Expanding the Mutation Spectrum in ABCA4: Sixty Novel Disease Causing Variants and Their Associated Phenotype in a Large French Stargardt Cohort

 , and
, and
Abstract
:1. Introduction
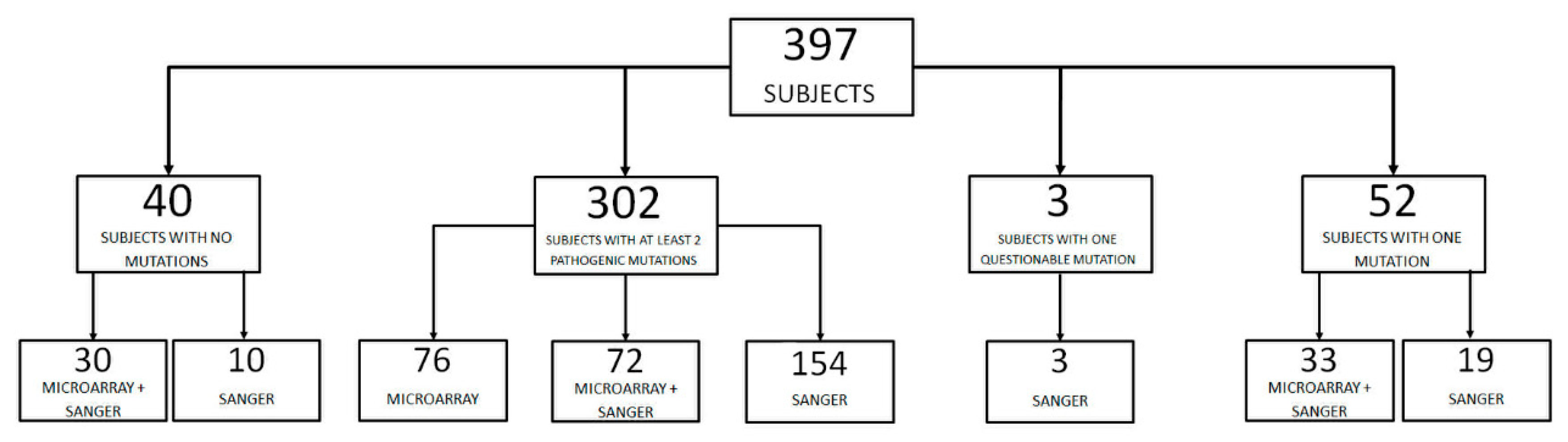
2. Results
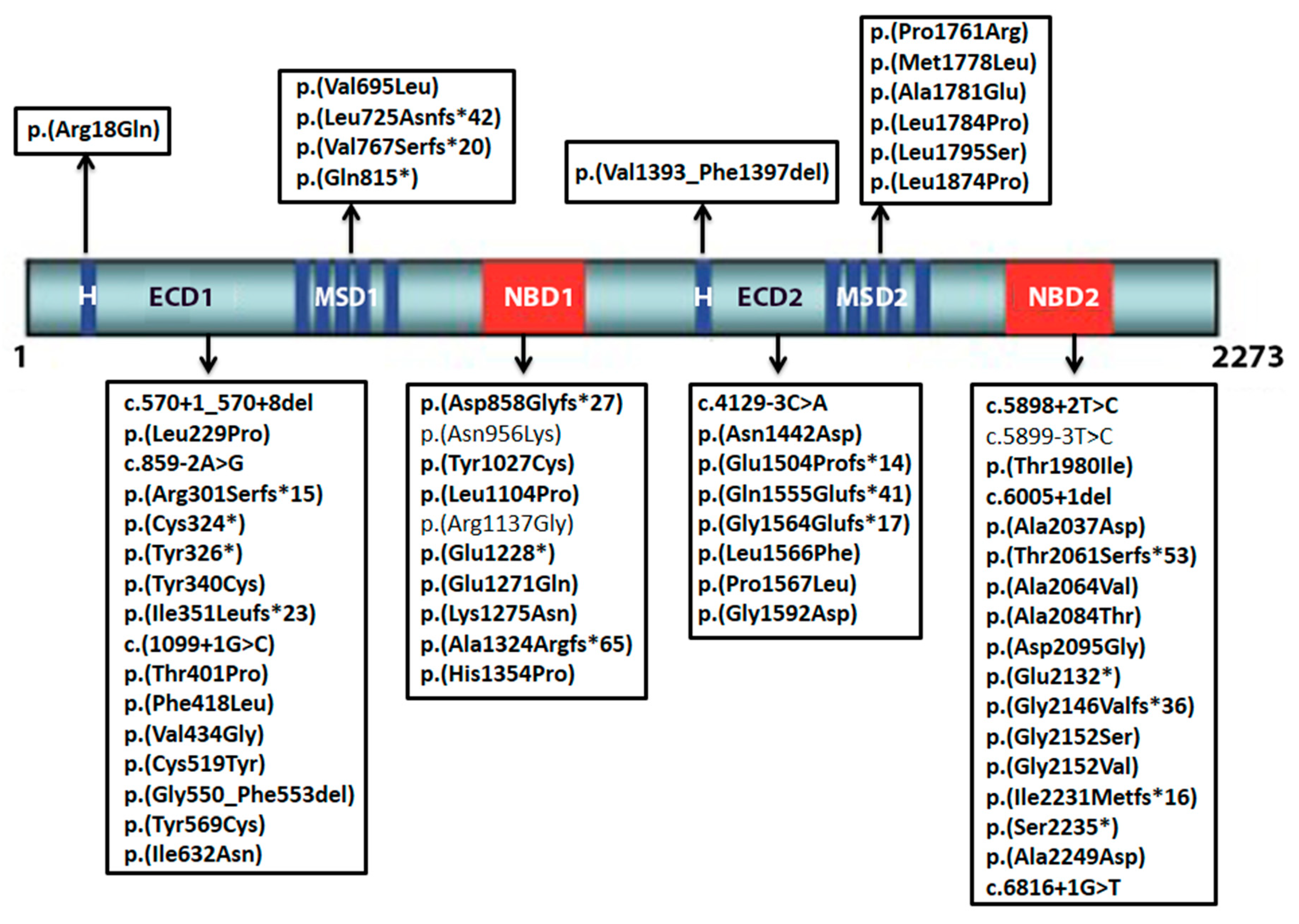
2.1. Novel Variants
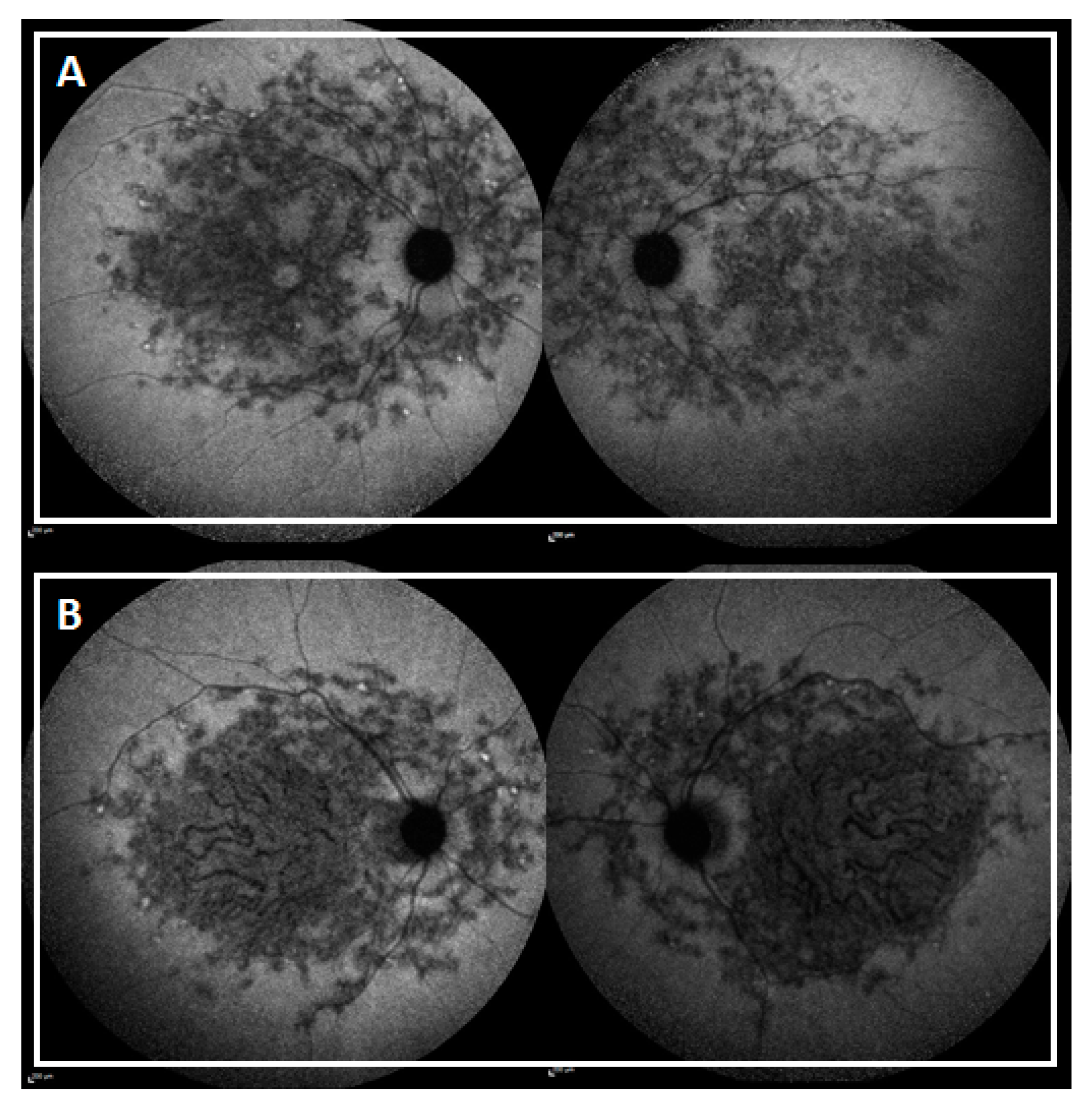
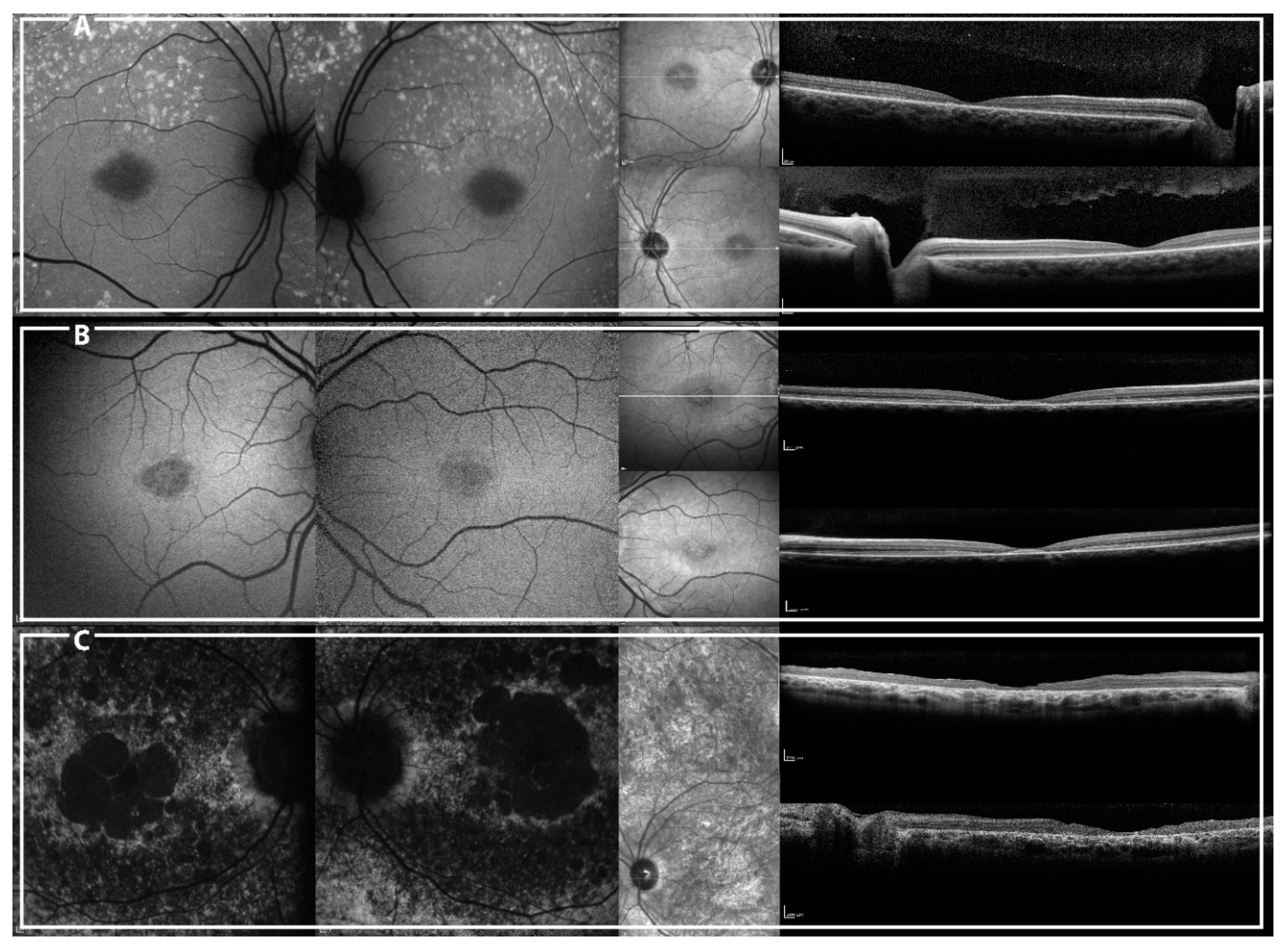
2.2. Genotype-Phenotype Correlation
3. Discussion
4. Materials and Methods
4.1. Mutation Analysis
4.2. Phenotypic Analysis
4.3. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| MDPI | Multidisciplinary Digital Publishing Institute |
| STGD-1 | Stargardt disease-1 |
| ABCA4 | ATP-binding cassette, subfamily A, member 4 |
| BCVA | Best Corrected Visual Acuity |
| ff-ERG | Full field electroretinography |
| mf-ERG | Multi focal electroretinography |
| SW-AF | Short-wavelength autofluorescence |
| NIR-AF | Near-infrared Autofluorescence |
| SD-OCT | Spectral Domain Optical Coherence Tomography |
| ISCEV | International Society for Clinical of Electrophysiology of Vision |
| ACMG | American College of Medical Genetics and Genomics |
| AMP | Association of Molecular Pathology |
| HGMD | Human Gene Mutation Database |
| ExAC | Exome Aggregation Consortium |
| PolyPhen2 | Polymorphism Phenotyping |
| UCSC | University of California Santa Cruz |
| SIFT | Sorting intolerant from tolerant |
| RPE | Retinal pigment epithelium |
| CRT | Central retinal thickness |
| TMV | Total macular volume |
| VUS | Variant of uncertain significance |
| MAF | Minor allele frequency |
| NGS | Next-generation sequencing |
| WES | Whole exome sequencing |
| WGS | Whole genome sequencing |
| EZ | Ellipsoid zone |
| FP | Fundus photograph |
| FS | Foveal sparing |
| MM | At least one missense variant in the genotype |
| NM | At least one null variant in the genotype |
References
- Molday, L.L.; Rabin, A.R.; Molday, R.S. ABCR expression in foveal cone photoreceptors and its role in Stargardt macular dystrophy. Nat. Genet. 2000, 25, 257–258. [Google Scholar] [CrossRef] [PubMed]
- Maeda, A.; Maeda, T.; Golczak, M.; Palczewski, K. Retinopathy in mice induced by disrupted all-trans-retinal clearance. J. Biol. Chem. 2008, 283, 26684–26693. [Google Scholar] [CrossRef] [PubMed]
- Maeda, T.; Maeda, A.; Matosky, M.; Okano, K.; Roos, S.; Tang, J.; Palczewski, K. Evaluation of potential therapies for a mouse model of human age-related macular degeneration caused by delayed all-trans-retinal clearance. Investig. Ophthalmol. Vis. Sci. 2009, 50, 4917–4925. [Google Scholar] [CrossRef] [PubMed]
- Koenekoop, R.K. The gene for Stargardt disease, ABCA4, is a major retinal gene: A mini-review. Ophthalmic Genet. 2003, 24, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Maia-Lopes, S.; Aguirre-Lamban, J.; Castelo-Branco, M.; Riveiro-Alvarez, R.; Ayuso, C.; Silva, E.D. ABCA4 mutations in Portuguese Stargardt patients: Identification of new mutations and their phenotypic analysis. Mol. Vis. 2009, 15, 584–591. [Google Scholar] [PubMed]
- Lewis, R.A.; Shroyer, N.F.; Singh, N.; Allikmets, R.; Hutchinson, A.; Li, Y.; Lupski, J.R.; Leppert, M.; Dean, M. Genotype/Phenotype analysis of a photoreceptor-specific ATP-binding cassette transporter gene, ABCR, in Stargardt disease. Am. J. Hum. Genet. 1999, 64, 422–434. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Mir, A.; Paloma, E.; Allikmets, R.; Ayuso, C.; del Rio, T.; Dean, M.; Vilageliu, L.; Gonzàlez-Duarte, R.; Balcells, S. Retinitis pigmentosa caused by a homozygous mutation in the Stargardt disease gene ABCR. Nat. Genet. 1998, 18, 11–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, X.; Yuan, L.; Xu, H.; Zheng, W.; Cao, Y.; Yi, J.; Guo, Y.; Yang, Z.; Li, Y.; Deng, H. Identification of a Novel Mutation in the ABCA4 Gene in a Chinese Family with Retinitis Pigmentosa Using Exome Sequencing. Biosci. Rep. 2018. [Google Scholar] [CrossRef] [PubMed]
- Audere, M.; Rutka, K.; Šepetiene, S.; Lāce, B. Presentation of Complex Homozygous Allele in ABCA4 Gene in a Patient with Retinitis Pigmentosa. Case Rep. Ophthalmol. Med. 2015, 2015, 452068. [Google Scholar] [CrossRef] [PubMed]
- Rozet, J.M.; Gerber, S.; Ghazi, I.; Perrault, I.; Ducroq, D.; Souied, E.; Cabot, A.; Dufier, J.L.; Munnich, A.; Kaplan, J. Mutations of the retinal specific ATP binding transporter gene (ABCR) in a single family segregating both autosomal recessive retinitis pigmentosa RP19 and Stargardt disease: Evidence of clinical heterogeneity at this locus. J. Med. Genet. 1999, 36, 447–451. [Google Scholar] [PubMed]
- Mullins, R.F.; Kuehn, M.H.; Radu, R.A.; Enriquez, G.S.; East, J.S.; Schindler, E.I.; Travis, G.H.; Stone, E.M. Autosomal recessive retinitis pigmentosa due to ABCA4 mutations: Clinical, pathologic, and molecular characterization. Investig. Ophthalmol. Vis. Sci. 2012, 53, 1883–1894. [Google Scholar] [CrossRef] [PubMed]
- Cornelis, S.S.; Bax, N.M.; Zernant, J.; Allikmets, R.; Fritsche, L.G.; den Dunnen, J.T.; Ajmal, M.; Hoyng, C.B.; Cremers, F.P.M. In Silico Functional Meta-Analysis of 5,962 ABCA4 Variants in 3,928 Retinal Dystrophy Cases. Hum. Mutat. 2017, 38, 400–408. [Google Scholar] [CrossRef] [PubMed]
- Allikmets, R. Simple and complex ABCR: Genetic predisposition to retinal disease. Am. J. Hum. Genet. 2000, 67, 793–799. [Google Scholar] [CrossRef] [PubMed]
- Passerini, I.; Sodi, A.; Giambene, B.; Mariottini, A.; Menchini, U.; Torricelli, F. Novel mutations in of the ABCR gene in Italian patients with Stargardt disease. Eye Lond. Engl. 2010, 24, 158–164. [Google Scholar] [CrossRef] [PubMed]
- Riveiro-Alvarez, R.; Lopez-Martinez, M.-A.; Zernant, J.; Aguirre-Lamban, J.; Cantalapiedra, D.; Avila-Fernandez, A.; Gimenez, A.; Lopez-Molina, M.-I.; Garcia-Sandoval, B.; Blanco-Kelly, F.; et al. Outcome of ABCA4 disease-associated alleles in autosomal recessive retinal dystrophies: Retrospective analysis in 420 Spanish families. Ophthalmology 2013, 120, 2332–2337. [Google Scholar] [CrossRef] [PubMed]
- Jiang, F.; Pan, Z.; Xu, K.; Tian, L.; Xie, Y.; Zhang, X.; Chen, J.; Dong, B.; Li, Y. Screening of ABCA4 Gene in a Chinese Cohort with Stargardt Disease or Cone-Rod Dystrophy with a Report on 85 Novel Mutations. Investig. Ophthalmol. Vis. Sci. 2016, 57, 145–152. [Google Scholar] [CrossRef] [PubMed]
- Bocquet, B.; Lacroux, A.; Surget, M.-O.; Baudoin, C.; Marquette, V.; Manes, G.; Hebrard, M.; Sénéchal, A.; Delettre, C.; Roux, A.-F.; et al. Relative frequencies of inherited retinal dystrophies and optic neuropathies in Southern France: Assessment of 21-year data management. Ophthalmic Epidemiol. 2013, 20, 13–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zernant, J.; Schubert, C.; Im, K.M.; Burke, T.; Brown, C.M.; Fishman, G.A.; Tsang, S.H.; Gouras, P.; Dean, M.; Allikmets, R. Analysis of the ABCA4 gene by next-generation sequencing. Investig. Ophthalmol. Vis. Sci. 2011, 52, 8479–8487. [Google Scholar] [CrossRef] [PubMed]
- Zernant, J.; Xie, Y.A.; Ayuso, C.; Riveiro-Alvarez, R.; Lopez-Martinez, M.-A.; Simonelli, F.; Testa, F.; Gorin, M.B.; Strom, S.P.; Bertelsen, M.; et al. Analysis of the ABCA4 genomic locus in Stargardt disease. Hum. Mol. Genet. 2014, 23, 6797–6806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braun, T.A.; Mullins, R.F.; Wagner, A.H.; Andorf, J.L.; Johnston, R.M.; Bakall, B.B.; Deluca, A.P.; Fishman, G.A.; Lam, B.L.; Weleber, R.G.; et al. Non-exomic and synonymous variants in ABCA4 are an important cause of Stargardt disease. Hum. Mol. Genet. 2013, 22, 5136–5145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bauwens, M.; De Zaeytijd, J.; Weisschuh, N.; Kohl, S.; Meire, F.; Dahan, K.; Depasse, F.; De Jaegere, S.; De Ravel, T.; De Rademaeker, M.; et al. An augmented ABCA4 screen targeting noncoding regions reveals a deep intronic founder variant in Belgian Stargardt patients. Hum. Mutat. 2015, 36, 39–42. [Google Scholar] [CrossRef] [PubMed]
- Bax, N.M.; Sangermano, R.; Roosing, S.; Thiadens, A.A.H.J.; Hoefsloot, L.H.; van den Born, L.I.; Phan, M.; Klevering, B.J.; Westeneng-van Haaften, C.; Braun, T.A.; et al. Heterozygous deep-intronic variants and deletions in ABCA4 in persons with retinal dystrophies and one exonic ABCA4 variant. Hum. Mutat. 2015, 36, 43–47. [Google Scholar] [CrossRef] [PubMed]
- Sangermano, R.; Khan, M.; Cornelis, S.S.; Richelle, V.; Albert, S.; Garanto, A.; Elmelik, D.; Qamar, R.; Lugtenberg, D.; van den Born, L.I.; et al. ABCA4 midigenes reveal the full splice spectrum of all reported noncanonical splice site variants in Stargardt disease. Genome Res. 2018, 28, 100–110. [Google Scholar] [CrossRef] [PubMed]
- Maugeri, A.; van Driel, M.A.; van de Pol, D.J.; Klevering, B.J.; van Haren, F.J.; Tijmes, N.; Bergen, A.A.; Rohrschneider, K.; Blankenagel, A.; Pinckers, A.J.; et al. The 2588G-->C mutation in the ABCR gene is a mild frequent founder mutation in the Western European population and allows the classification of ABCR mutations in patients with Stargardt disease. Am. J. Hum. Genet. 1999, 64, 1024–1035. [Google Scholar] [CrossRef] [PubMed]
- Allikmets, R.; Singh, N.; Sun, H.; Shroyer, N.F.; Hutchinson, A.; Chidambaram, A.; Gerrard, B.; Baird, L.; Stauffer, D.; Peiffer, A.; et al. A photoreceptor cell-specific ATP-binding transporter gene (ABCR) is mutated in recessive Stargardt macular dystrophy. Nat. Genet. 1997, 15, 236–246. [Google Scholar] [CrossRef] [PubMed]
- Rozet, J.M.; Gerber, S.; Souied, E.; Perrault, I.; Châtelin, S.; Ghazi, I.; Leowski, C.; Dufier, J.L.; Munnich, A.; Kaplan, J. Spectrum of ABCR gene mutations in autosomal recessive macular dystrophies. Eur. J. Hum. Genet. 1998, 6, 291–295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schulz, H.L.; Grassmann, F.; Kellner, U.; Spital, G.; Rüther, K.; Jägle, H.; Hufendiek, K.; Rating, P.; Huchzermeyer, C.; Baier, M.J.; et al. Mutation Spectrum of the ABCA4 Gene in 335 Stargardt Disease Patients from a Multicenter German Cohort-Impact of Selected Deep Intronic Variants and Common SNPs. Investig. Ophthalmol. Vis. Sci. 2017, 58, 394–403. [Google Scholar] [CrossRef] [PubMed]
- Salles, M.V.; Motta, F.L.; Dias da Silva, E.; Varela, P.; Costa, K.A.; Filippelli-Silva, R.; Martin, R.P.; Chiang, J.P.-W.; Pesquero, J.B.; Sallum, J.M.F. Novel Complex ABCA4 Alleles in Brazilian Patients with Stargardt Disease: Genotype-Phenotype Correlation. Investig. Ophthalmol. Vis. Sci. 2017, 58, 5723–5730. [Google Scholar] [CrossRef] [PubMed]
- Zernant, J.; Lee, W.; Collison, F.T.; Fishman, G.A.; Sergeev, Y.V.; Schuerch, K.; Sparrow, J.R.; Tsang, S.H.; Allikmets, R. Frequent hypomorphic alleles account for a significant fraction of ABCA4 disease and distinguish it from age-related macular degeneration. J. Med. Genet. 2017, 54, 404–412. [Google Scholar] [CrossRef] [PubMed]
- Burke, T.R.; Fishman, G.A.; Zernant, J.; Schubert, C.; Tsang, S.H.; Smith, R.T.; Ayyagari, R.; Koenekoop, R.K.; Umfress, A.; Ciccarelli, M.L.; et al. Retinal phenotypes in patients homozygous for the G1961E mutation in the ABCA4 gene. Investig. Ophthalmol. Vis. Sci. 2012, 53, 4458–4467. [Google Scholar] [CrossRef] [PubMed]
- Cella, W.; Greenstein, V.C.; Zernant-Rajang, J.; Smith, T.R.; Barile, G.; Allikmets, R.; Tsang, S.H. G1961E mutant allele in the Stargardt disease gene ABCA4 causes bull’s eye maculopathy. Exp. Eye Res. 2009, 89, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Campa, C.; Gallenga, C.E.; Bolletta, E.; Perri, P. The Role of Gene Therapy in the Treatment of Retinal Diseases: A Review. Curr. Gene Ther. 2017, 17, 194–213. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.J.; Liu, J.; Adelman, R.A. Novel therapeutics for Stargardt disease. Graefes Arch. Clin. Exp. Ophthalmol. 2017, 255, 1057–1062. [Google Scholar] [CrossRef] [PubMed]
- Webster, A.R.; Héon, E.; Lotery, A.J.; Vandenburgh, K.; Casavant, T.L.; Oh, K.T.; Beck, G.; Fishman, G.A.; Lam, B.L.; Levin, A.; et al. An analysis of allelic variation in the ABCA4 gene. Investig. Ophthalmol. Vis. Sci. 2001, 42, 1179–1189. [Google Scholar]
- Allikmets, R.; Shroyer, N.F.; Singh, N.; Seddon, J.M.; Lewis, R.A.; Bernstein, P.S.; Peiffer, A.; Zabriskie, N.A.; Li, Y.; Hutchinson, A.; et al. Mutation of the Stargardt disease gene (ABCR) in age-related macular degeneration. Science 1997, 277, 1805–1807. [Google Scholar] [CrossRef] [PubMed]
- Ernest, P.J.G.; Boon, C.J.F.; Klevering, B.J.; Hoefsloot, L.H.; Hoyng, C.B. Outcome of ABCA4 microarray screening in routine clinical practice. Mol. Vis. 2009, 15, 2841–2847. [Google Scholar] [PubMed]
- Jimenez-Rolando, B.; Noval, S.; Rosa-Perez, I.; Mata Diaz, E.; Del Pozo, A.; Ibañez, C.; Silla, J.C.; Montaño, V.E.F.; Martin-Arenas, R.; Vallespin, E. Next generation sequencing in the diagnosis of Stargardt’s disease. Arch. Soc. Espanola Oftalmol. 2017. [Google Scholar] [CrossRef]
- Fujinami, K.; Sergouniotis, P.I.; Davidson, A.E.; Wright, G.; Chana, R.K.; Tsunoda, K.; Tsubota, K.; Egan, C.A.; Robson, A.G.; Moore, A.T.; et al. Clinical and molecular analysis of Stargardt disease with preserved foveal structure and function. Am. J. Ophthalmol. 2013, 156, 487–501.e1. [Google Scholar] [CrossRef] [PubMed]
- Souied, E.H.; Ducroq, D.; Rozet, J.M.; Gerber, S.; Perrault, I.; Munnich, A.; Coscas, G.; Soubrane, G.; Kaplan, J. ABCR gene analysis in familial exudative age-related macular degeneration. Investig. Ophthalmol. Vis. Sci. 2000, 41, 244–247. [Google Scholar]
- Rivera, A.; White, K.; Stöhr, H.; Steiner, K.; Hemmrich, N.; Grimm, T.; Jurklies, B.; Lorenz, B.; Scholl, H.P.; Apfelstedt-Sylla, E.; Weber, B.H. A comprehensive survey of sequence variation in the ABCA4 (ABCR) gene in Stargardt disease and age-related macular degeneration. Am. J. Hum. Genet. 2000, 67, 800–813. [Google Scholar] [CrossRef] [PubMed]
- Sangermano, R.; Bax, N.M.; Bauwens, M.; van den Born, L.I.; De Baere, E.; Garanto, A.; Collin, R.W.J.; Goercharn-Ramlal, A.S.A.; den Engelsman-van Dijk, A.H.A.; Rohrschneider, K.; et al. Photoreceptor Progenitor mRNA Analysis Reveals Exon Skipping Resulting from the ABCA4 c.5461-10T→C Mutation in Stargardt Disease. Ophthalmology 2016, 123, 1375–1385. [Google Scholar] [CrossRef] [PubMed]
- Birch, D.G.; Peters, A.Y.; Locke, K.L.; Spencer, R.; Megarity, C.F.; Travis, G.H. Visual function in patients with cone-rod dystrophy (CRD) associated with mutations in the ABCA4(ABCR) gene. Exp. Eye Res. 2001, 73, 877–886. [Google Scholar] [CrossRef] [PubMed]
- Simonelli, F.; Testa, F.; Zernant, J.; Nesti, A.; Rossi, S.; Allikmets, R.; Rinaldi, E. Genotype-phenotype correlation in Italian families with Stargardt disease. Ophthalmic Res. 2005, 37, 159–167. [Google Scholar] [CrossRef] [PubMed]
- Briggs, C.E.; Rucinski, D.; Rosenfeld, P.J.; Hirose, T.; Berson, E.L.; Dryja, T.P. Mutations in ABCR (ABCA4) in patients with Stargardt macular degeneration or cone-rod degeneration. Investig. Ophthalmol. Vis. Sci. 2001, 42, 2229–2236. [Google Scholar]
- Klevering, B.J.; Deutman, A.F.; Maugeri, A.; Cremers, F.P.M.; Hoyng, C.B. The spectrum of retinal phenotypes caused by mutations in the ABCA4 gene. Graefes Arch. Clin. Exp. Ophthalmol. 2005, 243, 90–100. [Google Scholar] [CrossRef] [PubMed]
- Fishman, G.A.; Stone, E.M.; Grover, S.; Derlacki, D.J.; Haines, H.L.; Hockey, R.R. Variation of clinical expression in patients with Stargardt dystrophy and sequence variations in the ABCR gene. Arch. Ophthalmol. 1999, 117, 504–510. [Google Scholar] [CrossRef] [PubMed]
- Stenirri, S.; Fermo, I.; Battistella, S.; Galbiati, S.; Soriani, N.; Paroni, R.; Manitto, M.P.; Martina, E.; Brancato, R.; Allikmets, R.; et al. Denaturing HPLC profiling of the ABCA4 gene for reliable detection of allelic variations. Clin. Chem. 2004, 50, 1336–1343. [Google Scholar] [CrossRef] [PubMed]
- Jaakson, K.; Zernant, J.; Külm, M.; Hutchinson, A.; Tonisson, N.; Glavac, D.; Ravnik-Glavac, M.; Hawlina, M.; Meltzer, M.R.; Caruso, R.C.; et al. Genotyping microarray (gene chip) for the ABCR (ABCA4) gene. Hum. Mutat. 2003, 22, 395–403. [Google Scholar] [CrossRef] [PubMed]
- Papaioannou, M.; Ocaka, L.; Bessant, D.; Lois, N.; Bird, A.; Payne, A.; Bhattacharya, S. An analysis of ABCR mutations in British patients with recessive retinal dystrophies. Investig. Ophthalmol. Vis. Sci. 2000, 41, 16–19. [Google Scholar]
- Testa, F.; Rossi, S.; Sodi, A.; Passerini, I.; Di Iorio, V.; Della Corte, M.; Banfi, S.; Surace, E.M.; Menchini, U.; Auricchio, A.; et al. Correlation between photoreceptor layer integrity and visual function in patients with Stargardt disease: Implications for gene therapy. Investig. Ophthalmol. Vis. Sci. 2012, 53, 4409–4415. [Google Scholar] [CrossRef] [PubMed]
- Aukrust, I.; Jansson, R.W.; Bredrup, C.; Rusaas, H.E.; Berland, S.; Jørgensen, A.; Haug, M.G.; Rødahl, E.; Houge, G.; Knappskog, P.M. The intronic ABCA4 c.5461-10T>C variant, frequently seen in patients with Stargardt disease, causes splice defects and reduced ABCA4 protein level. Acta Ophthalmol. (Copenh.) 2017, 95, 240–246. [Google Scholar] [CrossRef] [PubMed]
- Fujinami, K.; Lois, N.; Mukherjee, R.; McBain, V.A.; Tsunoda, K.; Tsubota, K.; Stone, E.M.; Fitzke, F.W.; Bunce, C.; Moore, A.T.; et al. A longitudinal study of Stargardt disease: Quantitative assessment of fundus autofluorescence, progression, and genotype correlations. Investig. Ophthalmol. Vis. Sci. 2013, 54, 8181–8190. [Google Scholar] [CrossRef] [PubMed]
- Lois, N.; Holder, G.E.; Bunce, C.; Fitzke, F.W.; Bird, A.C. Phenotypic subtypes of Stargardt macular dystrophy-fundus flavimaculatus. Arch. Ophthalmol. 2001, 119, 359–369. [Google Scholar] [CrossRef] [PubMed]
- Fishman, G.A. Fundus flavimaculatus. A clinical classification. Arch. Ophthalmol. 1976, 94, 2061–2067. [Google Scholar] [CrossRef] [PubMed]
- Duncker, T.; Marsiglia, M.; Lee, W.; Zernant, J.; Tsang, S.H.; Allikmets, R.; Greenstein, V.C.; Sparrow, J.R. Correlations among near-infrared and short-wavelength autofluorescence and spectral-domain optical coherence tomography in recessive Stargardt disease. Investig. Ophthalmol. Vis. Sci. 2014, 55, 8134–8143. [Google Scholar] [CrossRef] [PubMed]
- Cideciyan, A.V.; Swider, M.; Aleman, T.S.; Sumaroka, A.; Schwartz, S.B.; Roman, M.I.; Milam, A.H.; Bennett, J.; Stone, E.M.; Jacobson, S.G. ABCA4-associated retinal degenerations spare structure and function of the human parapapillary retina. Investig. Ophthalmol. Vis. Sci. 2005, 46, 4739–4746. [Google Scholar] [CrossRef] [PubMed]
- Parodi, M.B.; Iacono, P.; Triolo, G.; La Spina, C.; Zucchiatti, I.; Cicinelli, M.V.; Borrelli, E.; Manitto, M.P.; Martina, E.; Bandello, F. Morpho-functional correlation of fundus autofluorescence in Stargardt disease. Br. J. Ophthalmol. 2015, 99, 1354–1359. [Google Scholar] [CrossRef] [PubMed]
- Ścieżyńska, A.; Oziębło, D.; Ambroziak, A.M.; Korwin, M.; Szulborski, K.; Krawczyński, M.; Stawiński, P.; Szaflik, J.; Szaflik, J.P.; Płoski, R.; et al. Next-generation sequencing of ABCA4: High frequency of complex alleles and novel mutations in patients with retinal dystrophies from Central Europe. Exp. Eye Res. 2016, 145, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Zaneveld, J.; Siddiqui, S.; Li, H.; Wang, X.; Wang, H.; Wang, K.; Li, H.; Ren, H.; Lopez, I.; Dorfman, A.; et al. Comprehensive analysis of patients with Stargardt macular dystrophy reveals new genotype-phenotype correlations and unexpected diagnostic revisions. Genet. Med. 2015, 17, 262–270. [Google Scholar] [CrossRef] [PubMed]
- Burke, T.R.; Allikmets, R.; Smith, R.T.; Gouras, P.; Tsang, S.H. Loss of peripapillary sparing in non-group I Stargardt disease. Exp. Eye Res. 2010, 91, 592–600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahn, J.; Beharry, S.; Molday, L.L.; Molday, R.S. Functional interaction between the two halves of the photoreceptor-specific ATP binding cassette protein ABCR (ABCA4). Evidence for a non-exchangeable ADP in the first nucleotide binding domain. J. Biol. Chem. 2003, 278, 39600–39608. [Google Scholar] [CrossRef] [PubMed]
- Audo, I.; Manes, G.; Mohand-Saïd, S.; Friedrich, A.; Lancelot, M.-E.; Antonio, A.; Moskova-Doumanova, V.; Poch, O.; Zanlonghi, X.; Hamel, C.P.; et al. Spectrum of rhodopsin mutations in French autosomal dominant rod-cone dystrophy patients. Investig. Ophthalmol. Vis. Sci. 2010, 51, 3687–3700. [Google Scholar] [CrossRef] [PubMed]
- Stenson, P.D.; Ball, E.V.; Mort, M.; Phillips, A.D.; Shiel, J.A.; Thomas, N.S.T.; Abeysinghe, S.; Krawczak, M.; Cooper, D.N. Human Gene Mutation Database (HGMD): 2003 update. Hum. Mutat. 2003, 21, 577–581. [Google Scholar] [CrossRef] [PubMed]
- Fokkema, I.F.A.C.; Taschner, P.E.M.; Schaafsma, G.C.P.; Celli, J.; Laros, J.F.J.; den Dunnen, J.T. LOVD v.2.0: The next generation in gene variant databases. Hum. Mutat. 2011, 32, 557–563. [Google Scholar] [CrossRef] [PubMed]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sim, N.-L.; Kumar, P.; Hu, J.; Henikoff, S.; Schneider, G.; Ng, P.C. SIFT web server: Predicting effects of amino acid substitutions on proteins. Nucleic Acids Res. 2012, 40, W452–W457. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, J.M.; Cooper, D.N.; Schuelke, M.; Seelow, D. MutationTaster2: Mutation prediction for the deep-sequencing age. Nat. Methods 2014, 11, 361–362. [Google Scholar] [CrossRef] [PubMed]
- Yeo, G.; Burge, C.B. Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. J. Comput. Biol. 2004, 11, 377–394. [Google Scholar] [CrossRef] [PubMed]
- Reese, M.G.; Eeckman, F.H.; Kulp, D.; Haussler, D. Improved splice site detection in Genie. J. Comput. Biol. 1997, 4, 311–323. [Google Scholar] [CrossRef] [PubMed]
- Desmet, F.-O.; Hamroun, D.; Lalande, M.; Collod-Béroud, G.; Claustres, M.; Béroud, C. Human Splicing Finder: An online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 2009, 37, e67. [Google Scholar] [CrossRef] [PubMed]
- Casper, J.; Zweig, A.S.; Villarreal, C.; Tyner, C.; Speir, M.L.; Rosenbloom, K.R.; Raney, B.J.; Lee, C.M.; Lee, B.T.; Karolchik, D.; et al. The UCSC Genome Browser database: 2018 update. Nucleic Acids Res. 2017. [Google Scholar] [CrossRef]
- Kent, W.J. BLAT—The BLAST-like alignment tool. Genome Res. 2002, 12, 656–664. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Marmor, M.F.; Fulton, A.B.; Holder, G.E.; Miyake, Y.; Brigell, M.; Bach, M.; International Society for Clinical Electrophysiology of Vision. ISCEV Standard for full-field clinical electroretinography (2008 update). Doc. Ophthalmol. 2009, 118, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Hood, D.C.; Bach, M.; Brigell, M.; Keating, D.; Kondo, M.; Lyons, J.S.; Palmowski-Wolfe, A.M. ISCEV guidelines for clinical multifocal electroretinography (2007 edition). Doc. Ophthalmol. 2008, 116, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Schmitz-Valckenberg, S.; Brinkmann, C.K.; Alten, F.; Herrmann, P.; Stratmann, N.K.; Göbel, A.P.; Fleckenstein, M.; Diller, M.; Jaffe, G.J.; Holz, F.G. Semiautomated image processing method for identification and quantification of geographic atrophy in age-related macular degeneration. Investig. Ophthalmol. Vis. Sci. 2011, 52, 7640–7646. [Google Scholar] [CrossRef] [PubMed]
- Strauss, R.W.; Muñoz, B.; Wolfson, Y.; Sophie, R.; Fletcher, E.; Bittencourt, M.G.; Scholl, H.P.N. Assessment of estimated retinal atrophy progression in Stargardt macular dystrophy using spectral-domain optical coherence tomography. Br. J. Ophthalmol. 2016, 100, 956–962. [Google Scholar] [CrossRef] [PubMed]
- Parker, M.A.; Choi, D.; Erker, L.R.; Pennesi, M.E.; Yang, P.; Chegarnov, E.N.; Steinkamp, P.N.; Schlechter, C.L.; Dhaenens, C.-M.; Mohand-Said, S.; et al. Test-Retest Variability of Functional and Structural Parameters in Patients with Stargardt Disease Participating in the SAR422459 Gene Therapy Trial. Transl. Vis. Sci. Technol. 2016, 5, 10. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.; Xie, Y.; Zernant, J.; Yuan, B.; Bearelly, S.; Tsang, S.H.; Lupski, J.R.; Allikmets, R. Complex inheritance of ABCA4 disease: Four mutations in a family with multiple macular phenotypes. Hum. Genet. 2016, 135, 9–19. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Exon/Intron | Variant | Protein Change | Classification |
|---|---|---|---|
| 1 | c.53G>A | p.(Arg18Gln) | likely pathogenic |
| IVS5 | c.570+1_570+8del | p.? | pathogenic |
| 6 | c.686T>C | p.(Leu229Pro) | likely pathogenic |
| IVS7 | c.859−2A>G | p.? | pathogenic |
| 8 | c.902del | p.(Arg301Serfs*15) | pathogenic |
| 8 | c.972_973delinsAT | p.(Cys324 *) | pathogenic |
| 8 | c.978C>A | p.(Tyr326 *) | pathogenic |
| 8 | c.1019A>G | p.(Tyr340Cys) | likely pathogenic |
| 8 | c.1050del | p.(Ile351Leufs*23) | pathogenic |
| IVS8 | c.1099+1G>C | p.? | pathogenic |
| 9 | c.1201A>C | p.(Thr401Pro) | likely pathogenic |
| 10 | c.1252T>C | p.(Phe418Leu) | likely pathogenic |
| 10 | c.1301T>G | p.(Val434Gly) | likely pathogenic |
| 12 | c.1556G>A | p.(Cys519Tyr) | likely pathogenic |
| 12 | c.1648_1659del | p.(Gly550_Phe553del) | likely pathogenic |
| 12 | c.1706A>G | p.(Tyr569Cys) | likely pathogenic |
| 13 | c.1895T>A | p.(Ile632Asn) | likely pathogenic |
| 14 | c.2083G>C | p.(Val695Leu) | likely pathogenic |
| 15 | c.2169_2172dup | p.(Leu725Asnfs*42) | pathogenic |
| 15 | c.2299del | p.(Val767Serfs*20) | pathogenic |
| 16 | c.2443C>T | p.(Gln815 *) | pathogenic |
| 16 | c.2572dup | p.(Asp858Glyfs*27) | pathogenic |
| 19 | c.2868C>A | p.(Asn956Lys) | uncertain |
| 21 | c.3080A>G | p.(Tyr1027Cys) | likely pathogenic |
| 22 | c.3311T>C | p.(Leu1104Pro) | likely pathogenic |
| 23 | c.3409A>G | p.(Arg1137Gly) | uncertain |
| 25 | c.3682G>T | p.(Glu1228 *) | likely pathogenic |
| 25 | c.3811G>C | p.(Glu1271Asp) | likely pathogenic |
| 26 | c.3825G>C | p.(Lys1275Asn) | likely pathogenic |
| 27 | c.3966del | p.(Ala1324Argfs*65) | pathogenic |
| 27 | c.4061A>C | p.(His1354Pro) | likely pathogenic |
| IVS27 | c.4129−3C>A | p.? | likely pathogenic |
| 28 | c.4178_4192del | p.(Val1393_Phe1397del) | likely pathogenic |
| 29 | c.4324A>G | p.(Asn1442Asp) | likely pathogenic |
| 30 | c.4510_4535del | p.(Glu1504Profs*42) | pathogenic |
| 32 | c.4663_4664del | p.(Gln1555Glufs*41) | pathogenic |
| 33 | c.4689del | p.(Gly1564Glufs*17) | pathogenic |
| 33 | c.4696C>T | p.(Leu1566Phe) | likely pathogenic |
| 33 | c.4700C>T | p.(Pro1567Leu) | likely pathogenic |
| 34 | c.4775G>A | p.(Gly1592Asp) | likely pathogenic |
| 37 | c.5282C>G | p.(Pro1761Arg) | likely pathogenic |
| 38 | c.5332A>T | p.(Met1778Leu) | likely pathogenic |
| 38 | c.5342C>A | p.(Ala1781Glu) | likely pathogenic |
| 38 | c.5351T>C | p.(Leu1784Pro) | likely pathogenic |
| 38 | c.5384T>C | p.(Leu1795Ser) | likely pathogenic |
| 40 | c.5621T>C | p.(Leu1874Pro) | likely pathogenic |
| IVS42 | c.5898+2T>C | p.? | pathogenic |
| IVS42 | c.5899−3T>C | p.? | uncertain |
| 43 | c.5939C>T | p.(Thr1980Ile) | likely pathogenic |
| IVS43 | c.6005+1del | p.? | pathogenic |
| 44 | c.6110C>A | p.(Ala2037Asp) | likely pathogenic |
| 45 | c.6181_6184del | p.(Thr2061Serfs*53) | pathogenic |
| 45 | c.6191C>T | p.(Ala2064Val) | likely pathogenic |
| 45 | c.6250G>A | p.(Ala2084Thr) | likely pathogenic |
| 46 | c.6284A>G | p.(Asp2095Gly) | likely pathogenic |
| 47 | c.6394G>T | p.(Glu2132 *) | pathogenic |
| 47 | c.6454G>A | p.(Gly2152Ser) | likely pathogenic |
| 47 | c.6455G>T | p.(Gly2152Val) | likely pathogenic |
| 47 | c.6436_6437insT | p.(Gly2146Valfs*36) | pathogenic |
| 48 | c.6693del | p.(Ile2231Metfs*16) | pathogenic |
| 48 | c.6704C>G | p.(Ser2235 *) | pathogenic |
| 49 | c.6746C>A | p.(Ala2249Asp) | likely pathogenic |
| IVS49 | c.6816+1G>T | p.? | pathogenic |
| Allele 1 | Allele 2 | Allele 1 or 2 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Patient ID | Family ID | Exon/Intron | Nucleotide Change | Protein Change | Exon/Intron | Nucleotide Change | Protein Change | Exon/Intron | Nucleotide Change | Protein Change | |
| CIC03734 | 1673 | Index | 1 | c.53G>A | p.(Arg18Gln) | 42 | c.5882G>A [25] | p.(Gly1961Glu) | |||
| CIC03735 | 1673 | Affected brother | 1 | c.53G>A | p.(Arg18Gln) | 42 | c.5882G>A [25] | p.(Gly1961Glu) | |||
| CIC06131 | 1673 | Unaffected father | 1 | c.53G>A | p.(Arg18Gln) | WILD TYPE | |||||
| CIC06908 | 3783 | Index | IVS5 | c.570+1_570+8del | p.? | 40 | c.5603A>T [34] | p.(Asn1868Ile) | |||
| 45 | c.6148G>C [25] | p.(Val2050Leu) | |||||||||
| CIC08771 | 3783 | Unaffected mother | WILD TYPE | 40 | c.5603A>T [34] | p.(Asn1868Ile) | |||||
| CIC08774 | 3783 | Unaffected brother | IVS5 | c.570+1_570+8del | - | WILD TYPE | |||||
| 45 | c.6148G>C [25] | p.(Val2050Leu) | |||||||||
| CIC07895 | 4412 | Index | 6 | c.686T>C | p.(Leu229Pro) | 24 | c.3602T>G [5] | p.(Leu1201Arg) | |||
| 40 | c.5621T>C | p.(Leu1874Pro) | |||||||||
| CIC07896 | 4412 | Unaffected mother | 6 | c.686T>C | p.(Leu229Pro) | WILD TYPE | |||||
| CIC07887 | 4407 | Index | 6 | c.686T>C | p.(Leu229Pro) | 42 | c.5882G>A [25] | p.(Gly1961Glu) | |||
| 48 | c.6529G>A [35] | p.(Asp2177Asn) | |||||||||
| CIC07888 | 4407 | Unaffected mother | 6 | c.686T>C | p.(Leu229Pro) | WILD TYPE | |||||
| 48 | c.6529G>A [35] | p.(Asp2177Asn) | |||||||||
| CIC09848 | 5658 | Index | IVS7 | c.859−2A>G | - | 8 | c.872C>T [36] | p.(Pro291Leu) | |||
| 11 | c.1531C>T [18] | p.(Arg511Cys) | |||||||||
| CIC09849 | 5658 | Unaffected mother | WILD TYPE | 8 | c.872C>T [37] | p.(Pro291Leu) | |||||
| 11 | c.1531C>T [18] | p.(Arg511Cys) | |||||||||
| CIC09382 | 5385 | Index | 8 | c.978C>A | p.(Tyr326 *) | IVS42 | c.5898+2T>C | p.? | |||
| CIC06749 | 3669 | Index | 8 | c.902del | p.(Arg301Serfs*15) | 46 | c.6320G>A [26] | p.(Arg2107His) | |||
| CIC06088 | 3203 | Index | 8 | c.972_973delinsAT | p.(Cys324 *) | 23 | c.3386G>T [38] | p.(Arg1129Leu) | |||
| CIC08477 | 3203 | Unaffected father | 8 | c.972_973delinsAT | p.(Cys324 *) | WILD TYPE | |||||
| CIC08478 | 3203 | Unaffected mother | WILD TYPE | 23 | c.3386G>T [38] | p.(Arg1129Leu) | |||||
| CIC07725 | 4301 | Index | 8 | c.1019A>G | p.(Tyr340Cys) | 8 | c.1019A>G | p.(Tyr340Cys) | |||
| CIC07726 | 4301 | Unaffected father | 8 | c.1019A>G | p.(Tyr340Cys) | WILD TYPE | |||||
| CICO7727 | 4301 | Unaffected mother | WILD TYPE | 8 | c.1019A>G | p.(Tyr340Cys) | |||||
| CIC04235 | 2021 | Index | 8 | c.1050del | p.(Ile351Leufs*23) | 38 | c.5351T>C | p.(Leu1784Pro) | |||
| CIC04236 | 2021 | Unaffected sister | 8 | c.1050del | p.(Ile351Leufs*23) | WILD TYPE | |||||
| CIC07308 | 4026 | Index | 8 | c.1050del | p.(Ile351Leufs*23) | 42 | c.5882G>A [25] | p.(Gly1961Glu) | |||
| CIC01080 | 659 | Index | 8 | c.1050del | p.(Ile351Leufs*23) | 19 | c.2819C>G [39] | p.(Pro940Arg) | |||
| 23 | c.3364G>A [6] | p.(Glu1122Lys) | |||||||||
| CIC03521 | 659 | Unaffected mother | WILD TYPE | 19 | c.2819C>G [39] | p.(Pro940Arg) | |||||
| 23 | c.3364G>A [6] | p.(Glu1122Lys) | |||||||||
| CIC03524 | 659 | Unaffected sister | WILD TYPE | WILD TYPE | |||||||
| CIC08372 | 4715 | Index | IVS8 | c.1099+1G>C | p.? | 22 | c.3322C>T [26] | p.(Arg1108Cys) | |||
| CIC05266 | 2672 | Index | 9 | c.1201A>C | p.(Thr401Pro) | 17 | c.2588G>C [25] | p.(Gly863Ala) | |||
| 40 | c.5603A>T [34] | p.(Asn1868Ile) | |||||||||
| CIC05267 | 2672 | Unaffected mother | 9 | c.1201A>C | p.(Thr401Pro) | WILD TYPE | |||||
| CIC05268 | 2672 | Unaffected father | WILD TYPE | 17 | c.2588G>C [25] | p.(Gly863Ala) | |||||
| 40 | c.5603A>T [34] | p.(Asn1868Ile) | |||||||||
| CIC02804 | 1037 | Index | 10 | c.1252T>C | p.(Phe418Leu) | IVS43 | c.6005+1del | p.? | 42 | c.5882G>A [25] | p.(Gly1961Glu) |
| CIC09625 | 5521 | Index | 10 | c.1301T>G | p.(Val434Gly) | 10 | c.1301T>G | p.(Val434Gly) | |||
| CIC09624 | 5521 | Affected brother | 10 | c.1301T>G | p.(Val434Gly) | 10 | c.1301T>G | p.(Val434Gly) | |||
| CIC09626 | 5521 | Unaffected father | 10 | c.1301T>G | p.(Val434Gly) | WILD TYPE | |||||
| CIC09628 | 5521 | Unaffected mother | WILD TYPE | 10 | c.1301T>G | p.(Val434Gly) | |||||
| CIC01301 | 784 | Index | 12 | c.1556G>A | p.(Cys519Tyr) | 22 | c.3292C>T [40] | p.(Arg1098Cys) | |||
| CIC09999 | 784 | Affected brother | 12 | c.1556G>A | p.(Cys519Tyr) | 22 | c.3292C>T [41] | p.(Arg1098Cys) | |||
| CIC06346 | 3356 | Index | 12 | c.1648_1659del | p.(Gly550_Phe553del) | 42 | c.5882G>A [25] | p.(Gly1961Glu) | |||
| CIC08884 | 3356 | Unaffected father | 12 | c.1648_1659del | p.(Gly550_Phe553del) | WILD TYPE | |||||
| CIC08949 | 3356 | Unaffected mother | WILD TYPE | 42 | c.5882G>A [25] | p.(Gly1961Glu) | |||||
| CIC04197 | 1992 | Index | 12 | c.1706A>G | p.(Tyr569Cys) | 40 | c.5603A>T [34] | p.(Asn1868Ile) | |||
| CIC07749 | 4314 | Index | 13 | c.1895T>A | p.(Ile632Asn) | 33 | c.4700C>T | p.(Pro1567Leu) | 24 | c.3602T>G [5] | p.(Leu1201Arg) |
| CIC08724 | 4948 | Index | 14 | c.2083G>C | p.(Val695Leu) | 47 | c.6445C>T [6] | p.(Arg2149 *) | |||
| 44 | c.6079C>T [29] | p.(Leu2027Phe) | |||||||||
| CIC08723 | 4948 | Affected sister | 14 | c.2083G>C | p.(Val695Leu) | 47 | c.6445C>T [6] | p.(Arg2149 *) | |||
| 44 | c.6079C>T [29] | p.(Leu2027Phe) | |||||||||
| CIC08858 | 4948 | Unaffected mother | 14 | c.2083G>C | p.(Val695Leu) | WILD TYPE | |||||
| 44 | c.6079C>T [29] | p.(Leu2027Phe) | |||||||||
| CIC08859 | 4948 | Unaffected father | WILD TYPE | 47 | c.6445C>T [6] | p.(Arg2149 *) | |||||
| CIC07985 | 4793 | Index | 15 | c.2169_2172dup | p.(Leu725Asnfs*42) | 47 | c.6454G>A | p.(Gly2152Ser) | 44 | c.6079C>T [25] | p.(Leu2027Phe) |
| 40 | c.5603A>T [34] | p.(Asn1868Ile) | |||||||||
| CIC06126 | 3227 | Index | 15 | c.2299del | p.(Val767Serfs*20) | 17 | c.2588G>C [25] | p.(Gly863Ala) | 40 | c.5603A>T [34] | p.(Asn1868Ile) |
| CIC09405 | 5401 | Index | 16 | c.2443C>T | p.(Gln815 *) | 42 | c.5882G>A [25] | p.(Gly1961Glu) | |||
| CIC09407 | 5401 | Unaffected mother | WILD TYPE | 42 | c.5882G>A [25] | p.(Gly1961Glu) | |||||
| CIC09408 | 5401 | Unaffected father | 16 | c.2443C>T | p.(Gln815 *) | WILD TYPE | |||||
| CIC00467 | 319 | Index | 16 | c.2572dup | p.(Asp858Glyfs*27) | 35 | c.4926C>G [42] | p.(Ser1642Arg) | |||
| 36 | c.5044_5058del [25] | p.(Val1682_Val1686del) | |||||||||
| CIC05014 | 319 | Unaffected sister | 16 | c.2572dup | p.(Asp858Glyfs*27) | WILD TYPE | |||||
| CIC05056 | 319 | Affected brother | 16 | c.2572dup | p.(Asp858Glyfs*27) | 35 | c.4926C>G [43] | p.(Ser1642Arg) | |||
| 36 | c.5044_5058del [25] | p.(Val1682_Val1686del) | |||||||||
| + CIC03678 | 1627 | Index | 19 | c.2868C>A | p.(Asn956Lys) | 43 | c.5939C>T | p.(Thr1980Ile) | |||
| CIC03679 | 1627 | Unaffected son | WILD TYPE | 43 | c.5939C>T | p.(Thr1980Ile) | |||||
| CIC04259 | 2036 | Index | 21 | c.3080A>G | p.(Tyr1027Cys) | 38 | c.5381C>A [24] | p.(Ala1794Asp) | |||
| CIC08538 | 4826 | Index | 22 | c.3311T>C | p.(Leu1104Pro) | IVS49 | c.6816+1G>A [6] | - | |||
| CIC04176 | 1973 | Index | 25 | c.3682G>T | p.(Glu1228*) | 22 | c.3322C>T [26] | p.(Arg1108Cys) | |||
| CIC02505 | 871 | Index | 25 | c.3811G>C | p.(Glu1271Gln) | 23 | c.3386G>T [38] | p.(Arg1129Leu) | |||
| CIC05899 | 3087 | Index | 25 | c.3811G>C | p.(Glu1271Gln) | 42 | c.5882G>A [25] | p.(Gly1961Glu) | |||
| CIC07120 | 3909 | Index | 26 | c.3825G>C | p.(Lys1275Asn) | 22 | c.3322C>T [26] | p.(Arg1108Cys) | 40 | c.5603A>T [34] | p.(Asn1868Ile) |
| 46 | c.6320G>A [26] | p.(Arg2107His) | |||||||||
| CIC01750 | 1242 | Index | 27 | c.3966del | p.(Ala1324Argfs*65) | 35 | c.4918C>T [26] | p.(Arg1640Trp) | |||
| 40 | c.5603A>T [34] | p.(Asn1868Ile) | |||||||||
| CIC02024 | 1242 | Affected sister | 27 | c.3966del | p.(Ala1324Argfs*65) | 35 | c.4918C>T [26] | p.(Arg1640Trp) | |||
| 40 | c.5603A>T [34] | p.(Asn1868Ile) | |||||||||
| CIC07100 | 1242 | Unaffected father | WILD TYPE | 35 | c.4918C>T [26] | p.(Arg1640Trp) | |||||
| 40 | c.5603A>T [34] | p.(Asn1868Ile) | |||||||||
| CIC07146 | 1242 | Unaffected mother | 27 | c.3966del | p.(Ala1324Argfs*65) | WILD TYPE | |||||
| CIC07744 | 4372 | Index | 27 | c.4061A>C | p.(His1354Pro) | 13 | c.1927G>A [44] | p.(Val643Met) | 8 | c.872C>T [37] | p.(Pro291Leu) |
| 24 | c.3602T>G [6] | p.(Leu1201Arg) | |||||||||
| CIC02690 | 961 | Index | IVS27 | c.4129−3C>A | p.? | IVS38 | c.5461-10T>C [45] | p. [Thr1821Valfs, Thr1821Aspfs] | |||
| 40 | c.5603A>T [34] | p.(Asn1868Ile) | |||||||||
| CIC02691 | 961 | Unaffected mother | WILD TYPE | IVS38 | c.5461-10T>C [45] | p. [Thr1821Valfs, Thr1821Aspfs] | |||||
| 40 | c.5603A>T [34] | p.(Asn1868Ile) | |||||||||
| CIC08194 | 4588 | Index | 28 | c.4178_4192del | p.(Val1393_Phe1397del) | 40 | c.5603A>T [34] | p.(Asn1868Ile) | 45 | c.6148G>C [25] | p.(Val2050Leu) |
| CIC07994 | 4463 | Index | 29 | c.4324A>G | p.(Asn1442Asp) | 3 | c.194G>A [46] | p.(Gly65Glu) | |||
| CIC07998 | 4463 | Unaffected mother | WILD TYPE | 3 | c.194G>A [46] | p.(Gly65Glu) | |||||
| CIC06727 | 3652 | Index | 30 | c.4510_4535del | p.(Glu1504Profs*42) | 28 | c.4139C>T [6] | p.(Pro1380Leu) | |||
| CIC06728 | 3652 | Unaffected father | WILD TYPE | 28 | c.4139C>T [6] | p.(Pro1380Leu) | |||||
| CIC06729 | 3652 | Unaffected brother | WILD TYPE | 28 | c.4139C>T [6] | p.(Pro1380Leu) | |||||
| CIC06730 | 3652 | Unaffected mother | 30 | c.4510_4535del | p.(Glu1504Profs*42) | WILD TYPE | |||||
| CIC08283 | 4647 | Index | 32 | c.4663_4664del | p.(Gln1555Glufs*41) | 40 | c.5603A>T [34] | p.(Asn1868Ile) | |||
| CIC03252 | 1375 | Index | 33 | c.4689del | p.(Gly1564Glufs*17) | 17 | c.2588G>C [25] | p.(Gly863Ala) | |||
| 40 | c.5603A>T [34] | p.(Asn1868Ile) | |||||||||
| CIC03253 | 1375 | Unaffected mother | WILD TYPE | 17 | c.2588G>C [25] | p.(Gly863Ala) | |||||
| 40 | c.5603A>T [34] | p.(Asn1868Ile) | |||||||||
| CIC09219 | 5282 | Index | 33 | c.4696C>T | p.(Leu1566Phe) | 21 | c.3056C>T [26] | p.(Thr1019Met) | |||
| 42 | c.5882G>A [25] | p.(Gly1961Glu) | |||||||||
| CIC10754 | 5282 | Unaffected mother | WILD TYPE | 21 | c.3056C>T [26] | p.(Thr1019Met) | |||||
| CIC10755 | 5282 | Unaffected father | 33 | c.4696C>T | p.(Leu1566Phe) | WILD TYPE | |||||
| 42 | c.5882G>A [25] | p.(Gly1961Glu) | |||||||||
| CIC08932 | 5089 | Index | 34 | c.4775G>A | p.(Gly1592Asp) | 3 | c.288C>A [47] | p.(Asn96Lys) | |||
| 42 | c.5882G>A [25] | p.(Gly1961Glu) | |||||||||
| ++ CIC08933 | 5089 | Unaffected father | WILD TYPE | WILD TYPE | |||||||
| CIC08934 | 5089 | Unaffected mother | WILD TYPE | 3 | c.288C>A [47] | p.(Asn96Lys) | |||||
| 42 | c.5882G>A [25] | p.(Gly1961Glu) | |||||||||
| CIC07036 | 3867 | Index | 37 | c.5282C>G | p.(Pro1761Arg) | 42 | c.5882G>A [25] | p.(Gly1961Glu) | |||
| CIC07960 | 4447 | Index | 37 | c.5282C>G | p.(Pro1761Arg) | 26 | c.3819dup [48] | p.(Leu1274Serfs*8) | |||
| 46 | c.6316C>T [25] | p.(Arg2106Cys) | |||||||||
| CIC07999 | 4447 | Unaffected sister | WILD TYPE | WILD TYPE | |||||||
| CIC08029 | 4447 | Unaffected mother | 37 | c.5282C>G | p.(Pro1761Arg) | WILD TYPE | |||||
| 46 | c.6316C>T [25] | p.(Arg2106Cys) | |||||||||
| CIC09601 | 5509 | Index | 37 | c.5282C>G | p.(Pro1761Arg) | 22 | c.3279C>A [18] | p.(Asp1093Glu) | |||
| 46 | c.6316C>T [25] | p.(Arg2106Cys) | |||||||||
| CIC09602 | 5509 | Unaffected daughter | 37 | c.5282C>G | p.(Pro1761Arg) | WILD TYPE | |||||
| 46 | c.6316C>T [25] | p.(Arg2106Cys) | |||||||||
| CIC07831 | 4373 | Index | 38 | c.5332A>T | p.(Met1778Leu) | 27 | c.3899G>A [44] | p.(Arg1300Gln) | |||
| CIC08439 | 4759 | Index | 38 | c.5332A>T | p.(Met1778Leu) | 47 | c.6394G>T | p.(Glu2132 *) | |||
| CIC08262 | 4633 | Index | 38 | c.5342C>A | p.(Ala1781Glu) | 3 | c.286A>G [49] | p.(Asn96Asp) | |||
| CIC08263 | 4633 | Unaffected mother | 38 | c.5342C>A | p.(Ala1781Glu) | WILD TYPE | |||||
| CIC09095 | 4633 | Unaffected father | WILD TYPE | 3 | c.286A>G [49] | p.(Asn96Asp) | |||||
| CIC08359 | 4702 | Index | 38 | c.5384T>C | p.(Leu1795Ser) | 17 | c.2588G>C [25] | p.(Gly863Ala) | 40 | c.5603A>T [34] | p.(Asn1868Ile) |
| CIC07436 | 4110 | Index | IVS42 | c.5898+2T>C | p.? | 40 | c.5642C>T [50] | p.(Ala1881Val) | |||
| CIC08523 | 4110 | Unaffected mother | WILD TYPE | 40 | c.5642C>T [50] | p.(Ala1881Val) | |||||
| CIC08524 | 4110 | Unaffected father | IVS42 | c.5898+2T>C | p.? | WILD TYPE | |||||
| CIC09857 | 5675 | Index | 44 | c.6110C>A | p.(Ala2037Asp) | IVS38 | c.5461−10T>C [45] | p. [Thr1821Valfs, Thr1821Aspfs] | |||
| 40 | c.5603A>T [34] | p.(Asn1868Ile) | |||||||||
| CIC09858 | 5675 | Unaffected mother | 44 | c.6110C>A | p.(Ala2037Asp) | WILD TYPE | |||||
| CIC10452 | 5675 | Unaffected father | WILD TYPE | IVS38 | c.5461−10T>C [45] | p. [Thr1821Valfs, Thr1821Aspfs] | |||||
| 40 | c.5603A>T [34] | p.(Asn1868Ile) | |||||||||
| CIC01199 | 719 | Index | 45 | c.6181_6184del | p.(Thr2061Serfs*53) | 42 | c.5882G>A [25] | p.(Gly1961Glu) | |||
| CIC08054 | 719 | Affected sister | 45 | c.6181_6184del | p.(Thr2061Serfs*53) | 42 | c.5882G>A [25] | p.(Gly1961Glu) | |||
| CIC08057 | 719 | Unaffected father | WILD TYPE | 42 | c.5882G>A [25] | p.(Gly1961Glu) | |||||
| CIC8058 | 719 | Unaffected mother | 45 | c.6181_6184del | p.(Thr2061Serfs*53) | WILD TYPE | |||||
| CIC03710 | 1657 | Index | 45 | c.6191C>T | p.(Ala2064Val) | 30 | c.4537dup [44] | p.(Gln1513Profs*42) | |||
| 40 | c.5603A>T [34] | p.(Asn1868Ile) | |||||||||
| CIC03711 | 1657 | Unaffected mother | 45 | c.6191C>T | p.(Ala2064Val) | WILD TYPE | |||||
| 40 | c.5603A>T [34] | p.(Asn1868Ile) | |||||||||
| CIC04571 | 2232 | Index | 45 | c.6250G>A | p.(Ala2084Thr) | 19 | c.2894A>G [25] | p.(Asn965Ser) | |||
| 42 | c.5882G>A [25] | p.(Gly1961Glu) | |||||||||
| CIC04572 | 2232 | Unaffected mother | WILD TYPE | 19 | c.2894A>G [25] | p.(Asn965Ser) | |||||
| CIC04573 | 2232 | Unaffected maternal aunt | WILD TYPE | 19 | c.2894A>G [25] | p.(Asn965Ser) | |||||
| CIC07568 | 2232 | Unaffected father | 45 | c.6250G>A | p.(Ala2084Thr) | WILD TYPE | |||||
| 42 | c.5882G>A [25] | p.(Gly1961Glu) | |||||||||
| CIC07748 | 4312 | Index | 46 | c.6284A>G | p.(Asp2095Gly) | 22 | c.3323G>A [34] | p.(Arg1108His) | |||
| CIC09593 | 4312 | Unaffected mother | 46 | c.6284A>G | p.(Asp2095Gly) | WILD TYPE | |||||
| CIC09594 | 4312 | Unaffected father | WILD TYPE | 22 | c.3323G>A [34] | p.(Arg1108His) | |||||
| CIC09374 | 5379 | Index | 47 | c.6455G>T | p.(Gly2152Val) | 9 | c.1222C>T [43] | p.(Arg408*) | |||
| CIC09375 | 5379 | Unaffected mother | 47 | c.6455G>T | p.(Gly2152Val) | WILD TYPE | |||||
| CIC06396 | 3389 | Index | 47 | c.6436_6437insT | p.(Gly2146Valfs*36) | 47 | c.6436_6437insT | p.( Gly2146Valfs*36) | |||
| CIC00130 | 102 | Index | 48 | c.6693del | p.(Ile2231Metfs*16) | 44 | c.6079C>T [25] | p.(Leu2027Phe) | |||
| CIC00131 | 102 | Unaffected father | 48 | c.6693del | p.(Ile2231Metfs*16) | WILD TYPE | |||||
| CIC00132 | 102 | Unaffected mother | WILD TYPE | 44 | c.6079C>T [25] | p.(Leu2027Phe) | |||||
| CIC09114 | 5205 | Index | 48 | c.6704C>G | p.(Ser2235 *) | 21 | c.3113C>T [24] | p.(Ala1038Val) | |||
| CIC08581 | 4855 | Index | 49 | c.6746C>A | p.(Ala2249Asp) | 13 | c.1819G>A [40] | p.(Gly607Arg) | 42 | c.5882G>A [25] | p.(Gly1961Glu) |
| CIC08381 | 4722 | Index | IVS49 | c.6816+1G>T | p.? | 14 | c.2023G>A [38] | p.(Val675Ile) | |||
| CIC08383 | 4722 | Affected sister | IVS49 | c.6816+1G>T | p.? | 14 | c.2023G>A [38] | p.(Val675Ile) | |||
| CIC08402 | 4722 | Unaffected mother | WILD TYPE | 14 | c.2023G>A [38] | p.(Val675Ile) | |||||
| Groups/Stages | Criteria | Reference | |
|---|---|---|---|
| Age of onset | n.a. | Age at which visual loss was first noticed | Lois et al., 2001 [53] |
| FP | Stage 1 | Central macular atrophy with parafoveal or perifoveal flecks | Fishman GA et al., 1976 [54] |
| Stage 2 | Numerous flecks extended anterior to the vascular arcades and/or nasal to the optic disc | ||
| Stage 3 | Desorbed flecks with choriocapillaris atrophy within the macula | ||
| Stage 4 | Widespread RPE and chorioretinal atrophy throughout the fundus defined stage | ||
| SW-AF and NIR-AF | Group 1 | Central lesion with jagged border | Duncker et al., 2014 [55] |
| Group 2 | Lesion with extensive fundus changes | ||
| Group 3 | Central lesion with smooth border and hyperautofluorescent SW-AF and NIR-AF ring | ||
| Group 4 | Central lesion with smooth border and no hyperautofluorescent NIR-AF ring | ||
| Group 5 | Discrete central lesions better visualized in NIR-AF images | ||
| Peripapillary area preserved | No alterations within an eccentricity of 0.6 mm from the optic disc | Cideciyan et al., 2005 [56] | |
| Flecks in the peripapillary area | Presence of flecks within an eccentricity of 0.6 mm from the optic disc | ||
| Peripapillary area not preserved | Absence of EZ band and/or EPR atrophy within an eccentricity of 0.6 mm from the optic disc | ||
| ERG | I | Normal scotopic and full-field ERG | Lois et al., 2001 [53] |
| II | Loss of photopic function | ||
| III | Loss of both photopic and scotopic function | ||
| SW-AF and OCT | FS-YES | Foveal sparing | Fujinami et al., 2013 [38] |
| FS-NO | Early onset foveal atrophy | ||
| OCT | EZ Absent | EZ band loss | Parodi et al., 2015 [57] |
| EZ Disrupted | EZ band disorganization | ||
| EZ Preserved | Identification of EZ band | ||
| Genotype | NM | At least one null or splice variant is present | Fujinami et al., 2013 [52] |
| MM | Two or more missense variants are present | ||
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nassisi, M.; Mohand-Saïd, S.; Dhaenens, C.-M.; Boyard, F.; Démontant, V.; Andrieu, C.; Antonio, A.; Condroyer, C.; Foussard, M.; Méjécase, C.; et al. Expanding the Mutation Spectrum in ABCA4: Sixty Novel Disease Causing Variants and Their Associated Phenotype in a Large French Stargardt Cohort. Int. J. Mol. Sci. 2018, 19, 2196. https://doi.org/10.3390/ijms19082196
Nassisi M, Mohand-Saïd S, Dhaenens C-M, Boyard F, Démontant V, Andrieu C, Antonio A, Condroyer C, Foussard M, Méjécase C, et al. Expanding the Mutation Spectrum in ABCA4: Sixty Novel Disease Causing Variants and Their Associated Phenotype in a Large French Stargardt Cohort. International Journal of Molecular Sciences. 2018; 19(8):2196. https://doi.org/10.3390/ijms19082196
Chicago/Turabian StyleNassisi, Marco, Saddek Mohand-Saïd, Claire-Marie Dhaenens, Fiona Boyard, Vanessa Démontant, Camille Andrieu, Aline Antonio, Christel Condroyer, Marine Foussard, Cécile Méjécase, and et al. 2018. "Expanding the Mutation Spectrum in ABCA4: Sixty Novel Disease Causing Variants and Their Associated Phenotype in a Large French Stargardt Cohort" International Journal of Molecular Sciences 19, no. 8: 2196. https://doi.org/10.3390/ijms19082196