Understanding the Role of Dysfunctional and Healthy Mitochondria in Stroke Pathology and Its Treatment



Abstract
1. Therapeutic Options for Stroke
2. Mitochondria and Stroke
3. Mitochondria, ETC, and OXPHOS
4. Mitochondria-Based Regenerative Medicine
4.1. SIRT1
4.2. Fission and Fusion Modulators
4.3. Purines
4.4. Methylene Blue
4.5. SOD Mimetics
4.6. Antioxidants
4.7. Exercise and Diet
5. Stem Cells as Source of Healthy Mitochondria
6. Stem Cells, Mitochondria, and Stroke
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Bansal, S.; Sangha, K.S.; Khatri, P. Drug treatment of acute ischemic stroke. Am. J. Cardiovasc. Drugs 2013, 13, 57–69. [Google Scholar] [CrossRef] [PubMed]
- Schiavone, S.; Trabace, L. Small molecules: Therapeutic application in neuropsychiatric and neurodegenerative disorders. Molecules 2018, 23, 411. [Google Scholar] [CrossRef] [PubMed]
- Sawada, H.; Nishimura, N.; Suzuki, E.; Zhuang, J.; Hasegawa, K.; Takamatsu, H.; Honda, K.; Hasumi, K. SMTP-7, a novel small-molecule thrombolytic for ischemic stroke: A study in rodents and primates. J. Cereb. Blood Flow Metab. 2014, 34, 235–241. [Google Scholar] [CrossRef] [PubMed]
- Tajiri, N.; Quach, D.M.; Kaneko, Y.; Wu, S.; Lee, D.; Lam, T.; Hayama, K.L.; Hazel, T.G.; Johe, K.; Wu, M.C.; et al. NSI-189, a small molecule with neurogenic properties, exerts behavioral, and neurostructural benefits in stroke rats. J. Cell. Physiol. 2017, 232, 2731–2740. [Google Scholar] [CrossRef] [PubMed]
- Napoli, E.; Song, G.; Schneider, A.; Hagerman, R.; Eldeeb, M.A.; Azarang, A.; Tassone, F.; Giulivi, C. Warburg effect linked to cognitive-executive deficits in FMR1 premutation. FASEB J. 2016, 30, 3334–3351. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.T.; Chen, P.; Ouyang, R.Y.; Song, L. Multifaceted role of prohibitin in cell survival and apoptosis. Apoptosis 2015, 20, 1135–1149. [Google Scholar] [CrossRef] [PubMed]
- Bergman, O.; Ben-Shachar, D. Mitochondrial oxidative phosphorylation system (OXPHOS) deficits in schizophrenia: Possible interactions with cellular processes. Can. J. Psychiatry 2016, 61, 457–469. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Mohsen, A.W.; Mihalik, S.J.; Goetzman, E.S.; Vockley, J. Evidence for physical association of mitochondrial fatty acid oxidation and oxidative phosphorylation complexes. J. Biol. Chem. 2010, 285, 29834–29841. [Google Scholar] [CrossRef] [PubMed]
- Bovo, E.; Mazurek, S.R.; de Tombe, P.P.; Zima, A.V. Increased energy demand during adrenergic receptor stimulation contributes to Ca(2+) wave generation. Biophys. J. 2015, 109, 1583–1591. [Google Scholar] [CrossRef] [PubMed]
- Muller, F.L.; Liu, Y.; Van Remmen, H. Complex III releases superoxide to both sides of the inner mitochondrial membrane. J. Biol. Chem. 2004, 279, 49064–49073. [Google Scholar] [CrossRef] [PubMed]
- Du, F.; Zhu, X.H.; Zhang, Y.; Friedman, M.; Zhang, N.; Ugurbil, K.; Chen, W. Tightly coupled brain activity and cerebral ATP metabolic rate. Proc. Natl. Acad. Sci. USA 2008, 105, 6409–6414. [Google Scholar] [CrossRef] [PubMed]
- Silzer, T.K.; Phillips, N.R. Etiology of type 2 diabetes and Alzheimer’s disease: Exploring the mitochondria. Mitochondrion 2018. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.S.; Geng, W.S.; Jia, J.J.; Chen, L.; Zhang, P.P. Cellular and molecular basis of neurodegeneration in Parkinson disease. Front. Aging Neurosci. 2018, 10, 109. [Google Scholar] [CrossRef] [PubMed]
- Karabatsiakis, A.; Bock, C.; Salinas-Manrique, J.; Kolassa, S.; Calzia, E.; Dietrich, D.E.; Kolassa, I.T. Mitochondrial respiration in peripheral blood mononuclear cells correlates with depressive subsymptoms and severity of major depression. Transl. Psychiatry 2014, 4, e397. [Google Scholar] [CrossRef] [PubMed]
- Prabakaran, S.; Swatton, J.E.; Ryan, M.M.; Huffaker, S.J.; Huang, J.T.; Griffin, J.L.; Wayland, M.; Freeman, T.; Dudbridge, F.; Lilley, K.S.; et al. Mitochondrial dysfunction in schizophrenia: Evidence for compromised brain metabolism and oxidative stress. Mol. Psychiatry 2004, 9, 684–697. [Google Scholar] [CrossRef] [PubMed]
- Napoli, E.; Wong, S.; Giulivi, C. Evidence of reactive oxygen species-mediated damage to mitochondrial DNA in children with typical autism. Mol. Autism 2013, 4, 2. [Google Scholar] [CrossRef] [PubMed]
- Napoli, E.; Wong, S.; Hertz-Picciotto, I.; Giulivi, C. Deficits in bioenergetics and impaired immune response in granulocytes from children with autism. Pediatrics 2014, 133, e1405–e1410. [Google Scholar] [CrossRef] [PubMed]
- Tuttolomondo, A.; Pecoraro, R.; Simonetta, I.; Miceli, S.; Arnao, V.; Licata, G.; Pinto, A. Neurological complications of Anderson-Fabry disease. Curr. Pharm. Des. 2013, 19, 6014–6030. [Google Scholar] [CrossRef] [PubMed]
- Tuttolomondo, A.; Pecoraro, R.; Simonetta, I.; Miceli, S.; Pinto, A.; Licata, G. Anderson-Fabry disease: A multiorgan disease. Curr. Pharm. Des. 2013, 19, 5974–5996. [Google Scholar] [CrossRef] [PubMed]
- Lücke, T.; Höppner, W.; Schmidt, E.; Illsinger, S.; Das, A.M. Fabry disease: Reduced activities of respiratory chain enzymes with decreased levels of energy-rich phosphates in fibroblasts. Mol. Genet. Metab. 2004, 82, 93–97. [Google Scholar] [CrossRef] [PubMed]
- Stonesifer, C.; Corey, S.; Ghanekar, S.; Diamandis, Z.; Acosta, S.A.; Borlongan, C.V. Stem cell therapy for abrogating stroke-induced neuroinflammation and relevant secondary cell death mechanisms. Prog. Neurobiol. 2017, 158, 94–131. [Google Scholar] [CrossRef] [PubMed]
- Jauch, E.C.; Saver, J.L.; Adams, H.P., Jr.; Bruno, A.; Connors, J.J.; Demaerschalk, B.M.; Khatri, P.; McMullan, P.W., Jr.; Qureshi, A.I.; Rosenfield, K.; et al. Guidelines for the early management of patients with acute ischemic stroke: A guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke 2013, 44, 870–947. [Google Scholar] [CrossRef] [PubMed]
- Kelly-Hayes, M. Influence of age and health behaviors on stroke risk: Lessons from longitudinal studies. J. Am. Geriatr. Soc. 2010, 58 (Suppl. 2), S325–S328. [Google Scholar] [CrossRef] [PubMed]
- Singhal, A.B.; Biller, J.; Elkind, M.S.; Fullerton, H.J.; Jauch, E.C.; Kittner, S.J.; Levine, D.A.; Levine, S.R. Recognition and management of stroke in young adults and adolescents. Neurology 2013, 81, 1089–1097. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.S.; Jin, H.; Sun, X.; Huang, S.; Zhang, F.L.; Guo, Z.N.; Yang, Y. Free radical damage in ischemia-reperfusion injury: An obstacle in acute ischemic stroke after revascularization therapy. Oxid. Med. Cell. Longev. 2018, 2018, 3804979. [Google Scholar] [CrossRef] [PubMed]
- Dalkara, T.; Arsava, E.M. Can restoring incomplete microcirculatory reperfusion improve stroke outcome after thrombolysis? J. Cereb. Blood Flow Metab. 2012, 32, 2091–2099. [Google Scholar] [CrossRef] [PubMed]
- Langhorne, P.; Bernhardt, J.; Kwakkel, G. Stroke rehabilitation. Lancet 2011, 377, 1693–1702. [Google Scholar] [CrossRef]
- Honda, H.M.; Korge, P.; Weiss, J.N. Mitochondria and ischemia/reperfusion injury. Ann. N. Y. Acad. Sci. 2005, 1047, 248–258. [Google Scholar] [CrossRef] [PubMed]
- Kann, O.; Kovacs, R. Mitochondria and neuronal activity. Am. J. Physiol. Cell Physiol. 2007, 292, C641–C657. [Google Scholar] [CrossRef] [PubMed]
- Brand, M.D.; Nicholls, D.G. Assessing mitochondrial dysfunction in cells. Biochem. J. 2011, 435, 297–312. [Google Scholar] [CrossRef] [PubMed]
- Shivakumar, A.; Yogendra Kumar, M.S. Critical review on the analytical mechanistic steps in the evaluation of antioxidant activity. Crit. Rev. Anal. Chem. 2018, 48, 214–236. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Yu, E.; Mercer, J.; Bennett, M. Mitochondria in vascular disease. Cardiovasc. Res. 2012, 95, 173–182. [Google Scholar] [CrossRef] [PubMed]
- Schagger, H.; de Coo, R.; Bauer, M.F.; Hofmann, S.; Godinot, C.; Brandt, U. Significance of respirasomes for the assembly/stability of human respiratory chain complex I. J. Biol. Chem. 2004, 279, 36349–36353. [Google Scholar] [CrossRef] [PubMed]
- Chaban, Y.; Boekema, E.J.; Dudkina, N.V. Structures of mitochondrial oxidative phosphorylation supercomplexes and mechanisms for their stabilisation. Biochim. Biophys. Acta 2014, 1837, 418–426. [Google Scholar] [CrossRef] [PubMed]
- Thorburn, D.R. Mitochondrial disorders: Prevalence, myths and advances. J. Inherit. Metab. Dis. 2004, 27, 349–362. [Google Scholar] [CrossRef] [PubMed]
- Distelmaier, F.; Koopman, W.J.; van den Heuvel, L.P.; Rodenburg, R.J.; Mayatepek, E.; Willems, P.H.; Smeitink, J.A. Mitochondrial complex I deficiency: From organelle dysfunction to clinical disease. Brain 2009, 132, 833–842. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, R.H. The neurodegenerative mitochondriopathies. J. Alzheimers Dis. 2009, 17, 737–751. [Google Scholar] [CrossRef] [PubMed]
- Smeitink, J.; van den Heuvel, L.; DiMauro, S. The genetics and pathology of oxidative phosphorylation. Nat. Rev. Genet. 2001, 2, 342–352. [Google Scholar] [CrossRef] [PubMed]
- Duchen, M.R. Mitochondria in health and disease: Perspectives on a new mitochondrial biology. Mol. Asp. Med. 2004, 25, 365–451. [Google Scholar] [CrossRef] [PubMed]
- Droge, W. Free radicals in the physiological control of cell function. Physiol. Rev. 2002, 82, 47–95. [Google Scholar] [CrossRef] [PubMed]
- Hoekstra, A.S.; Bayley, J.P. The role of complex II in disease. Biochim. Biophys. Acta 2013, 1827, 543–551. [Google Scholar] [CrossRef] [PubMed]
- Lemarie, A.; Huc, L.; Pazarentzos, E.; Mahul-Mellier, A.L.; Grimm, S. Specific disintegration of complex II succinate: Ubiquinone oxidoreductase links pH changes to oxidative stress for apoptosis induction. Cell Death Differ. 2011, 18, 338–349. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.; Kim, J.Y.; Hwang, J.; Shin, K.S.; Kang, S.J. Heptachlor induced mitochondria-mediated cell death via impairing electron transport chain complex III. Biochem. Biophys. Res. Commun. 2013, 437, 632–636. [Google Scholar] [CrossRef] [PubMed]
- Freire, C.; Koifman, S. Pesticide exposure and Parkinson’s disease: Epidemiological evidence of association. Neurotoxicology 2012, 33, 947–971. [Google Scholar] [CrossRef] [PubMed]
- Andreu, A.L.; Hanna, M.G.; Reichmann, H.; Bruno, C.; Penn, A.S.; Tanji, K.; Pallotti, F.; Iwata, S.; Bonilla, E.; Lach, B.; et al. Exercise intolerance due to mutations in the cytochrome b gene of mitochondrial DNA. N. Engl. J. Med. 1999, 341, 1037–1044. [Google Scholar] [CrossRef] [PubMed]
- Marin-Garcia, J.; Hu, Y.; Ananthakrishnan, R.; Pierpont, M.E.; Pierpont, G.L.; Goldenthal, M.J. A point mutation in the cytb gene of cardiac mtDNA associated with complex III deficiency in ischemic cardiomyopathy. Biochem. Mol. Biol. Int. 1996, 40, 487–495. [Google Scholar] [CrossRef] [PubMed]
- Shoubridge, E.A. Cytochrome c oxidase deficiency. Am. J. Med. Genet. 2001, 106, 46–52. [Google Scholar] [CrossRef] [PubMed]
- Diaz, F. Cytochrome c oxidase deficiency: Patients and animal models. Biochim. Biophys. Acta 2010, 1802, 100–110. [Google Scholar] [CrossRef] [PubMed]
- Pieczenik, S.R.; Neustadt, J. Mitochondrial dysfunction and molecular pathways of disease. Exp. Mol. Pathol. 2007, 83, 84–92. [Google Scholar] [CrossRef] [PubMed]
- Bonora, M.; Wieckowsk, M.R.; Chinopoulos, C.; Kepp, O.; Kroemer, G.; Galluzzi, L.; Pinton, P. Molecular mechanisms of cell death: Central implication of ATP synthase in mitochondrial permeability transition. Oncogene 2015, 34, 1608. [Google Scholar] [CrossRef] [PubMed]
- Paumard, P.; Vaillier, J.; Coulary, B.; Schaeffer, J.; Soubannier, V.; Mueller, D.M.; Brethes, D.; di Rago, J.P.; Velours, J. The ATP synthase is involved in generating mitochondrial cristae morphology. EMBO J. 2002, 21, 221–230. [Google Scholar] [CrossRef] [PubMed]
- Rodenburg, R.J. Biochemical diagnosis of mitochondrial disorders. J. Inherit. Metab. Dis. 2011, 34, 283–292. [Google Scholar] [CrossRef] [PubMed]
- Houstek, J.; Pickova, A.; Vojtiskova, A.; Mracek, T.; Pecina, P.; Jesina, P. Mitochondrial diseases and genetic defects of ATP synthase. Biochim. Biophys. Acta 2006, 1757, 1400–1405. [Google Scholar] [CrossRef] [PubMed]
- Schon, E.A.; Santra, S.; Pallotti, F.; Girvin, M.E. Pathogenesis of primary defects in mitochondrial ATP synthesis. Semin. Cell Dev. Biol. 2001, 12, 441–448. [Google Scholar] [CrossRef] [PubMed]
- Tuppen, H.A.; Blakely, E.L.; Turnbull, D.M.; Taylor, R.W. Mitochondrial DNA mutations and human disease. Biochim. Biophys. Acta 2010, 1797, 113–128. [Google Scholar] [CrossRef] [PubMed]
- Reeve, A.K.; Krishnan, K.J.; Turnbull, D. Mitochondrial DNA mutations in disease, aging, and neurodegeneration. Ann. N. Y. Acad. Sci. 2008, 1147, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Radak, D.; Resanovic, I.; Isenovic, E.R. Link between oxidative stress and acute brain ischemia. Angiology 2014, 65, 667–676. [Google Scholar] [CrossRef] [PubMed]
- El-Benna, J.; Dang, P.M.; Gougerot-Pocidalo, M.A.; Marie, J.C.; Braut-Boucher, F. p47phox, the phagocyte NADPH oxidase/NOX2 organizer: Structure, phosphorylation and implication in diseases. Exp. Mol. Med. 2009, 41, 217–225. [Google Scholar] [CrossRef] [PubMed]
- Carbone, F.; Teixeira, P.C.; Braunersreuther, V.; Mach, F.; Vuilleumier, N.; Montecucco, F. Pathophysiology and treatments of oxidative injury in ischemic stroke: Focus on the phagocytic NADPH oxidase 2. Antioxid. Redox Signal. 2015, 23, 460–489. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Youle, R. Cell biology: Form follows function for mitochondria. Nature 2016, 530, 288–289. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Yuan, J. Caspases in apoptosis and beyond. Oncogene 2008, 27, 6194–6206. [Google Scholar] [CrossRef] [PubMed]
- Krautwald, S.; Ziegler, E.; Rolver, L.; Linkermann, A.; Keyser, K.A.; Steen, P.; Wollert, K.C.; Korf-Klingebiel, M.; Kunzendorf, U. Effective blockage of both the extrinsic and intrinsic pathways of apoptosis in mice by TAT-crmA. J. Biol. Chem. 2010, 285, 19997–20005. [Google Scholar] [CrossRef] [PubMed]
- Giorgi, C.; Baldassari, F.; Bononi, A.; Bonora, M.; De Marchi, E.; Marchi, S.; Missiroli, S.; Patergnani, S.; Rimessi, A.; Suski, J.M.; et al. Mitochondrial Ca(2+) and apoptosis. Cell Calcium 2012, 52, 36–43. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Zhao, X.; Xu, H.; Chen, F.; Xu, Y.; Li, Z.; Sanchis, D.; Jin, L.; Zhang, Y.; Ye, J. AKT2 blocks nucleus translocation of apoptosis-inducing factor (AIF) and endonuclease G (EndoG) while promoting caspase activation during cardiac ischemia. Int. J. Mol. Sci. 2017, 18. [Google Scholar] [CrossRef] [PubMed]
- Susin, S.A.; Lorenzo, H.K.; Zamzami, N.; Marzo, I.; Snow, B.E.; Brothers, G.M.; Mangion, J.; Jacotot, E.; Costantini, P.; Loeffler, M.; et al. Molecular characterization of mitochondrial apoptosis-inducing factor. Nature 1999, 397, 441–446. [Google Scholar] [CrossRef] [PubMed]
- Lauber, K.; Bohn, E.; Krober, S.M.; Xiao, Y.J.; Blumenthal, S.G.; Lindemann, R.K.; Marini, P.; Wiedig, C.; Zobywalski, A.; Baksh, S.; et al. Apoptotic cells induce migration of phagocytes via caspase-3-mediated release of a lipid attraction signal. Cell 2003, 113, 717–730. [Google Scholar] [CrossRef]
- Shklyar, B.; Levy-Adam, F.; Mishnaevski, K.; Kurant, E. Caspase activity is required for engulfment of apoptotic cells. Mol. Cell. Biol. 2013, 33, 3191–3201. [Google Scholar] [CrossRef] [PubMed]
- Kober, A.M.; Legewie, S.; Pforr, C.; Fricker, N.; Eils, R.; Krammer, P.H.; Lavrik, I.N. Caspase-8 activity has an essential role in CD95/Fas-mediated MAPK activation. Cell Death Dis. 2011, 2, e212. [Google Scholar] [CrossRef] [PubMed]
- Golstein, P.; Griffiths, G.M. An early history of T cell-mediated cytotoxicity. Nat. Rev. Immunol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Formigli, L.; Papucci, L.; Tani, A.; Schiavone, N.; Tempestini, A.; Orlandini, G.E.; Capaccioli, S.; Orlandini, S.Z. Aponecrosis: Morphological and biochemical exploration of a syncretic process of cell death sharing apoptosis and necrosis. J. Cell. Physiol. 2000, 182, 41–49. [Google Scholar] [CrossRef]
- Crowley, M.G.; Liska, M.G.; Borlongan, C.V. Stem cell therapy for sequestering neuroinflammation in traumatic brain injury: An update on exosome-targeting to the spleen. J. Neurosurg. Sci. 2017, 61, 291–302. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, H.; Wang, H.G. The protein kinase PKB/Akt regulates cell survival and apoptosis by inhibiting Bax conformational change. Oncogene 2001, 20, 7779–7786. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.G.; Pathan, N.; Ethell, I.M.; Krajewski, S.; Yamaguchi, Y.; Shibasaki, F.; McKeon, F.; Bobo, T.; Franke, T.F.; Reed, J.C. Ca2+-induced apoptosis through calcineurin dephosphorylation of BAD. Science 1999, 284, 339–343. [Google Scholar] [CrossRef] [PubMed]
- Chandra, D.; Liu, J.W.; Tang, D.G. Early mitochondrial activation and cytochrome c up-regulation during apoptosis. J. Biol. Chem. 2002, 277, 50842–50854. [Google Scholar] [CrossRef] [PubMed]
- Cereghetti, G.M.; Costa, V.; Scorrano, L. Inhibition of Drp1-dependent mitochondrial fragmentation and apoptosis by a polypeptide antagonist of calcineurin. Cell Death Differ. 2010, 17, 1785–1794. [Google Scholar] [CrossRef] [PubMed]
- Gao, W.; Pu, Y.; Luo, K.Q.; Chang, D.C. Temporal relationship between cytochrome c release and mitochondrial swelling during UV-induced apoptosis in living HeLa cells. J. Cell Sci. 2001, 114, 2855–2862. [Google Scholar] [PubMed]
- Cha, M.Y.; Kim, D.K.; Mook-Jung, I. The role of mitochondrial DNA mutation on neurodegenerative diseases. Exp. Mol. Med. 2015, 47, e150. [Google Scholar] [CrossRef] [PubMed]
- Kwong, J.Q.; Beal, M.F.; Manfredi, G. The role of mitochondria in inherited neurodegenerative diseases. J. Neurochem. 2006, 97, 1659–1675. [Google Scholar] [CrossRef] [PubMed]
- Ou, X.; Lee, M.R.; Huang, X.; Messina-Graham, S.; Broxmeyer, H.E. SIRT1 positively regulates autophagy and mitochondria function in embryonic stem cells under oxidative stress. Stem Cells 2014, 32, 1183–1194. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Auwerx, J. Protein deacetylation by SIRT1: An emerging key post-translational modification in metabolic regulation. Pharmacol. Res. 2010, 62, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Liang, F.; Kume, S.; Koya, D. SIRT1 and insulin resistance. Nat. Rev. Endocrinol. 2009, 5, 367–373. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Sarruf, D.A.; Li, P.; Osborn, O.; Sanchez-Alavez, M.; Talukdar, S.; Chen, A.; Bandyopadhyay, G.; Xu, J.; Morinaga, H.; et al. Neuronal SIRT1 deficiency increases insulin sensitivity in both brain and peripheral tissues. J. Biol. Chem. 2013, 288, 10722–10735. [Google Scholar] [CrossRef] [PubMed]
- Rodgers, J.T.; Lerin, C.; Gerhart-Hines, Z.; Puigserver, P. Metabolic adaptations through the PGC-1 alpha and SIRT1 pathways. FEBS Lett. 2008, 582, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Chong, Z.Z.; Shang, Y.C.; Wang, S.; Maiese, K. SIRT1: New avenues of discovery for disorders of oxidative stress. Expert Opin. Ther. Targets 2012, 16, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Borra, M.T.; Smith, B.C.; Denu, J.M. Mechanism of human SIRT1 activation by resveratrol. J. Biol. Chem. 2005, 280, 17187–17195. [Google Scholar] [CrossRef] [PubMed]
- Della-Morte, D.; Dave, K.R.; DeFazio, R.A.; Bao, Y.C.; Raval, A.P.; Perez-Pinzon, M.A. Resveratrol pretreatment protects rat brain from cerebral ischemic damage via a sirtuin 1-uncoupling protein 2 pathway. Neuroscience 2009, 159, 993–1002. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Karamanlidis, G.; Tian, R. Novel targets for mitochondrial medicine. Sci. Transl. Med. 2016, 8, 326rv3. [Google Scholar] [CrossRef] [PubMed]
- Reddy, P.H. Inhibitors of mitochondrial fission as a therapeutic strategy for diseases with oxidative stress and mitochondrial dysfunction. J. Alzheimers Dis. 2014, 40, 245–256. [Google Scholar] [CrossRef] [PubMed]
- Miret-Casals, L.; Sebastian, D.; Brea, J.; Rico-Leo, E.M.; Palacin, M.; Fernandez-Salguero, P.M.; Loza, M.I.; Albericio, F.; Zorzano, A. Identification of new activators of mitochondrial fusion reveals a link between mitochondrial morphology and pyrimidine metabolism. Cell Chem. Biol. 2018, 25, 268–278.e4. [Google Scholar] [CrossRef] [PubMed]
- Szabo, A.; Sumegi, K.; Fekete, K.; Hocsak, E.; Debreceni, B.; Setalo, G., Jr.; Kovacs, K.; Deres, L.; Kengyel, A.; Kovacs, D.; et al. Activation of mitochondrial fusion provides a new treatment for mitochondria-related diseases. Biochem. Pharmacol. 2018, 150, 86–96. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, A.; Vera, J.; Wolkenhauer, O. The systems biology of mitochondrial fission and fusion and implications for disease and aging. Biogerontology 2014, 15, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Lim To, W.K.; Kumar, P.; Marshall, J.M. Hypoxia is an effective stimulus for vesicular release of ATP from human umbilical vein endothelial cells. Placenta 2015, 36, 759–766. [Google Scholar] [CrossRef] [PubMed]
- Gerasimovskaya, E.V.; Woodward, H.N.; Tucker, D.A.; Stenmark, K.R. Extracellular ATP is a pro-angiogenic factor for pulmonary artery vasa vasorum endothelial cells. Angiogenesis 2008, 11, 169–182. [Google Scholar] [CrossRef] [PubMed]
- Lindberg, D.; Shan, D.; Ayers-Ringler, J.; Oliveros, A.; Benitez, J.; Prieto, M.; McCullumsmith, R.; Choi, D.S. Purinergic signaling and energy homeostasis in psychiatric disorders. Curr. Mol. Med. 2015, 15, 275–295. [Google Scholar] [CrossRef] [PubMed]
- Fields, R.D.; Burnstock, G. Purinergic signalling in neuron-glia interactions. Nat. Rev. Neurosci. 2006, 7, 423–436. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Talley Watts, L.; Holstein, D.M.; Wewer, J.; Lechleiter, J.D. P2Y1R-initiated, IP3R-dependent stimulation of astrocyte mitochondrial metabolism reduces and partially reverses ischemic neuronal damage in mouse. J. Cereb. Blood Flow Metab. 2013, 33, 600–611. [Google Scholar] [CrossRef] [PubMed]
- Sperlagh, B.; Illes, P. P2X7 receptor: An emerging target in central nervous system diseases. Trends Pharmacol. Sci. 2014, 35, 537–547. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.; Shen, T.; Hu, J.; Zhang, L.; Zhang, Y.; Bao, L.; Cui, C.; Jin, G.; Zan, K.; Zhang, Z.; et al. Purinergic 2X7 receptor/NLRP3 pathway triggers neuronal apoptosis after ischemic stroke in the mouse. Exp. Neurol. 2017, 292, 46–55. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Duong, T.Q. Methylene blue treatment in experimental ischemic stroke: A mini review. Brain Circ. 2016, 2, 48–53. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.; Li, W.; Poteet, E.C.; Xie, L.; Tan, C.; Yan, L.J.; Ju, X.; Liu, R.; Qian, H.; Marvin, M.A.; et al. Alternative mitochondrial electron transfer as a novel strategy for neuroprotection. J. Biol. Chem. 2011, 286, 16504–16515. [Google Scholar] [CrossRef] [PubMed]
- Poteet, E.; Winters, A.; Yan, L.J.; Shufelt, K.; Green, K.N.; Simpkins, J.W.; Wen, Y.; Yang, S.H. Neuroprotective actions of methylene blue and its derivatives. PLoS ONE 2012, 7, e48279. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Du, F.; Shih, Y.Y.; Shen, Q.; Gonzalez-Lima, F.; Duong, T.Q. Methylene blue potentiates stimulus-evoked fMRI responses and cerebral oxygen consumption during normoxia and hypoxia. Neuroimage 2013, 72, 237–242. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, T.; Imai, H. Hydrogen peroxide produced by superoxide dismutase SOD-2 activates sperm in Caenorhabditis elegans. J. Biol. Chem. 2017, 292, 14804–14813. [Google Scholar] [CrossRef] [PubMed]
- Van Raamsdonk, J.M.; Hekimi, S. Superoxide dismutase is dispensable for normal animal lifespan. Proc. Natl. Acad. Sci. USA 2012, 109, 5785–5790. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Yoshioka, H.; Kim, G.S.; Jung, J.E.; Okami, N.; Sakata, H.; Maier, C.M.; Narasimhan, P.; Goeders, C.E.; Chan, P.H. Oxidative stress in ischemic brain damage: Mechanisms of cell death and potential molecular targets for neuroprotection. Antioxid. Redox Signal. 2011, 14, 1505–1517. [Google Scholar] [CrossRef] [PubMed]
- Coucha, M.; Li, W.; Hafez, S.; Abdelsaid, M.; Johnson, M.H.; Fagan, S.C.; Ergul, A. SOD1 overexpression prevents acute hyperglycemia-induced cerebral myogenic dysfunction: Relevance to contralateral hemisphere and stroke outcomes. Am. J. Physiol. Heart Circ. Physiol. 2015, 308, H456–H466. [Google Scholar] [CrossRef] [PubMed]
- Muscoli, C.; Cuzzocrea, S.; Riley, D.P.; Zweier, J.L.; Thiemermann, C.; Wang, Z.Q.; Salvemini, D. On the selectivity of superoxide dismutase mimetics and its importance in pharmacological studies. Br. J. Pharmacol. 2003, 140, 445–460. [Google Scholar] [CrossRef] [PubMed]
- Batinic-Haberle, I.; Reboucas, J.S.; Spasojevic, I. Superoxide dismutase mimics: Chemistry, pharmacology, and therapeutic potential. Antioxid. Redox Signal. 2010, 13, 877–918. [Google Scholar] [CrossRef] [PubMed]
- Kelso, G.F.; Maroz, A.; Cocheme, H.M.; Logan, A.; Prime, T.A.; Peskin, A.V.; Winterbourn, C.C.; James, A.M.; Ross, M.F.; Brooker, S.; et al. A mitochondria-targeted macrocyclic Mn(II) superoxide dismutase mimetic. Chem. Biol. 2012, 19, 1237–1246. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.F.; Guo, F.; Cao, Y.Z.; Shi, W.; Xia, Q. Neuroprotection by manganese superoxide dismutase (MnSOD) mimics: Antioxidant effect and oxidative stress regulation in acute experimental stroke. CNS Neurosci. Ther. 2012, 18, 811–818. [Google Scholar] [CrossRef] [PubMed]
- Hirschberg, K.; Radovits, T.; Korkmaz, S.; Loganathan, S.; Zollner, S.; Seidel, B.; Pali, S.; Barnucz, E.; Merkely, B.; Karck, M.; et al. Combined superoxide dismutase mimetic and peroxynitrite scavenger protects against neointima formation after endarterectomy in association with decreased proliferation and nitro-oxidative stress. Eur. J. Vasc. Endovasc. Surg. 2010, 40, 168–175. [Google Scholar] [CrossRef] [PubMed]
- Szabo, A.; Balog, M.; Mark, L.; Montsko, G.; Turi, Z.; Gallyas, F., Jr.; Sumegi, B.; Kalai, T.; Hideg, K.; Kovacs, K. Induction of mitochondrial destabilization and necrotic cell death by apolar mitochondria-directed SOD mimetics. Mitochondrion 2011, 11, 476–487. [Google Scholar] [CrossRef] [PubMed]
- Sheu, S.S.; Nauduri, D.; Anders, M.W. Targeting antioxidants to mitochondria: A new therapeutic direction. Biochim. Biophys. Acta 2006, 1762, 256–265. [Google Scholar] [CrossRef] [PubMed]
- Hu, Q.; Ren, J.; Li, G.; Wu, J.; Wu, X.; Wang, G.; Gu, G.; Ren, H.; Hong, Z.; Li, J. The mitochondrially targeted antioxidant MitoQ protects the intestinal barrier by ameliorating mitochondrial DNA damage via the Nrf2/ARE signaling pathway. Cell Death Dis. 2018, 9, 403. [Google Scholar] [CrossRef] [PubMed]
- Graham, D.; Huynh, N.N.; Hamilton, C.A.; Beattie, E.; Smith, R.A.; Cocheme, H.M.; Murphy, M.P.; Dominiczak, A.F. Mitochondria-targeted antioxidant MitoQ10 improves endothelial function and attenuates cardiac hypertrophy. Hypertension 2009, 54, 322–328. [Google Scholar] [CrossRef] [PubMed]
- Skulachev, V.P.; Anisimov, V.N.; Antonenko, Y.N.; Bakeeva, L.E.; Chernyak, B.V.; Erichev, V.P.; Filenko, O.F.; Kalinina, N.I.; Kapelko, V.I.; Kolosova, N.G.; et al. An attempt to prevent senescence: A mitochondrial approach. Biochim. Biophys. Acta 2009, 1787, 437–461. [Google Scholar] [CrossRef] [PubMed]
- Ojano-Dirain, C.P.; Antonelli, P.J.; Le Prell, C.G. Mitochondria-targeted antioxidant MitoQ reduces gentamicin-induced ototoxicity. Otol. Neurotol. 2014, 35, 533–539. [Google Scholar] [CrossRef] [PubMed]
- Gane, E.J.; Weilert, F.; Orr, D.W.; Keogh, G.F.; Gibson, M.; Lockhart, M.M.; Frampton, C.M.; Taylor, K.M.; Smith, R.A.; Murphy, M.P. The mitochondria-targeted anti-oxidant mitoquinone decreases liver damage in a phase II study of hepatitis C patients. Liver Int. 2010, 30, 1019–1026. [Google Scholar] [CrossRef] [PubMed]
- Snow, B.J.; Rolfe, F.L.; Lockhart, M.M.; Frampton, C.M.; O’Sullivan, J.D.; Fung, V.; Smith, R.A.; Murphy, M.P.; Taylor, K.M. ProtecT Study Group. A double-blind, placebo-controlled study to assess the mitochondria-targeted antioxidant MitoQ as a disease-modifying therapy in Parkinson’s disease. Mov. Disord. 2010, 25, 1670–1674. [Google Scholar] [CrossRef] [PubMed]
- Oyewole, A.O.; Birch-Machin, M.A. Mitochondria-targeted antioxidants. FASEB J. 2015, 29, 4766–4771. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Hu, X.H.; Jia, Z.G.; Xu, M.H.; Guo, Z.Y.; Gao, F.H. Tiron protects against UVB-induced senescence-like characteristics in human dermal fibroblasts by the inhibition of superoxide anion production and glutathione depletion. Australas. J. Dermatol. 2012, 53, 172–180. [Google Scholar] [CrossRef] [PubMed]
- Mao, G.; Kraus, G.A.; Kim, I.; Spurlock, M.E.; Bailey, T.B.; Zhang, Q.; Beitz, D.C. A mitochondria-targeted vitamin E derivative decreases hepatic oxidative stress and inhibits fat deposition in mice. J. Nutr. 2010, 140, 1425–1431. [Google Scholar] [CrossRef] [PubMed]
- Filipovska, A.; Kelso, G.F.; Brown, S.E.; Beer, S.M.; Smith, R.A.; Murphy, M.P. Synthesis and characterization of a triphenylphosphonium-conjugated peroxidase mimetic. Insights into the interaction of ebselen with mitochondria. J. Biol. Chem. 2005, 280, 24113–24126. [Google Scholar] [CrossRef] [PubMed]
- Mailloux, R.J. Application of mitochondria-targeted pharmaceuticals for the treatment of heart disease. Curr. Pharm. Des. 2016, 22, 4763–4779. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.; Manczak, M.; Reddy, P.H. Mitochondria-targeted molecules MitoQ and SS31 reduce mutant huntingtin-induced mitochondrial toxicity and synaptic damage in Huntington’s disease. Hum. Mol. Genet. 2016, 25, 1739–1753. [Google Scholar] [CrossRef] [PubMed]
- Manczak, M.; Mao, P.; Calkins, M.J.; Cornea, A.; Reddy, A.P.; Murphy, M.P.; Szeto, H.H.; Park, B.; Reddy, P.H. Mitochondria-targeted antioxidants protect against amyloid-beta toxicity in Alzheimer’s disease neurons. J. Alzheimers Dis. 2010, 20 (Suppl. 2), S609–S631. [Google Scholar] [CrossRef] [PubMed]
- Bath, P.M.; Gray, L.J.; Bath, A.J.; Buchan, A.; Miyata, T.; Green, A.R. Effects of NXY-059 in experimental stroke: An individual animal meta-analysis. Br. J. Pharmacol. 2009, 157, 1157–1171. [Google Scholar] [CrossRef] [PubMed]
- Shuaib, A.; Lees, K.R.; Lyden, P.; Grotta, J.; Davalos, A.; Davis, S.M.; Diener, H.C.; Ashwood, T.; Wasiewski, W.W.; Emeribe, U.; et al. NXY-059 for the treatment of acute ischemic stroke. N. Engl. J. Med. 2007, 357, 562–571. [Google Scholar] [CrossRef] [PubMed]
- Diener, H.C.; Lees, K.R.; Lyden, P.; Grotta, J.; Davalos, A.; Davis, S.M.; Shuaib, A.; Ashwood, T.; Wasiewski, W.; Alderfer, V.; et al. NXY-059 for the treatment of acute stroke: Pooled analysis of the SAINT I and II trials. Stroke 2008, 39, 1751–1758. [Google Scholar] [CrossRef] [PubMed]
- Ley, J.J.; Vigdorchik, A.; Belayev, L.; Zhao, W.; Busto, R.; Khoutorova, L.; Becker, D.A.; Ginsberg, M.D. Stilbazulenyl nitrone, a second-generation azulenyl nitrone antioxidant, confers enduring neuroprotection in experimental focal cerebral ischemia in the rat: Neurobehavior, histopathology, and pharmacokinetics. J. Pharmacol. Exp. Ther. 2005, 313, 1090–1100. [Google Scholar] [CrossRef] [PubMed]
- Becker, D.A.; Ley, J.J.; Echegoyen, L.; Alvarado, R. Stilbazulenyl nitrone (STAZN): A nitronyl-substituted hydrocarbon with the potency of classical phenolic chain-breaking antioxidants. J. Am. Chem. Soc. 2002, 124, 4678–4684. [Google Scholar] [CrossRef] [PubMed]
- Steiner, J.L.; Murphy, E.A.; McClellan, J.L.; Carmichael, M.D.; Davis, J.M. Exercise training increases mitochondrial biogenesis in the brain. J. Appl. Physiol. 2011, 111, 1066–1071. [Google Scholar] [CrossRef] [PubMed]
- Vincent, G.; Lamon, S.; Gant, N.; Vincent, P.J.; MacDonald, J.R.; Markworth, J.F.; Edge, J.A.; Hickey, A.J. Changes in mitochondrial function and mitochondria associated protein expression in response to 2-weeks of high intensity interval training. Front. Physiol. 2015, 6, 51. [Google Scholar] [CrossRef] [PubMed]
- Richter, E.A.; Ruderman, N.B. AMPK and the biochemistry of exercise: Implications for human health and disease. Biochem. J. 2009, 418, 261–275. [Google Scholar] [CrossRef] [PubMed]
- Jager, S.; Handschin, C.; St-Pierre, J.; Spiegelman, B.M. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1alpha. Proc. Natl. Acad. Sci. USA 2007, 104, 12017–12022. [Google Scholar] [CrossRef] [PubMed]
- Lumini, J.A.; Magalhaes, J.; Oliveira, P.J.; Ascensao, A. Beneficial effects of exercise on muscle mitochondrial function in diabetes mellitus. Sports Med. 2008, 38, 735–750. [Google Scholar] [CrossRef] [PubMed]
- Huertas, J.R.; Al Fazazi, S.; Hidalgo-Gutierrez, A.; Lopez, L.C.; Casuso, R.A. Antioxidant effect of exercise: Exploring the role of the mitochondrial complex I superassembly. Redox Biol. 2017, 13, 477–481. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Triolo, M.; Hood, D.A. Impact of aging and exercise on mitochondrial quality control in skeletal muscle. Oxid. Med. Cell. Longev. 2017, 2017, 3165396. [Google Scholar] [CrossRef] [PubMed]
- Redman, L.M.; Ravussin, E. Caloric restriction in humans: Impact on physiological, psychological, and behavioral outcomes. Antioxid. Redox Signal. 2011, 14, 275–287. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Lluch, G.; Hunt, N.; Jones, B.; Zhu, M.; Jamieson, H.; Hilmer, S.; Cascajo, M.V.; Allard, J.; Ingram, D.K.; Navas, P.; et al. Calorie restriction induces mitochondrial biogenesis and bioenergetic efficiency. Proc. Natl. Acad. Sci. USA 2006, 103, 1768–1773. [Google Scholar] [CrossRef] [PubMed]
- Canto, C.; Auwerx, J. Caloric restriction, SIRT1 and longevity. Trends Endocrinol. Metab. 2009, 20, 325–331. [Google Scholar] [CrossRef] [PubMed]
- Tang, B.L. SIRT1 and the mitochondria. Mol. Cells 2016, 39, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Hayakawa, K.; Esposito, E.; Wang, X.; Terasaki, Y.; Liu, Y.; Xing, C.; Ji, X.; Lo, E.H. Transfer of mitochondria from astrocytes to neurons after stroke. Nature 2016, 535, 551–555. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Xu, K.; Nguyen, H.; Guedes, V.A.; Borlongan, C.V.; Acosta, S.A. Stem cell-induced biobridges as possible tools to aid neuroreconstruction after CNS injury. Front. Cell Dev. Biol. 2017, 5, 51. [Google Scholar] [CrossRef] [PubMed]
- Hayakawa, K.; Chan, S.J.; Mandeville, E.T.; Park, J.H.; Bruzzese, M.; Montaner, J.; Arai, K.; Rosell, A.; Lo, E.H. Protective effects of endothelial progenitor cell-derived extracellular mitochondria in brain endothelium. Stem Cells 2018. [Google Scholar] [CrossRef] [PubMed]
- Chou, S.H.; Lan, J.; Esposito, E.; Ning, M.; Balaj, L.; Ji, X.; Lo, E.H.; Hayakawa, K. Extracellular mitochondria in cerebrospinal fluid and neurological recovery after subarachnoid hemorrhage. Stroke 2017, 48, 2231–2237. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.Y.; Liou, C.W.; Chen, S.D.; Hsu, T.Y.; Chuang, J.H.; Wang, P.W.; Huang, S.T.; Tiao, M.M.; Chen, J.B.; Lin, T.K.; et al. Mitochondrial transfer from Wharton’s jelly-derived mesenchymal stem cells to mitochondria-defective cells recaptures impaired mitochondrial function. Mitochondrion 2015, 22, 31–44. [Google Scholar] [CrossRef] [PubMed]
- Acquistapace, A.; Bru, T.; Lesault, P.F.; Figeac, F.; Coudert, A.E.; le Coz, O.; Christov, C.; Baudin, X.; Auber, F.; Yiou, R.; et al. Human mesenchymal stem cells reprogram adult cardiomyocytes toward a progenitor-like state through partial cell fusion and mitochondria transfer. Stem Cells 2011, 29, 812–824. [Google Scholar] [CrossRef] [PubMed]
- Rogers, R.S.; Bhattacharya, J. When cells become organelle donors. Physiology (Bethesda) 2013, 28, 414–422. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.M.; Kim, J.H.; Kim, M.; Park, S.J.; Koh, S.H.; Ahn, H.S.; Kang, G.H.; Lee, J.B.; Park, K.S.; Lee, H.K. Mesenchymal stem cells transfer mitochondria to the cells with virtually no mitochondrial function but not with pathogenic mtDNA mutations. PLoS ONE 2012, 7, e32778. [Google Scholar] [CrossRef] [PubMed]
- Spees, J.L.; Olson, S.D.; Whitney, M.J.; Prockop, D.J. Mitochondrial transfer between cells can rescue aerobic respiration. Proc. Natl. Acad. Sci. USA 2006, 103, 1283–1288. [Google Scholar] [CrossRef] [PubMed]
- Torralba, D.; Baixauli, F.; Sanchez-Madrid, F. Mitochondria know no boundaries: Mechanisms and functions of intercellular mitochondrial transfer. Front. Cell Dev. Biol. 2016, 4, 107. [Google Scholar] [CrossRef] [PubMed]
- Berridge, M.V.; McConnell, M.J.; Grasso, C.; Bajzikova, M.; Kovarova, J.; Neuzil, J. Horizontal transfer of mitochondria between mammalian cells: Beyond co-culture approaches. Curr. Opin. Genet. Dev. 2016, 38, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Ji, K.; Guo, L.; Wu, W.; Lu, H.; Shan, P.; Yan, C. Mesenchymal stem cells rescue injured endothelial cells in an in vitro ischemia-reperfusion model via tunneling nanotube like structure-mediated mitochondrial transfer. Microvasc. Res. 2014, 92, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Han, H.; Hu, J.; Yan, Q.; Zhu, J.; Zhu, Z.; Chen, Y.; Sun, J.; Zhang, R. Bone marrow-derived mesenchymal stem cells rescue injured H9c2 cells via transferring intact mitochondria through tunneling nanotubes in an in vitro simulated ischemia/reperfusion model. Mol. Med. Rep. 2016, 13, 1517–1524. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhang, Y.; Yeung, S.C.; Liang, Y.; Liang, X.; Ding, Y.; Ip, M.S.; Tse, H.F.; Mak, J.C.; Lian, Q. Mitochondrial transfer of induced pluripotent stem cell-derived mesenchymal stem cells to airway epithelial cells attenuates cigarette smoke-induced damage. Am. J. Respir. Cell Mol. Biol. 2014, 51, 455–465. [Google Scholar] [CrossRef] [PubMed]
- Plotnikov, E.Y.; Khryapenkova, T.G.; Vasileva, A.K.; Marey, M.V.; Galkina, S.I.; Isaev, N.K.; Sheval, E.V.; Polyakov, V.Y.; Sukhikh, G.T.; Zorov, D.B. Cell-to-cell cross-talk between mesenchymal stem cells and cardiomyocytes in co-culture. J. Cell. Mol. Med. 2008, 12, 1622–1631. [Google Scholar] [CrossRef] [PubMed]
- Mahrouf-Yorgov, M.; Augeul, L.; Da Silva, C.C.; Jourdan, M.; Rigolet, M.; Manin, S.; Ferrera, R.; Ovize, M.; Henry, A.; Guguin, A.; et al. Mesenchymal stem cells sense mitochondria released from damaged cells as danger signals to activate their rescue properties. Cell Death Differ. 2017, 24, 1224–1238. [Google Scholar] [CrossRef] [PubMed]
- Hayakawa, K.; Bruzzese, M.; Chou, S.H.; Ning, M.; Ji, X.; Lo, E.H. Extracellular mitochondria for therapy and diagnosis in acute central nervous system injury. JAMA Neurol. 2018, 75, 119–122. [Google Scholar] [CrossRef] [PubMed]
- Maki, T.; Morancho, A.; Martinez-San Segundo, P.; Hayakawa, K.; Takase, H.; Liang, A.C.; Gabriel-Salazar, M.; Medina-Gutierrez, E.; Washida, K.; Montaner, J.; et al. Endothelial progenitor cell secretome and oligovascular repair in a mouse model of prolonged cerebral hypoperfusion. Stroke 2018, 49, 1003–1010. [Google Scholar] [CrossRef] [PubMed]
- Islam, M.N.; Das, S.R.; Emin, M.T.; Wei, M.; Sun, L.; Westphalen, K.; Rowlands, D.J.; Quadri, S.K.; Bhattacharya, S.; Bhattacharya, J. Mitochondrial transfer from bone-marrow-derived stromal cells to pulmonary alveoli protects against acute lung injury. Nat. Med. 2012, 18, 759–765. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, T.; Mukherjee, S.; Pattnaik, B.; Kumar, M.; Singh, S.; Kumar, M.; Rehman, R.; Tiwari, B.K.; Jha, K.A.; Barhanpurkar, A.P.; et al. Miro1 regulates intercellular mitochondrial transport & enhances mesenchymal stem cell rescue efficacy. EMBO J. 2014, 33, 994–1010. [Google Scholar] [CrossRef] [PubMed]
- Moschoi, R.; Imbert, V.; Nebout, M.; Chiche, J.; Mary, D.; Prebet, T.; Saland, E.; Castellano, R.; Pouyet, L.; Collette, Y.; et al. Protective mitochondrial transfer from bone marrow stromal cells to acute myeloid leukemic cells during chemotherapy. Blood 2016, 128, 253–264. [Google Scholar] [CrossRef] [PubMed]
- Bukoreshtliev, N.V.; Wang, X.; Hodneland, E.; Gurke, S.; Barroso, J.F.; Gerdes, H.H. Selective block of tunneling nanotube (TNT) formation inhibits intercellular organelle transfer between PC12 cells. FEBS Lett. 2009, 583, 1481–1488. [Google Scholar] [CrossRef] [PubMed]
- Rustom, A.; Saffrich, R.; Markovic, I.; Walther, P.; Gerdes, H.H. Nanotubular highways for intercellular organelle transport. Science 2004, 303, 1007–1010. [Google Scholar] [CrossRef] [PubMed]
- He, K.; Shi, X.; Zhang, X.; Dang, S.; Ma, X.; Liu, F.; Xu, M.; Lv, Z.; Han, D.; Fang, X.; et al. Long-distance intercellular connectivity between cardiomyocytes and cardiofibroblasts mediated by membrane nanotubes. Cardiovasc. Res. 2011, 92, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Wang, Y.; Zhang, J.; Tu, J.; Wang, X.J.; Su, X.D.; Wang, L.; Zhang, Y. Tunneling-nanotube direction determination in neurons and astrocytes. Cell Death Dis. 2012, 3, e438. [Google Scholar] [CrossRef] [PubMed]
- Lou, E.; Fujisawa, S.; Morozov, A.; Barlas, A.; Romin, Y.; Dogan, Y.; Gholami, S.; Moreira, A.L.; Manova-Todorova, K.; Moore, M.A. Tunneling nanotubes provide a unique conduit for intercellular transfer of cellular contents in human malignant pleural mesothelioma. PLoS ONE 2012, 7, e33093. [Google Scholar] [CrossRef] [PubMed]
- Mittelbrunn, M.; Sanchez-Madrid, F. Intercellular communication: Diverse structures for exchange of genetic information. Nat. Rev. Mol. Cell Biol. 2012, 13, 328–335. [Google Scholar] [CrossRef] [PubMed]
- Pitt, J.M.; Kroemer, G.; Zitvogel, L. Extracellular vesicles: Masters of intercellular communication and potential clinical interventions. J. Clin. Investig. 2016, 126, 1139–1143. [Google Scholar] [CrossRef] [PubMed]
- Spees, J.L.; Olson, S.D.; Ylostalo, J.; Lynch, P.J.; Smith, J.; Perry, A.; Peister, A.; Wang, M.Y.; Prockop, D.J. Differentiation, cell fusion, and nuclear fusion during ex vivo repair of epithelium by human adult stem cells from bone marrow stroma. Proc. Natl. Acad. Sci. USA 2003, 100, 2397–2402. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Dolado, M.; Pardal, R.; Garcia-Verdugo, J.M.; Fike, J.R.; Lee, H.O.; Pfeffer, K.; Lois, C.; Morrison, S.J.; Alvarez-Buylla, A. Fusion of bone-marrow-derived cells with Purkinje neurons, cardiomyocytes and hepatocytes. Nature 2003, 425, 968–973. [Google Scholar] [CrossRef] [PubMed]
- Oh, H.; Bradfute, S.B.; Gallardo, T.D.; Nakamura, T.; Gaussin, V.; Mishina, Y.; Pocius, J.; Michael, L.H.; Behringer, R.R.; Garry, D.J.; et al. Cardiac progenitor cells from adult myocardium: Homing, differentiation, and fusion after infarction. Proc. Natl. Acad. Sci. USA 2003, 100, 12313–12318. [Google Scholar] [CrossRef] [PubMed]
- Vassilopoulos, G.; Wang, P.R.; Russell, D.W. Transplanted bone marrow regenerates liver by cell fusion. Nature 2003, 422, 901–904. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Willenbring, H.; Akkari, Y.; Torimaru, Y.; Foster, M.; Al-Dhalimy, M.; Lagasse, E.; Finegold, M.; Olson, S.; Grompe, M. Cell fusion is the principal source of bone-marrow-derived hepatocytes. Nature 2003, 422, 897–901. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, A.; Kurihara, H.; Yagita, H.; Okumura, K.; Nakano, H. Mitochondrial extrusion through the cytoplasmic vacuoles during cell death. J. Biol. Chem. 2008, 283, 24128–24135. [Google Scholar] [CrossRef] [PubMed]
- Caielli, S.; Athale, S.; Domic, B.; Murat, E.; Chandra, M.; Banchereau, R.; Baisch, J.; Phelps, K.; Clayton, S.; Gong, M.; et al. Oxidized mitochondrial nucleoids released by neutrophils drive type I interferon production in human lupus. J. Exp. Med. 2016, 213, 697–713. [Google Scholar] [CrossRef] [PubMed]
- Napoli, E.; Lippert, T.; Borlongan, C.V. Stem cell therapy: Repurposing cell-based regenerative medicine beyond cell replacement. Adv. Exp. Med. Biol. 2018. [Google Scholar] [CrossRef]
- Babenko, V.A.; Silachev, D.N.; Popkov, V.A.; Zorova, L.D.; Pevzner, I.B.; Plotnikov, E.Y.; Sukhikh, G.T.; Zorov, D.B. Miro1 enhances mitochondria transfer from multipotent mesenchymal stem cells (MMSC) to neural cells and improves the efficacy of cell recovery. Molecules 2018, 23, 687. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Topic | Key Points |
|---|---|
| Therapeutic options for stroke | Few treatments for stroke exist, which include intravenous thrombolysis and endovascular thrombectomy [1]. Small molecules such as Stachybotrys microspora triprenyl phenol-7 and NSI-189 show promise for treating stroke [3,4]. |
| Mitochondria and stroke | Mitochondria may generate reactive oxygen species that may contribute to diseases such as myocardial infarction and inflammatory conditions [5,9]. Dysfunctional mitochondrial energy generation may lead to Anderson-Fabry disease, which may cause an ischemic stroke [18,19,20]. |
| Mitochondria, ETC, and OXPHOS | The electron transport chain and oxidative phosphorylation processes that occur within the mitochondria are crucial for cellular energy, and thus require optimal function [30,31,32,33]. Defects in the various electron transport chain complex enzymes that facilitate oxidative phosphorylation may lead to different disease pathologies [36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57]. Altered mitochondrial conditions, such as cytochrome c release, electron transport modifications, and changed cellular redox states, may cause downstream pathways to initiate cell death [61,62,63,64,65,66,67,68,69,70,71,72,73,74,75,76,77]. |
| Mitochondria-based regenerative medicine | Mitochondria are a promising therapeutic target for treating stroke, neurodegenerative diseases, aging, and other metabolic disorders. Sirtuin 1, mitochondrial fission and fusion modulators, purinergic agonists, methylene blue, superoxide dismutase mimetics, antioxidants, and proper diet and exercise can improve mitochondrial function and potentially treat diseases associated with mitochondrial dysfunction [78,79,80,81,82,83,84,85,86,87,88,89,90,91,92,93,94,95,96,97,98,99,100,101,102,103,104,105,106,107,108,109,110,111,112,113,114,115,116,117,118,119,120,121,122,123,124,125,126,127,128,129,130,131,132,133,134,135,136,137,138,139,140,141,142,143]. |
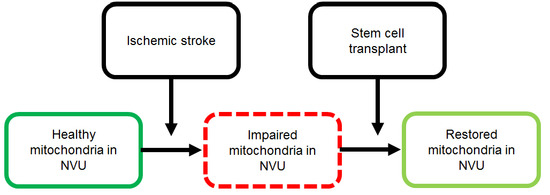
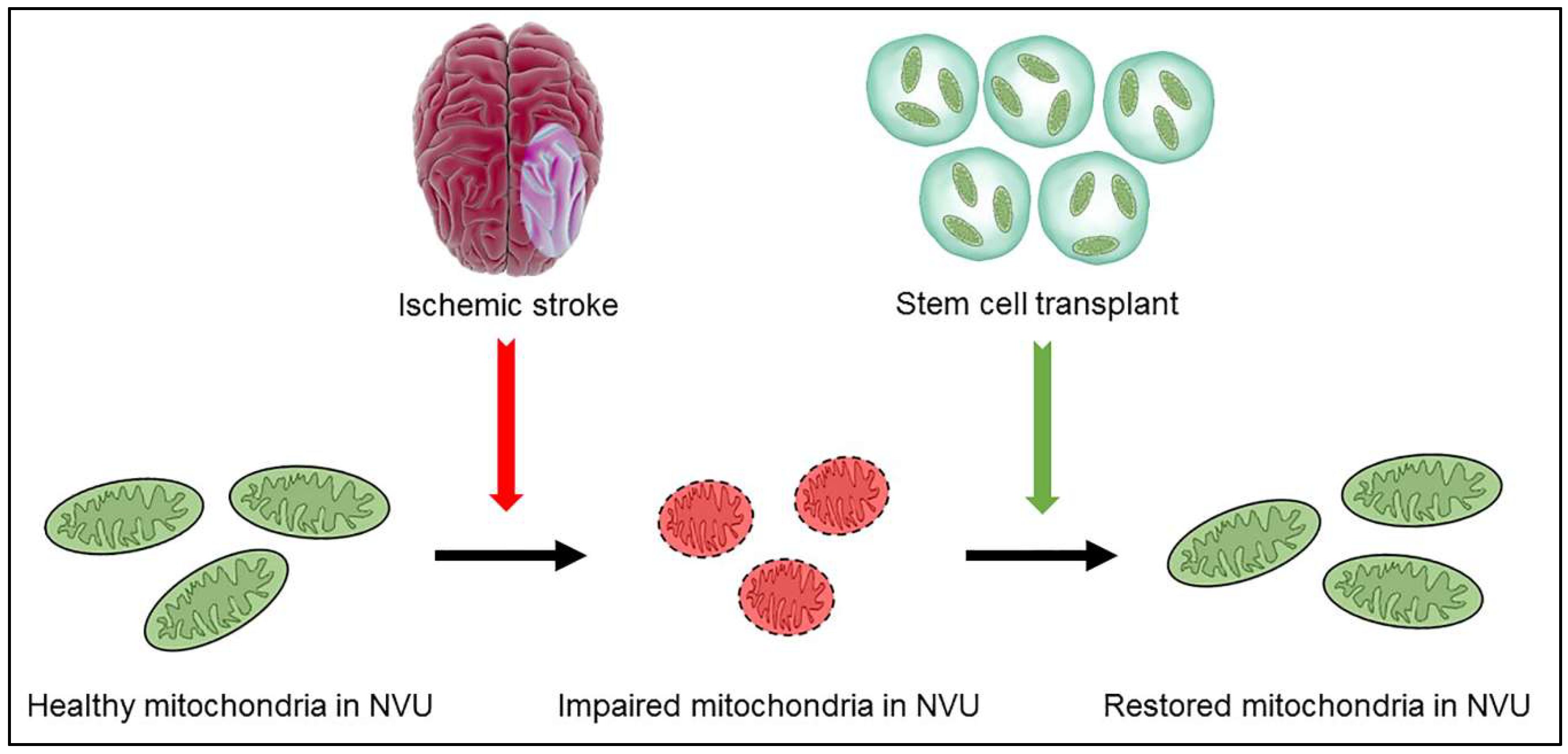
| Stem cells as source of healthy mitochondria | Stem cells may be able to transfer healthy mitochondria to ischemic neurons with impaired mitochondria, restoring mitochondrial function in ischemic neurons and rescuing dying neurons after ischemic stroke [146,147,148,149]. Mesenchymal stem cells have successfully transferred healthy mitochondria to various types of impaired cells and repaired cellular damage [155,156,157,158,159]. Mitochondrial transfer may be facilitated by recognition of injured cells and may occur via tunneling nanotubes, extracellular vesicles, or cell fusion [151,152,153,160,161,162,163,164,165,166,167,168,169,170,171,172,173,174,175,176,177,178]. |
| Stem cells, mitochondria, and stroke | Stem cell transfer of viable mitochondria to ischemic cells may be a possible method for treating ischemic stroke. Mitochondrial transfer restores the bioenergetics of the receiving cells and promotes their proliferation [180]. The Miro1 protein facilitates mitochondrial transfer and overexpression of Miro1 may enhance mitochondrial transfer to effectively treat stroke [180]. |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nguyen, H.; Zarriello, S.; Rajani, M.; Tuazon, J.; Napoli, E.; Borlongan, C.V. Understanding the Role of Dysfunctional and Healthy Mitochondria in Stroke Pathology and Its Treatment. Int. J. Mol. Sci. 2018, 19, 2127. https://doi.org/10.3390/ijms19072127
Nguyen H, Zarriello S, Rajani M, Tuazon J, Napoli E, Borlongan CV. Understanding the Role of Dysfunctional and Healthy Mitochondria in Stroke Pathology and Its Treatment. International Journal of Molecular Sciences. 2018; 19(7):2127. https://doi.org/10.3390/ijms19072127
Chicago/Turabian StyleNguyen, Hung, Sydney Zarriello, Mira Rajani, Julian Tuazon, Eleonora Napoli, and Cesar V. Borlongan. 2018. "Understanding the Role of Dysfunctional and Healthy Mitochondria in Stroke Pathology and Its Treatment" International Journal of Molecular Sciences 19, no. 7: 2127. https://doi.org/10.3390/ijms19072127
APA StyleNguyen, H., Zarriello, S., Rajani, M., Tuazon, J., Napoli, E., & Borlongan, C. V. (2018). Understanding the Role of Dysfunctional and Healthy Mitochondria in Stroke Pathology and Its Treatment. International Journal of Molecular Sciences, 19(7), 2127. https://doi.org/10.3390/ijms19072127