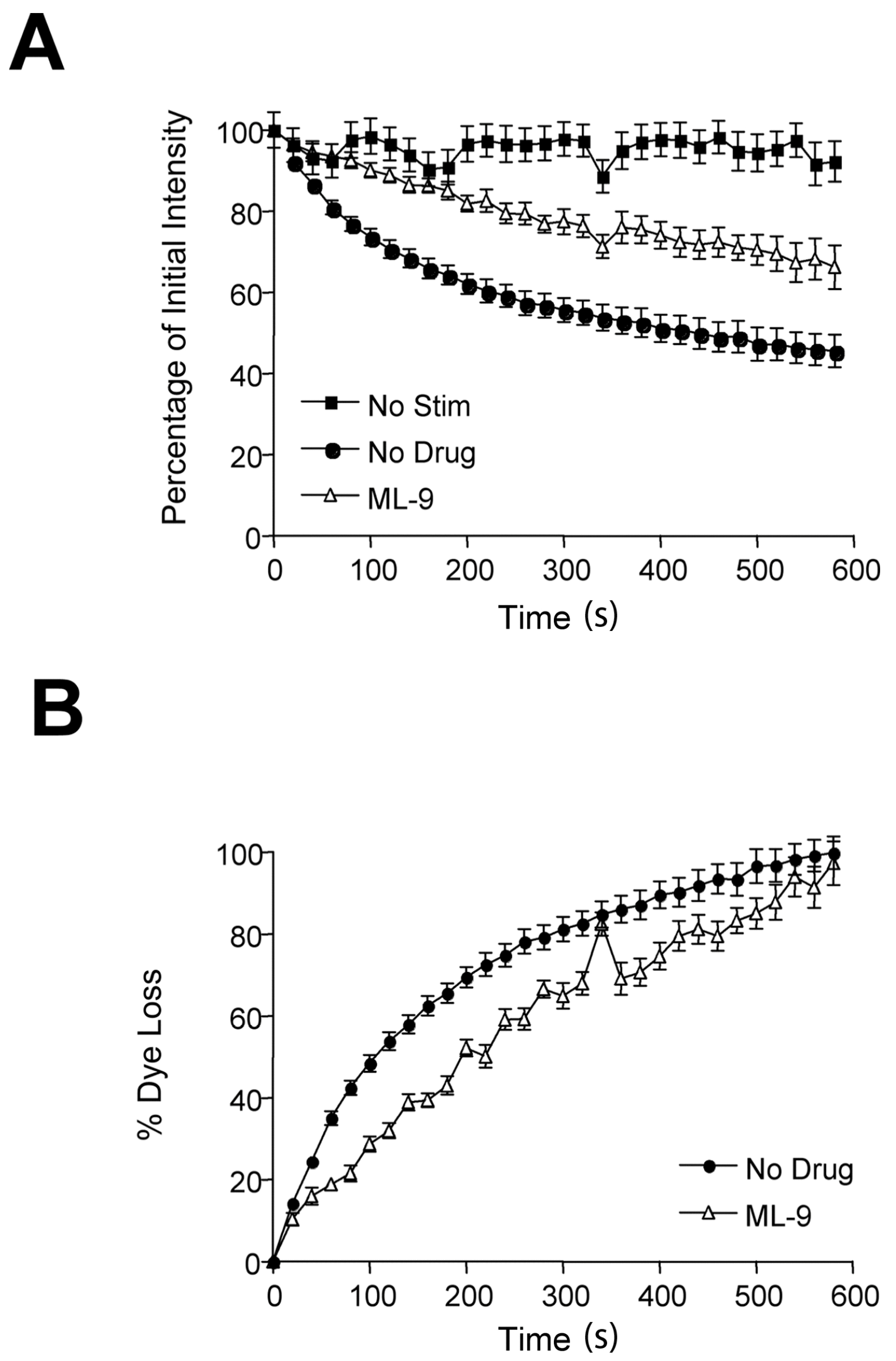
2.1. MLCK Inhibitors Block FM1-43 Release in Rat Motor Nerve Terminals
2.2. MLCK Inhibitors Do Not Impair Neurotransmitter Release in EDL NMJs at 20 Hz
2.3. A Component of FM1-43 Release Persists During Kinase Inhibition
2.4. Estimating the Size of the Inhibitor-Resistant Vesicle Pool Releasing FM1-43
2.5. MLCK Inhibition Slows, but Does Not Block, FM 2–10 Release
2.6. Sustained Transmitter Release from Fatigue-Resistant Terminals Is More Sensitive to Kinase Inhibition
3. Discussion
3.1. Kinase Inhibitors
3.2. Vesicle Exocytosis in Kinase-Blocked Terminals
4. Methods
4.1. Dissection
4.2. Styryl Dye Labelling and Imaging
4.3. Intracellular Recording
4.4. Data Analysis
4.5. Statistics
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Ryan, T.A. Inhibitors of myosin light chain kinase block synaptic vesicle pool mobilization during action potential firing. J. Neurosci. 1999, 19, 1317–1323. [Google Scholar] [CrossRef] [PubMed]
- Rizzoli, S.O.; Betz, W.J. Synaptic vesicle pools. Nat. Rev. Neurosci. 2005, 6, 57–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richards, D.A.; Guatimosim, C.; Betz, W.J. Two endocytic recycling routes selectively fill two vesicle pools in frog motor nerve terminals. Neuron 2000, 27, 551–559. [Google Scholar] [CrossRef]
- Betz, W.J.; Bewick, G.S. Optical analysis of synaptic vesicle recycling at the frog neuromuscular junction. Science 1992, 255, 200–203. [Google Scholar] [CrossRef] [PubMed]
- Reid, B.; Slater, C.R.; Bewick, G.S. Synaptic vesicle dynamics in rat fast and slow motor nerve terminals. J. Neurosci. 1999, 19, 2511–2521. [Google Scholar] [CrossRef] [PubMed]
- Jordan, R.; Lemke, E.A.; Klingauf, J. Visualisation of synaptic vesicle movement in intact synaptic boutons using fluorescence fluctuation spectroscopy. Biophys. J. 2005, 89, 2091–2102. [Google Scholar] [CrossRef] [PubMed]
- Polo-Parada, L.; Plattner, F.; Bose, C.; Landmesser, L.T. NCAM 180 acting via a conserved C-terminal domain and MLCK is essential for effective transmission with repetitive stimulation. Neuron 2005, 46, 917–931. [Google Scholar] [CrossRef] [PubMed]
- Henkel, A.W.; Betz, W.J. Staurosporine blocks evoked release of FM1-43 but not acetylcholine from frog motor nerve terminals. J. Neurosci. 1995, 15, 8246–8258. [Google Scholar] [CrossRef] [PubMed]
- Becherer, U.; Guatimosim, C.; Betz, W. Effects of staurosporine on exocytosis and endocytosis at frog motor nerve terminals. J. Neurosci. 2001, 21, 782–787. [Google Scholar] [CrossRef] [PubMed]
- Maeno-Hikichi, Y.; Polo-Parada, L.; Kastanenka, K.V.; Landmesser, L.T. Frequency-dependent modes of synaptic vesicle endocytosis and exocytosis at adult mouse neuromuscular junctions. J. Neurosci. 2011, 31, 1093–1105. [Google Scholar] [CrossRef] [PubMed]
- Hennig, R.; Lømo, T. Firing patterns of motor units in normal rats. Nature 1985, 314, 164–166. [Google Scholar] [CrossRef] [PubMed]
- Rudling, J.E.; Cuthbert, A.; McWilliam, R.; Reid, B.; Bewick, G.S. Protein kinase inhibitors decrease vesicle recycle time at the rat neuromuscular junction. Soc. Neurosci. Abstr. 2001, 27, 273. [Google Scholar]
- Reid, B.; Martinov, V.N.; Nja, A.; Lømo, T.; Bewick, G.S. Activity-dependent plasticity of transmitter release from nerve terminals in rat fast and slow muscles. J. Neurosci. 2003, 23, 9340–9348. [Google Scholar] [CrossRef] [PubMed]
- Reinhold, S.L.; Prescott, S.M.; Zimmerman, G.A.; McIntyre, T.M. Activation of human neutrophil phospholipase D by three separable mechanisms. FASEB J. 1990, 4, 208–214. [Google Scholar] [CrossRef] [PubMed]
- Ozawa, K.; Masujima, T.; Ikeda, K.; Kodama, Y.; Nonomura, Y. Different pathways of inhibitory effects of wortmannin on exocytosis are revealed by video-enhanced light microscope. Biochem. Biophys. Res. Commun. 1996, 222, 243–248. [Google Scholar] [CrossRef] [PubMed]
- Rao, K.; Paik, W.Y.; Zheng, L.; Jobin, R.M.; Tomic, M.; Jiang, H.; Nakanishi, S.; Stojilkovic, S.S. Wortmannin-sensitive and -insensitive steps in calcium-controlled exocytosis in pituitary gonadotrophs: Evidence that myosin light chain kinase mediates calcium-dependent and wortmannin-sensitive gonadotropin secretion. Endocrinology 1997, 138, 1440–1449. [Google Scholar] [CrossRef] [PubMed]
- Ribchester, R.R.; Mao, F.; Betz, W.J. Optical measurements of activity-dependent membrane recycling in motor nerve terminals of mammalian skeletal muscle. Proc. Biol. Sci. 1994, 255, 61–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klingauf, J.; Kavalali, E.T.; Tsien, R.W. Kinetics and regulation of fast endocytosis at hippocampal synapses. Nature 1998, 394, 581–585. [Google Scholar] [CrossRef] [PubMed]
- Kay, A.R.; Alfonso, A.; Alford, S.; Cline, H.T.; Holgado, A.M.; Sakmann, B.; Snitsarev, V.A.; Stricker, T.P.; Takahashi, M.; Wu, L.G. Imaging synaptic activity in intact brain and slices with FM1-43 in C. elegans, lamprey, and rat. Neuron 1999, 24, 809–817. [Google Scholar] [CrossRef]
- Polo-Parada, L.; Bose, C.M.; Landmesser, L.T. Alterations in transmission, vesicle dynamics, and transmitter release machinery at NCAM-deficient neuromuscular junctions. Neuron 2001, 32, 815–828. [Google Scholar] [CrossRef]
- Betz, W.J.; Bewick, G.S. Optical monitoring of transmitter release and synaptic vesicle recycling at the frog neuromuscular junction. J. Physiol. 1993, 460, 287–309. [Google Scholar] [CrossRef] [PubMed]
- Rizzoli, S.O.; Betz, W.J. Effects of 2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one on synaptic vesicle cycling at the frog neuromuscular junction. J. Neurosci. 2002, 22, 10680–10689. [Google Scholar] [CrossRef] [PubMed]
- Richards, D.A.; Rizzoli, S.O.; Betz, W.J. Effects of wortmannin and latrunculin A on slow endocytosis at the frog neuromuscular junction. J. Physiol. 2004, 557, 77–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, S.J.; Chang, C.C. Inhibition of quantal release from motor nerve by wortmannin. Br. J. Pharmacol. 1999, 128, 142–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tokuoka, H.; Goda, Y. Myosin light chain kinase is not a regulator of synaptic vesicle trafficking during repetitive exocytosis in cultured hippocampal neurons. J. Neurosci. 2006, 26, 11606–11614. [Google Scholar] [CrossRef] [PubMed]
- Vlahos, C.J.; Matter, W.F.; Hui, K.Y.; Brown, R.F. A specific inhibitor of phosphatidylinositol 3-kinase, 2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one (LY294002). J. Biol. Chem. 1994, 269, 5241–5248. [Google Scholar] [PubMed]
- Arcaro, A.; Wymann, M.P. Wortmannin is a potent phosphatidylinositol 3-kinase inhibitor: The role of phosphatidylinositol 3,4,5-trisphosphate in neutrophil responses. Biochem. J. 1993, 296, 297–301. [Google Scholar] [CrossRef] [PubMed]
- Kraszewski, K.; Daniell, L.; Mundigl, O.; De Camilli, P. Mobility of synaptic vesicles in nerve endings monitored by recovery from photobleaching of synaptic vesicle-associated fluorescence. J. Neurosci. 1996, 16, 5905–5913. [Google Scholar] [CrossRef] [PubMed]
- Wood, S.J.; Slater, C.R. Safety factor at the neuromuscular junction. Prog. Neurobiol. 2001, 64, 393–429. [Google Scholar] [CrossRef]
- Wood, S.J.; Slater, C.R. The contribution of postsynaptic folds to the safety factor for neuromuscular transmission in rat fast- and slow-twitch muscles. J. Physiol. 1997, 500 Pt 1, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Liley, A.W. An investigation of spontaneous activity at the neuromuscular junction of the rat. J. Physiol. 1956, 132, 650–666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gray, W.R.; Olivera, B.M.; Cruz, L.J. Peptide toxins from venomous conus snails. Annu. Rev. Biochem. 1988, 57, 665–700. [Google Scholar] [CrossRef] [PubMed]
- Plomp, J.J.; van Kempen, G.T.; Molenaar, P.C. Adaptation of quantal content to decreased postsynaptic sensitivity at single endplates in alpha-bungarotoxin-treated rats. J. Physiol. 1992, 458, 487–499. [Google Scholar] [CrossRef] [PubMed]
- McLachlan, E.M.; Martin, A.R. Non-linear summation of end-plate potentials in the frog and mouse. J. Physiol. 1981, 311, 307–324. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug | n | Deviation Time (s) | Destaining Pool Size |
|---|---|---|---|
| No Drug | 6 | 125 ± 15 | 154,918 ± 7625 |
| Staurosporine | 5 | 14 ± 5 * | 11,250 ± 3049 ** |
| Wortmannin | 3 | 13 ± 4 * | 12,257 ± 3192 ** |
| ML-9 | 3 | 30 ± 7 * | 36,592 ± 7451 ** |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rudling, J.E.; Drever, B.D.; Reid, B.; Bewick, G.S. Importance of Full-Collapse Vesicle Exocytosis for Synaptic Fatigue-Resistance at Rat Fast and Slow Muscle Neuromuscular Junctions. Int. J. Mol. Sci. 2018, 19, 1936. https://doi.org/10.3390/ijms19071936
Rudling JE, Drever BD, Reid B, Bewick GS. Importance of Full-Collapse Vesicle Exocytosis for Synaptic Fatigue-Resistance at Rat Fast and Slow Muscle Neuromuscular Junctions. International Journal of Molecular Sciences. 2018; 19(7):1936. https://doi.org/10.3390/ijms19071936
Chicago/Turabian StyleRudling, Jane E., Benjamin D. Drever, Brian Reid, and Guy S. Bewick. 2018. "Importance of Full-Collapse Vesicle Exocytosis for Synaptic Fatigue-Resistance at Rat Fast and Slow Muscle Neuromuscular Junctions" International Journal of Molecular Sciences 19, no. 7: 1936. https://doi.org/10.3390/ijms19071936