Maintenance of Kidney Metabolic Homeostasis by PPAR Gamma


{kind=link}
{kind=link}
Abstract
:1. Introduction
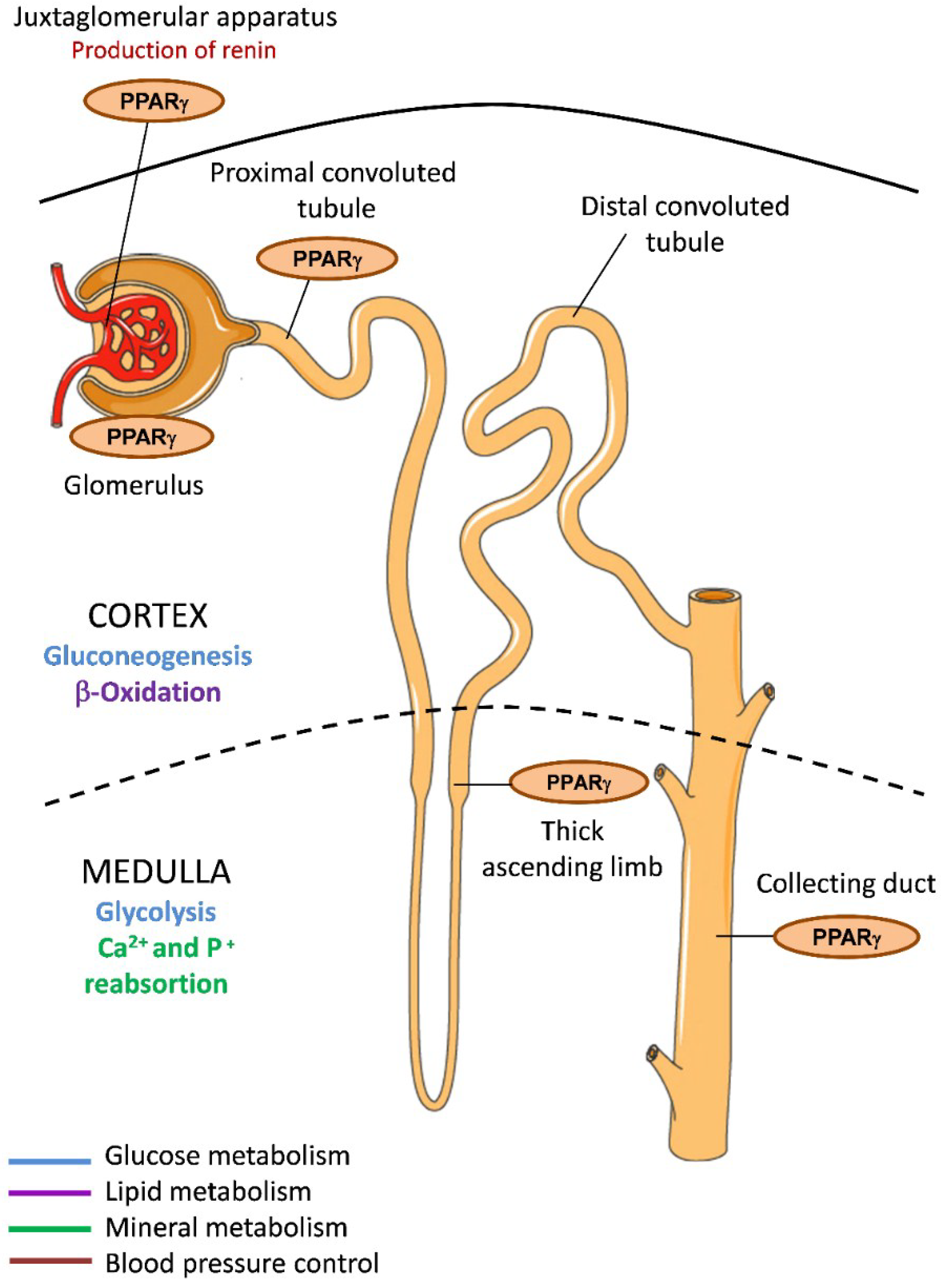
2. PPARγ in Renal Lipid Metabolism
3. PPARγ in Renal Glucose Metabolism
4. PPARγ in Renal Mineral Metabolism
5. PPARγ in Systemic Blood Pressure Control
6. PPARγ and Circadian Rhythm
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| CKD | chronic kidney disease |
| EMT | epithelial-mesenchymal transition |
| FFA | free fatty acids |
| FGF23 | fibroblast growth factor 23 |
| GLUT | glucose transporter |
| PPAR | Peroxisome proliferator-activated receptors |
| PPRE | PPAR response element |
| RAAS | Renin Angiotensin Aldosterone System |
| RXR | retinoid X receptors |
| SGLT | sodium-dependent glucose cotransporter |
| TGFβ | transforming growth factor beta |
| TZD | thiazolidinedione |
References
- Khurana, S.; Bruggeman, L.A.; Kao, H.-Y. Nuclear hormone receptors in podocytes. Cell Biosci. 2012, 2, 33. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.C.; Jandeleit-Dahm, K.A.; Tikellis, C. The renoprotective actions of peroxisome proliferator-activated receptors agonists in diabetes. PPAR Res. 2012, 2012, 456529. [Google Scholar] [CrossRef] [PubMed]
- Higgins, L.S.; Depaoli, A.M. Selective peroxisome proliferator-activated receptor gamma (PPAR gamma) modulation as a strategy for safer therapeutic PPAR gamma activation 1–3. Am. J. Clin. Nutr. 2010, 91, 267–272. [Google Scholar] [CrossRef] [PubMed]
- Vitale, S.G.; Laganà, A.S.; Nigro, A.; La Rosa, V.L.; Rossetti, P.; Rapisarda, A.M.C.; La Vignera, S.; Condorelli, R.A.; Corrado, F.; Buscema, M.; et al. Peroxisome Proliferator-Activated Receptor Modulation during Metabolic Diseases and Cancers: Master Minions. PPAR Res. 2016, 2016, 6517313. [Google Scholar] [CrossRef] [PubMed]
- Tovar-Palacio, C.; Torres, N.; Diaz-Villaseñor, A.; Tovar, A.R. The role of nuclear receptors in the kidney in obesity and metabolic syndrome. Genes Nutr. 2012, 7, 483–498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Escher, P.; Braissant, O.; Basu-Modak, S.; Michalik, L.; Wahli, W.; Desvergne, B. Rat PPARs: Quantitative Analysis in Adult Rat Tissues and Regulation in Fasting and Refeeding. Endocrinology 2001, 142, 4195–4202. [Google Scholar] [CrossRef] [PubMed]
- Guan, Y.; Zhang, Y.; Davis, L.; Breyer, M.D. Expression of peroxisome proliferator-activated receptors in urinary tract of rabbits and humans. Am. J. Physiol. 1997, 273, F1013–F1022. [Google Scholar] [CrossRef] [PubMed]
- Beamer, B.A.; Negri, C.; Yen, C.J.; Gavrilova, O.; Rumberger, J.M.; Durcan, M.J.; Yarnall, D.P.; Hawkins, A.L.; Griffin, C.A.; Burns, D.K.; et al. Chromosomal localization and partial genomic structure of the human peroxisome proliferator activated receptor-gamma (hPPAR gamma) gene. Biochem. Biophys. Res. Commun. 1997, 233, 756–759. [Google Scholar] [CrossRef] [PubMed]
- Martin, G.; Schoonjans, K.; Staels, B.; Auwerx, J. PPARγ activators improve glucose-homeostasis by stimulating fatty-acid uptake in the adipocytes. Atherosclerosis 1998, 137, S75–S80. [Google Scholar] [CrossRef]
- Astarci, E.; Banerjee, S. PPARG (peroxisome proliferator-activated receptor gamma). Atlas Genet. Cytogenet. Oncol. Heamatol. 2009, 13, 417–421. [Google Scholar] [CrossRef]
- Navidshad, B.; Royan, M. Ligands and Regulatory Modes of Peroxisome Proliferator-Activated Receptor Gamma (PPARγ) in Avians. Crit. Rev. Eukaryot. Gene Expr. 2015, 25, 287–292. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Janesick, A.; Wu, L.; Hu, L.; Tang, W.; Blumberg, B.; Shi, H. The unexpected teratogenicity of RXR antagonist UVI3003 via activation of PPARγ in Xenopus tropicalis. Toxicol. Appl. Pharmacol. 2017, 314, 91–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ristow, M.; Muller-Wieland, D.; Pfeiffer, A.; Krone, W.; Kahn, C.R. Obesity associated with a mutation in a genetic regulator of adipocite diferentiation. Engl. J. Med. 1998, 339, 953. [Google Scholar] [CrossRef] [PubMed]
- Barroso, I.; Gurnell, M.; Crowley, V.E.F.; Agostini, M.; Schwabe, J.W.; Soos, M.A.; Maslen, G.L.; Williams, T.D.M.; Lewis, H.; Schafer, A.J.; et al. Dominant negative mutations in human PPARgamma associated with severe insulin resistance, diabetes mellitus and hypertension. Nature 1999, 402, 880–883. [Google Scholar] [CrossRef] [PubMed]
- Nikiforova, M.N.; Lynch, R.A.; Biddinger, P.W.; Alexander, E.K.; Dorn, G.W.; Tallini, G.; Kroll, T.G.; Nikiforov, Y.E. RAS point mutations and PAX8-PPARγ Rearrangement in thyroid tumors: Evidence for distinct molecular pathways in thyroid follicular carcinoma. J. Clin. Endocrinol. Metab. 2003, 88, 2318–2326. [Google Scholar] [CrossRef] [PubMed]
- Marques, A.R.; Espadinha, C.; Catarino, A.L.; Moniz, S.; Pereira, T.; Sobrinho, L.G.; Leite, V. Expression of PAX8-PPAR gamma 1 rearrangements in both follicular thyroid carcinomas and adenomas. J. Clin. Endocrinol. Metab. 2002, 87, 3947–3952. [Google Scholar] [CrossRef] [PubMed]
- Rosen, E.D.; Spiegelman, B.M. PPARgamma: A nuclear regulator of metabolism, differentiation, and cell growth. J. Biol. Chem. 2001, 276, 37731–37734. [Google Scholar] [CrossRef] [PubMed]
- Sugawara, A.; Uruno, A.; Kudo, M.; Matsuda, K.; Yang, C.W.; Ito, S. Effects of PPARγ on hypertension, atherosclerosis, and chronic kidney disease. Endocr. J. 2010, 57, 847–852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwanishi, M.; Ebihara, K.; Kusakabe, T.; Chen, W.; Ito, J.; Masuzaki, H.; Hosoda, K.; Nakao, K. Clinical characteristics and efficacy of pioglitazone in a Japanese diabetic patient with an unusual type of familial partial lipodystrophy. Metabolism 2009, 58, 1681–1687. [Google Scholar] [CrossRef] [PubMed]
- Al-Shali, K.; Cao, H.; Knoers, N.; Hermus, A.R.; Tack, C.J.; Hegele, R.A. A single-base mutation in the peroxisome proliferator-activated receptor gamma4 promoter associated with altered in vitro expression and partial lipodystrophy. J. Clin. Endocrinol. Metab. 2004, 89, 5655–5660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmadian, M.; Suh, J.M.; Hah, N.; Liddle, C.; Atkins, A.R.; Downes, M.; Evans, R.M. PPARγ signaling and metabolism: The good, the bad and the future. Nat. Med. 2013, 19, 557–566. [Google Scholar] [CrossRef] [PubMed]
- Burns, K.A.; Vanden Heuvel, J.P. Modulation of PPAR activity via phosphorylation. Biochim. Biophys. Acta 2007, 1771, 952–960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berrabah, W.; Aumercier, P.; Lefebvre, P.; Staels, B. Control of nuclear receptor activities in metabolism by post-translational modifications. FEBS Lett. 2011, 585, 1640–1650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, J.H.; Banks, A.S.; Estall, J.L.; Kajimura, S.; Boström, P.; Laznik, D.; Ruas, J.L.; Chalmers, M.J.; Kamenecka, T.M.; Blüher, M.; et al. Anti-diabetic drugs inhibit obesity-linked phosphorylation of PPARgamma by Cdk5. Nature 2010, 466, 451–456. [Google Scholar] [CrossRef] [PubMed]
- Povero, D.; Panera, N.; Eguchi, A.; Johnson, C.D.; Papouchado, B.G.; de Araujo Horcel, L.; Pinatel, E.M.; Alisi, A.; Nobili, V.; Feldstein, A.E. Lipid-Induced Hepatocyte-Derived Extracellular Vesicles Regulate Hepatic Stellate Cells via MicroRNA Targeting Peroxisome Proliferator-Activated Receptor-γ. Cell. Mol. Gastroenterol. Hepatol. 2015, 1, 646–663. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Michele, D.E.; Park, J.; Smart, A.M.; Lin, Z.; Brosius, F.C.; Schnermann, J.B.; Briggs, J.P. Expression of peroxisomal proliferator-activated receptors and retinoid X receptors in the kidney. Am. J. Physiol. 1999, 277, F966–F973. [Google Scholar] [CrossRef] [PubMed]
- Kiss-Tóth, É.; Röszer, T. PPAR γ in kidney physiology and pathophysiology. PPAR Res. 2008, 2008, 183108. [Google Scholar] [CrossRef] [PubMed]
- Sarafidis, P.A.; Bakris, G.L. Protection of the kidney by thiazolidinediones: An assessment from bench to bedside. Kidney Int. 2006, 70, 1223–1233. [Google Scholar] [CrossRef] [PubMed]
- Druilhet, R.E.; Overturf, M.L.; Kirkendall, W.M. Cortical and medullary lipids of normal and nephrosclerotic human kidney. Int. J. Biochem. 1978, 9, 729–734. [Google Scholar] [CrossRef]
- Bobulescu, I.A.; Lotan, Y.; Zhang, J.; Rosenthal, T.R.; Rogers, J.T.; Adams-Huet, B.; Sakhaee, K.; Moe, O.W. Triglycerides in the human kidney cortex: Relationship with body size. PLoS ONE 2014, 9, e101285. [Google Scholar] [CrossRef] [PubMed]
- Moorhead, J.F.; Chan, M.K.; El-Nahas, M.; Varghese, Z. Lipid nephrotoxicity in chronic progressive glomerular and tubulo-interstitial disease. Lancet 1982, 2, 1309–1311. [Google Scholar] [CrossRef]
- Izquierdo-Lahuerta, A.; Martínez-García, C.; Medina-Gómez, G. Lipotoxicity as a trigger factor of renal disease. J. Nephrol. 2016, 29, 603–610. [Google Scholar] [CrossRef] [PubMed]
- Kume, S.; Uzu, T.; Araki, S.I.; Sugimoto, T.; Isshiki, K.; Chin-Kanasaki, M.; Sakaguchi, M.; Kubota, N.; Terauchi, Y.; Kadowaki, T.; et al. Role of Altered Renal Lipid Metabolism in the Development of Renal Injury Induced by a High-Fat Diet. J. Am. Soc. Nephrol. 2007, 18, 2715–2723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martínez-García, C.; Izquierdo, A.; Velagapudi, V.; Vivas, Y.; Velasco, I.; Campbell, M.; Burling, K.; Cava, F.; Ros, M.; Orešič, M.; et al. Accelerated renal disease is associated with the development of metabolic syndrome in a glucolipotoxic mouse model. Dis. Models Mech. 2012, 5, 636–648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Medina-Gomez, G.; Yetukuri, L.; Velagapudi, V.; Campbell, M.; Blount, M.; Jimenez-Linan, M.; Ros, M.; Orešič, M.; Vidal-Puig, A. Adaptation and failure of pancreatic cells in murine models with different degrees of metabolic syndrome. Dis. Models Mech. 2009, 2, 582–592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martínez-García, C.; Izquierdo-Lahuerta, A.; Vivas, Y.; Velasco, I.; Yeo, T.K.; Chen, S.; Medina-Gomez, G. Renal Lipotoxicity-Associated Inflammation and Insulin Resistance Affects Actin Cytoskeleton Organization in Podocytes. PLoS ONE 2015, 10, e0142291. [Google Scholar] [CrossRef] [PubMed]
- Berg, A.H.; Combs, T.P.; Scherer, P.E. ACRP30/adiponectin: An adipokine regulating glucose and lipid metabolism. Trends Endocrinol. Metab. 2002, 13, 84–89. [Google Scholar] [CrossRef]
- Long, Q.; Lei, T.; Feng, B.; Yin, C.; Jin, D.; Wu, Y.; Zhu, X.; Chen, X.; Gan, L.; Yang, Z. Peroxisome proliferator-activated receptor-γ increases adiponectin secretion via transcriptional repression of endoplasmic reticulum chaperone protein ERp44. Endocrinology 2010, 151, 3195–3203. [Google Scholar] [CrossRef] [PubMed]
- Rutkowski, J.M.; Wang, Z.V.; Park, A.S.D.; Zhang, J.; Zhang, D.; Hu, M.C.; Moe, O.W.; Susztak, K.; Scherer, P.E. Adiponectin Promotes Functional Recovery after Podocyte Ablation. J. Am. Soc. Nephrol. 2013, 24, 268–282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guettier, J.M.; Park, J.Y.; Cochran, E.K.; Poitou, C.; Basdevant, A.; Meier, M.; Clément, K.; Magré, J.; Gorden, P. Leptin therapy for partial lipodystrophy linked to a PPAR-γ mutation. Clin. Endocrinol. 2008, 68, 547–554. [Google Scholar] [CrossRef] [PubMed]
- Simonds, S.E.; Pryor, J.T.; Ravussin, E.; Greenway, F.L.; Dileone, R.; Allen, A.M.; Bassi, J.; Elmquist, J.K.; Keogh, J.M.; Henning, E.; et al. Leptin mediates the increase in blood pressure associated with obesity. Cell 2014, 159, 1404–1416. [Google Scholar] [CrossRef] [PubMed]
- Rőszer, T.; Menéndez-Gutiérrez, M.P.; Lefterova, M.I.; Alameda, D.; Núñez, V.; Lazar, M.A.; Fischer, T.; Ricote, M. Autoimmune Kidney Disease and Impaired Engulfment of Apoptotic Cells in Mice with Macrophage Peroxisome Proliferator-Activated Receptor or Retinoid X Receptor Deficiency. J. Immunol. 2011, 186, 621–631. [Google Scholar] [CrossRef] [PubMed]
- Luzar, B.; Ferluga, D. Role of lipids in the progression of renal disease in systemic lupus erythematosus patients. Wiener Klinische Wochenschrift 2000, 112, 716–721. [Google Scholar] [PubMed]
- Mather, A.; Pollock, C. Glucose handling by the kidney. Kidney Int. Suppl. 2011, 79, S1–S6. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.J.; Han, H.J. Troglitazone ameliorates high glucose-induced EMT and dysfunction of SGLTs through PI3K/Akt, GSK-3, Snail1, and -catenin in renal proximal tubule cells. AJP Renal Physiol. 2010, 298, F1263–F1275. [Google Scholar] [CrossRef] [PubMed]
- Derlacz, R.A.; Hyc, K.; Usarek, M.; Jagielski, A.K.; Drozak, J.; Jarzyna, R. PPAR-gamma-independent inhibitory effect of rosiglitazone on glucose synthesis in primary cultured rabbit kidney-cortex tubules. Biochem. Cell. Biol. 2008, 86, 396–404. [Google Scholar] [CrossRef] [PubMed]
- Kung, J.; Henry, R.R. Thiazolidinedione safety. Expert Opin. Drug Saf. 2012, 11, 565–579. [Google Scholar] [CrossRef] [PubMed]
- Pap, A.; Cuaranta-Monroy, I.; Peloquin, M.; Nagy, L. Is the mouse a good model of human PPAR??-related metabolic diseases? Int. J. Mol. Sci. 2016, 17, 1236. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zhu, S.; Chen, J.; Tang, Y.; Hu, H.; Mohan, V.; Venkatesan, R.; Wang, J.; Chen, H. Peroxisome proliferator-activated receptor γ polymorphism Pro12Ala is associated with nephropathy in type 2 diabetes: Evidence from meta-analysis of 18 studies. Diabetes Care 2012, 35, 1388–1393. [Google Scholar] [CrossRef] [PubMed]
- Agostini, M.; Schoenmakers, E.; Mitchell, C.; Szatmari, I.; Savage, D.; Smith, A.; Rajanayagam, O.; Semple, R.; Luan, J.A.; Bath, L.; et al. Non-DNA binding, dominant-negative, human PPARγ mutations cause lipodystrophic insulin resistance. Cell Metab. 2006, 4, 303–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dyment, D.A.; Gibson, W.T.; Huang, L.; Bassyouni, H.; Hegele, R.A.; Innes, A.M. Biallelic mutations at PPARG cause a congenital, generalized lipodystrophy similar to the Berardinelli–Seip syndrome. Eur. J. Genet. 2014, 57, 524–526. [Google Scholar] [CrossRef] [PubMed]
- Savage, D.B.; Tan, G.D.; Acerini, C.L.; Jebb, S.A.; Agostini, M.; Gurnell, M.; Williams, R.L.; Umpleby, A.M.; Thomas, E.L.; Bell, J.D.; et al. Human metabolic syndrome resulting from dominant-negative mutations in the nuclear receptor peroxisome proliferator-activated receptor-gamma. Diabetes 2003, 52, 910–917. [Google Scholar] [CrossRef] [PubMed]
- Hegele, R.A.; Cao, H.; Frankowski, C.; Mathews, S.T.; Leff, T. PPARG F388L, a transactivation-deficient mutant, in familial partial lipodystrophy. Diabetes 2002, 51, 3586–3590. [Google Scholar] [CrossRef] [PubMed]
- Auclair, M.; Vigouroux, C.; Boccara, F.; Capel, E.; Vigeral, C.; Guerci, B.; Lascols, O.; Capeau, J.; Caron-Debarle, M. Peroxisome proliferator-activated receptor-γ mutations responsible for lipodystrophy with severe hypertension activate the cellular renin-angiotensin system. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 829–838. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.C.; Kuro-o, M.; Moe, O.W. Klotho and Chronic Kidney Disease. Contrib. Nephrol. 2013, 180, 47–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akune, T.; Ohba, S.; Kamekura, S.; Yamaguchi, M.; Chung, U.I.; Kubota, N.; Terauchi, Y.; Harada, Y.; Azuma, Y.; Nakamura, K.; et al. PPARγ insufficiency enhances osteogenesis through osteoblast formation from bone marrow progenitors. J. Clin. Investig. 2004, 113, 846–855. [Google Scholar] [CrossRef] [PubMed]
- Takada, I.; Kouzmenko, A.P.; Kato, S. Wnt and PPARγ signaling in osteoblastogenesis and adipogenesis. Nat. Rev. Rheumatol. 2009, 5, 442–447. [Google Scholar] [CrossRef] [PubMed]
- Lecarpentier, Y.; Claes, V.; Duthoit, G.; Hébert, J.-L. Circadian rhythms, Wnt/beta-catenin pathway and PPAR alpha/gamma profiles in diseases with primary or secondary cardiac dysfunction. Front. Physiol. 2014, 5, 429. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Li, Y.; Fan, Y.; Wu, J.; Zhao, B.; Guan, Y.; Chien, S.; Wang, N. Klotho is a target gene of PPAR-gamma. Kidney Int. 2008, 74, 732–739. [Google Scholar] [CrossRef] [PubMed]
- Kuro-o, M. Klotho and the Aging Process. Korean J. Intern. Med. 2011, 26, 113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Izquierdo, M.C.; Perez-Gomez, M.V.; Sanchez-Niño, M.D.; Sanz, A.B.; Ruiz-Andres, O.; Poveda, J.; Moreno, J.A.; Egido, J.; Ortiz, A. Klotho, phosphate and inflammation/ageing in chronic kidney disease. Nephrol. Dial. Transplant. 2012, 27, 6–10. [Google Scholar] [CrossRef] [PubMed]
- Yamagishi, T.; Saito, Y.; Nakamura, T.; Takeda, S.I.; Kanai, H.; Sumino, H.; Kuro-o, M.; Nabeshima, Y.I.; Kurabayashi, M.; Nagai, R. Troglitazone improves endothelial function and augments renal klotho mRNA expression in Otsuka Long-Evans Tokushima Fatty (OLETF) rats with multiple atherogenic risk factors. Hypertens. Res. 2001, 24, 705–709. [Google Scholar] [CrossRef] [PubMed]
- Dominick, M.A.; White, M.R.; Sanderson, T.P.; Van Vleet, T.; Cohen, S.M.; Arnold, L.E.; Cano, M.; Tannehill-Gregg, S.; Moehlenkamp, J.D.; Waites, C.R.; et al. Urothelial carcinogenesis in the urinary bladder of male rats treated with muraglitazar, a PPAR alpha/gamma agonist: Evidence for urolithiasis as the inciting event in the mode of action. Toxicol. Pathol. 2006, 34, 903–920. [Google Scholar] [CrossRef] [PubMed]
- Monami, M.; Dicembrini, I.; Mannucci, E. Thiazolidinediones and cancer: Results of a meta-analysis of randomized clinical trials. Acta Diabetol. 2014, 51, 91–101. [Google Scholar] [CrossRef] [PubMed]
- Halabi, C.M.; Beyer, A.M.; de Lange, W.J.; Keen, H.L.; Baumbach, G.L.; Faraci, F.M.; Sigmund, C.D. Interference with PPARγ Function in Smooth Muscle Causes Vascular Dysfunction and Hypertension. Cell Metab. 2008, 7, 215–226. [Google Scholar] [CrossRef] [PubMed]
- Todorov, V.T.; Desch, M.; Schmitt-Nilson, N.; Todorova, A.; Kurtz, A. Peroxisome proliferator-activated receptor-gamma is involved in the control of renin gene expression. Hypertension 2007, 50, 939–944. [Google Scholar] [CrossRef] [PubMed]
- Benson, S.C.; Pershadsingh, H.A.; Ho, C.I.; Chittiboyina, A.; Desai, P.; Pravenec, M.; Qi, N.; Wang, J.; Avery, M.A.; Kurtz, T.W. Identification of Telmisartan as a Unique Angiotensin II Receptor Antagonist with Selective PPARγ-Modulating Activity. Hypertension 2004, 43, 993–1002. [Google Scholar] [CrossRef] [PubMed]
- Schupp, M.; Janke, J.; Clasen, R.; Unger, T.; Kintscher, U. Angiotensin Type 1 Receptor Blockers Induce Peroxisome Proliferator-Activated Receptor-g Activity. Circulation 2004, 109, 2054–2057. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Luo, Q.; Xiong, Z.; Liang, W.; Chen, L.; Xiong, Z. Telmisartan counteracts TGF-β1 induced epithelial-to-mesenchymal transition via PPAR-γ in human proximal tubule epithelial cells. Int. J. Clin. Exp. Pathol. 2012, 5, 522–529. [Google Scholar] [PubMed]
- Raptis, A.E.; Bacharaki, D.; Mazioti, M.; Marathias, K.P.; Markakis, K.P.; Raptis, S.A.; Dimitriadis, G.D.; Vlahakos, D.V. Anemia due to coadministration of renin-angiotensin-system inhibitors and PPARγ agonists in uncomplicated diabetic patients. Exp. Clin. Endocrinol. Diabetes 2012, 120, 416–419. [Google Scholar] [CrossRef] [PubMed]
- Arima, S.; Kohagura, K.; Takeuchi, K.; Taniyama, Y.; Sugawara, A.; Ikeda, Y.; Abe, M.; Omata, K.; Ito, S. Biphasic vasodilator action of troglitazone on the renal microcirculation. J. Am. Soc. Nephrol. 2002, 13, 342–349. [Google Scholar] [PubMed]
- Guan, Y.; Hao, C.; Cha, D.R.; Rao, R.; Lu, W.; Kohan, D.E.; Magnuson, M.A.; Redha, R.; Zhang, Y.; Breyer, M.D. Thiazolidinediones expand body fluid volume through PPARgamma stimulation of ENaC-mediated renal salt absorption. Nat. Med. 2005, 11, 861–866. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zhang, A.; Kohan, D.E.; Nelson, R.D.; Gonzalez, F.J.; Yang, T. Collecting duct-specific deletion of peroxisome proliferator-activated receptor blocks thiazolidinedione-induced fluid retention. Proc. Natl. Acad. Sci. USA 2005, 102, 9406–9411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, N.; Yang, G.; Jia, Z.; Zhang, H.; Aoyagi, T.; Soodvilai, S.; Symons, J.D.; Schnermann, J.B.; Gonzalez, F.J.; Litwin, S.E. Vascular PPARγ Controls Circadian Variation in Blood Pressure and Heart Rate through Bmal1. Cell Metab. 2008, 8, 482–491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawai, M.; Rosen, C.J. PPARγ: A circadian transcription factor in adipogenesis and osteogenesis. Nat. Rev. Endocrinol. 2010, 6, 629–636. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Jia, Z.; Aoyagi, T.; McClain, D.; Mortensen, R.M.; Yang, T. Systemic PPARγ Deletion Impairs Circadian Rhythms of Behavior and Metabolism. PLoS ONE 2012, 7, e38117. [Google Scholar] [CrossRef] [PubMed]
- Stubblefield, J.J.; Terrien, J.; Green, C.B. Nocturnin: At the crossroads of clocks and metabolism. Trends Endocrinol. Metab. 2012, 23, 326–333. [Google Scholar] [CrossRef] [PubMed]


© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Corrales, P.; Izquierdo-Lahuerta, A.; Medina-Gómez, G. Maintenance of Kidney Metabolic Homeostasis by PPAR Gamma. Int. J. Mol. Sci. 2018, 19, 2063. https://doi.org/10.3390/ijms19072063
Corrales P, Izquierdo-Lahuerta A, Medina-Gómez G. Maintenance of Kidney Metabolic Homeostasis by PPAR Gamma. International Journal of Molecular Sciences. 2018; 19(7):2063. https://doi.org/10.3390/ijms19072063
Chicago/Turabian StyleCorrales, Patricia, Adriana Izquierdo-Lahuerta, and Gema Medina-Gómez. 2018. "Maintenance of Kidney Metabolic Homeostasis by PPAR Gamma" International Journal of Molecular Sciences 19, no. 7: 2063. https://doi.org/10.3390/ijms19072063
APA StyleCorrales, P., Izquierdo-Lahuerta, A., & Medina-Gómez, G. (2018). Maintenance of Kidney Metabolic Homeostasis by PPAR Gamma. International Journal of Molecular Sciences, 19(7), 2063. https://doi.org/10.3390/ijms19072063