Overexpression of Lilium formosanum MADS-box (LFMADS) Causing Floral Defects While Promoting Flowering in Arabidopsis thaliana, Whereas Only Affecting Floral Transition Time in Nicotiana tabacum

 , ,
, ,
Abstract
:
1. Introduction
2. Results and Discussions
2.1. Illumina Sequencing, De Novo Assembly, and Functional Annotation of the L. formosanum Transcriptome
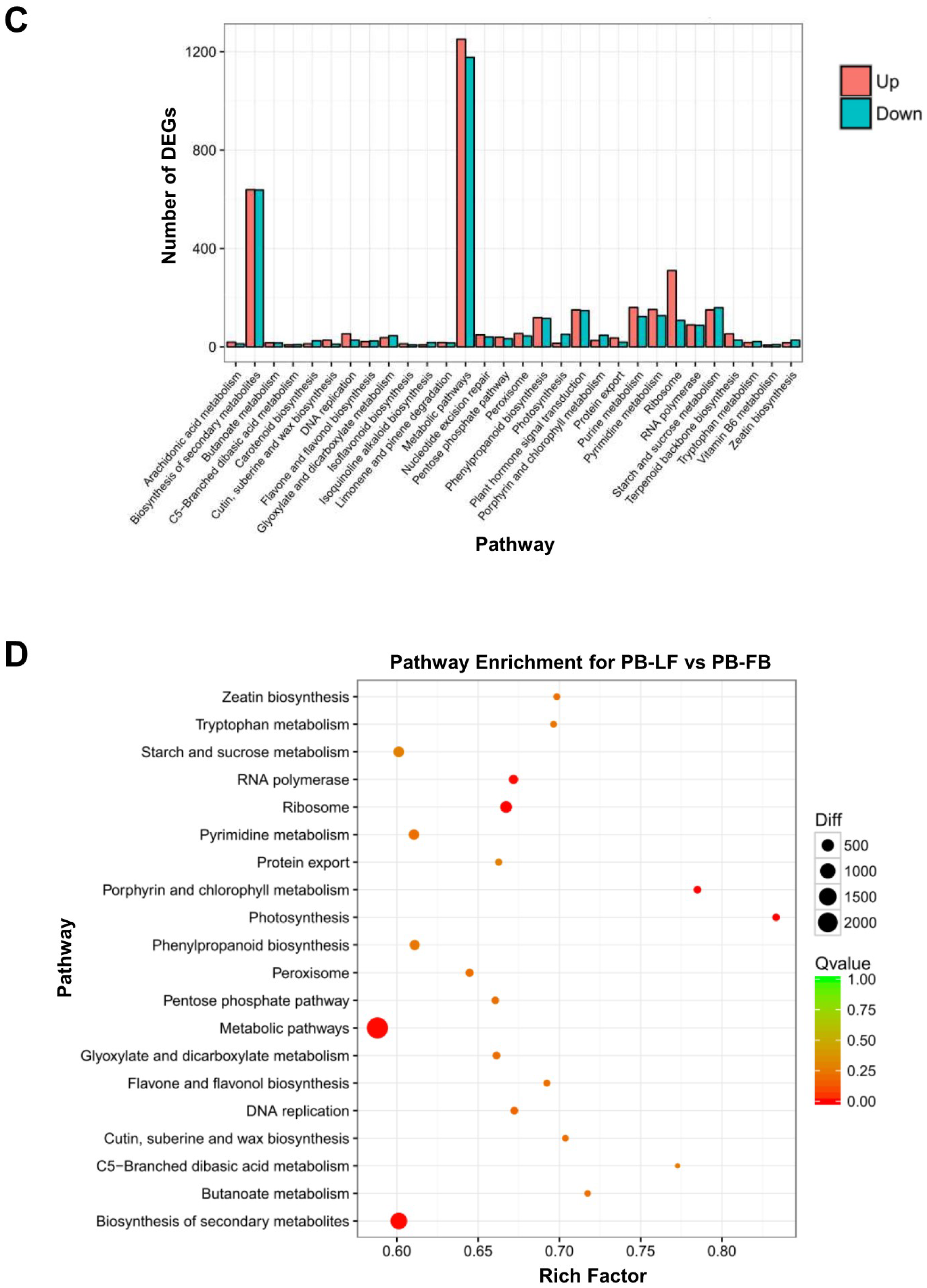
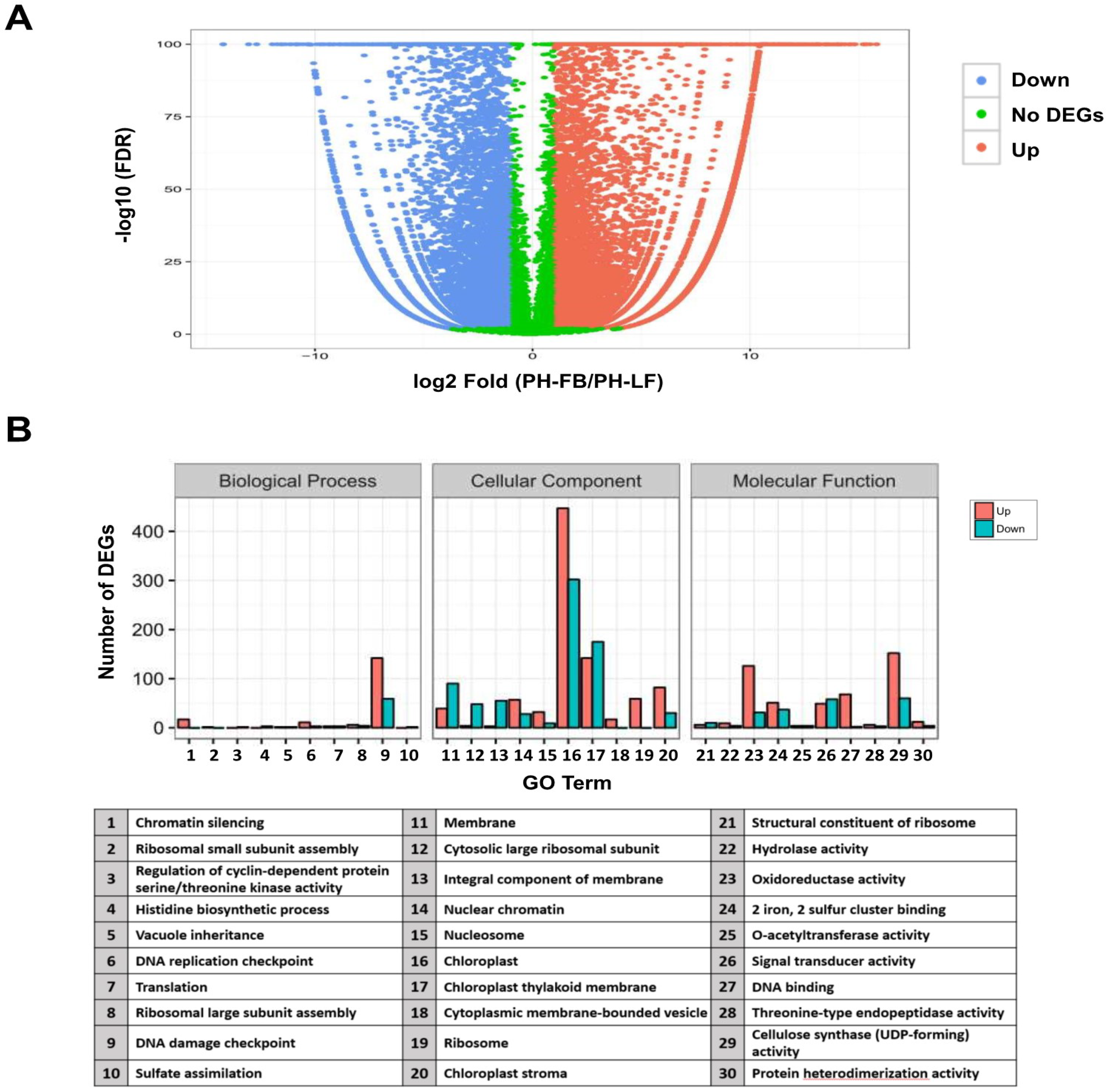
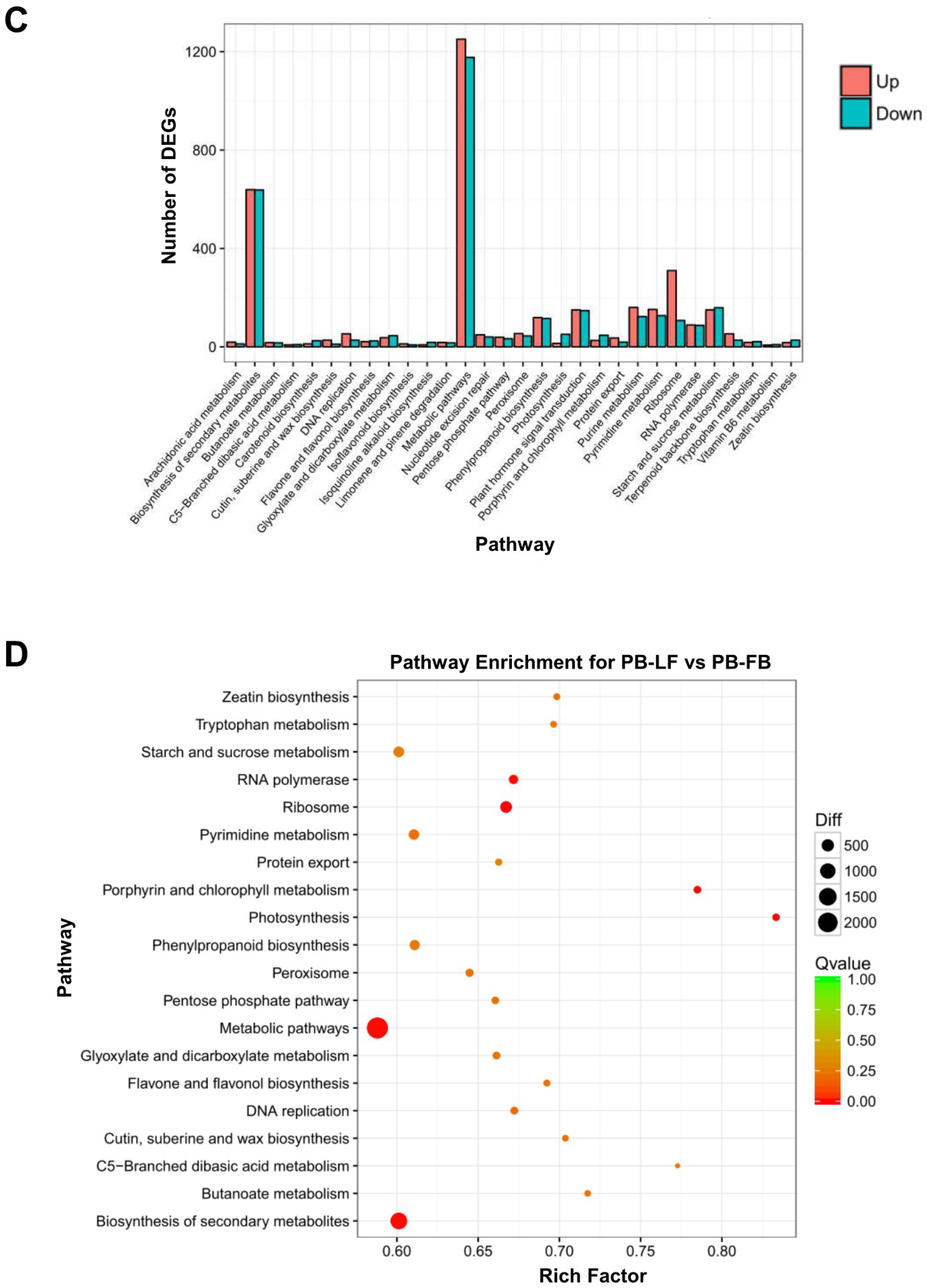
2.2. Analysis of Differential Gene Expression in Developing Floral Buds and Vegetative Leaves in L. formosanum Transcriptome
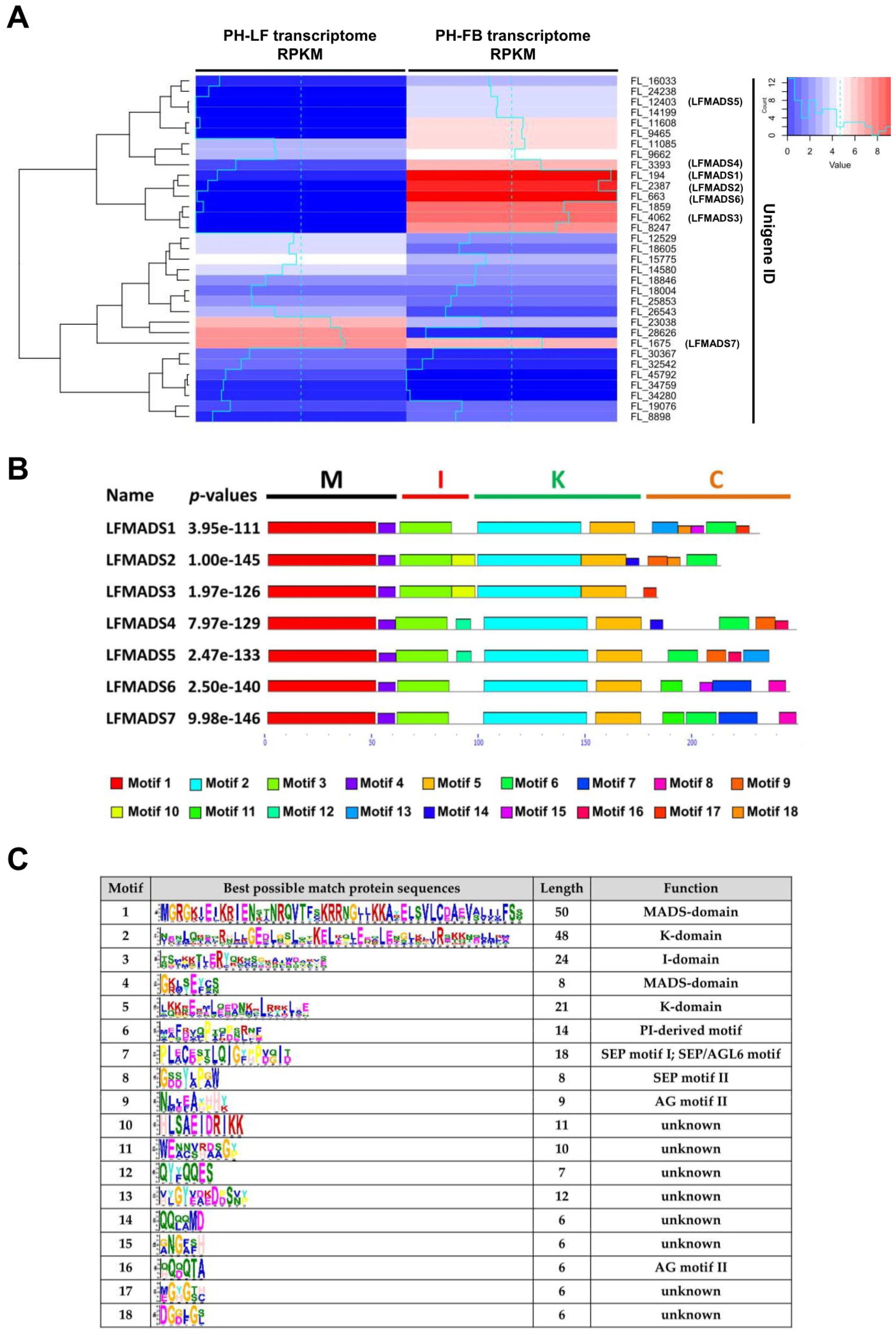
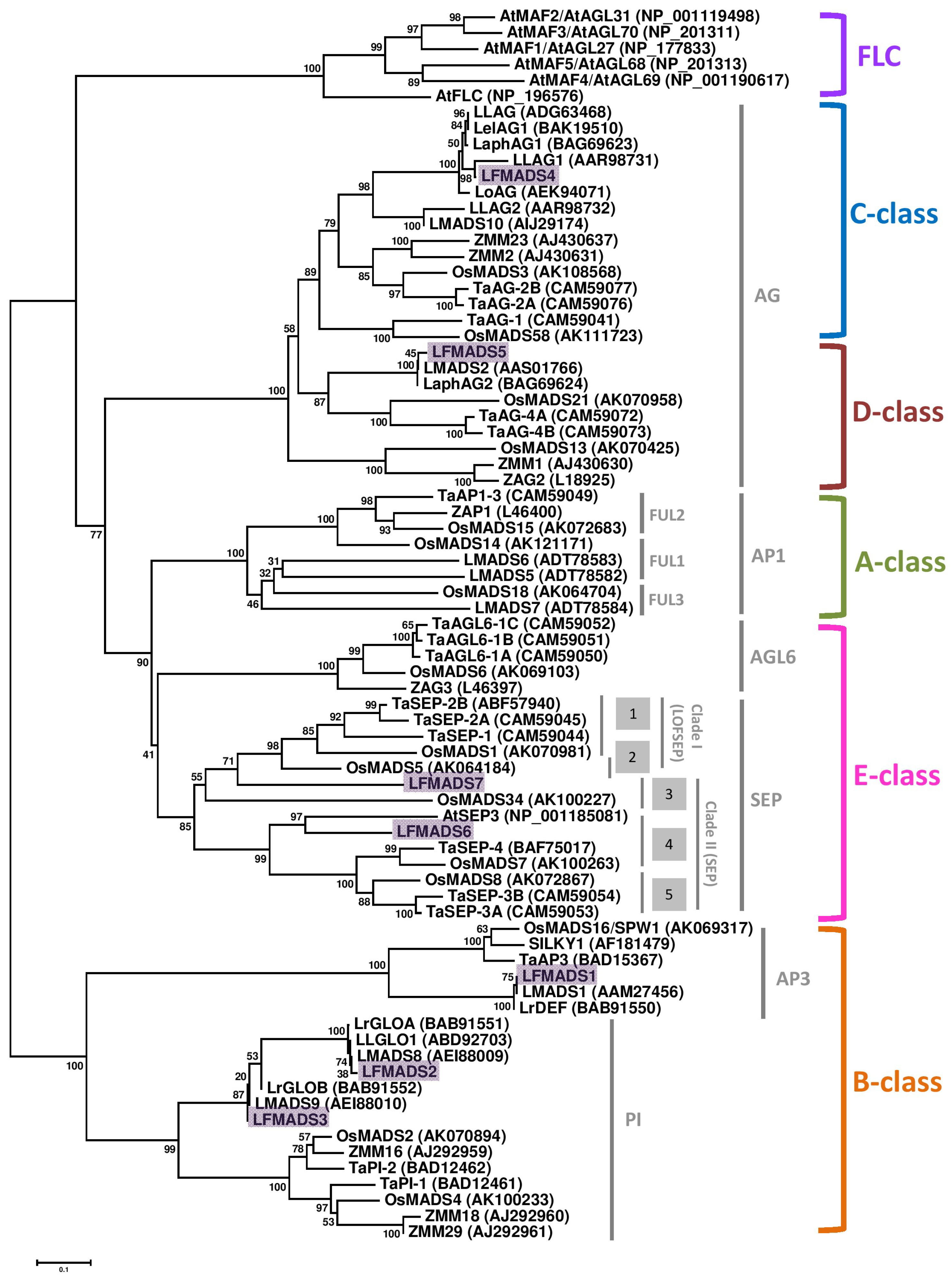
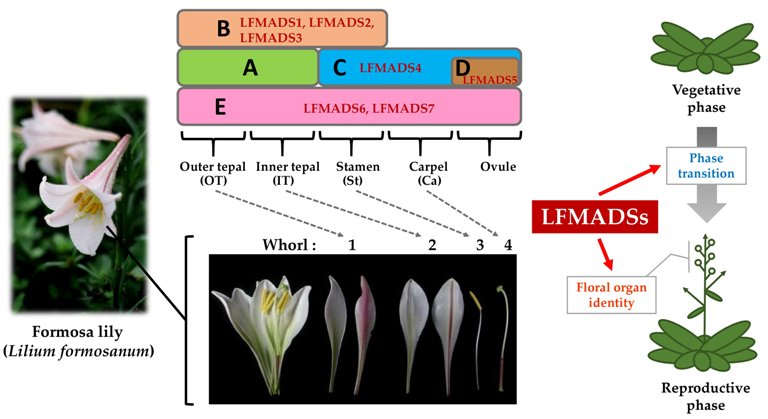
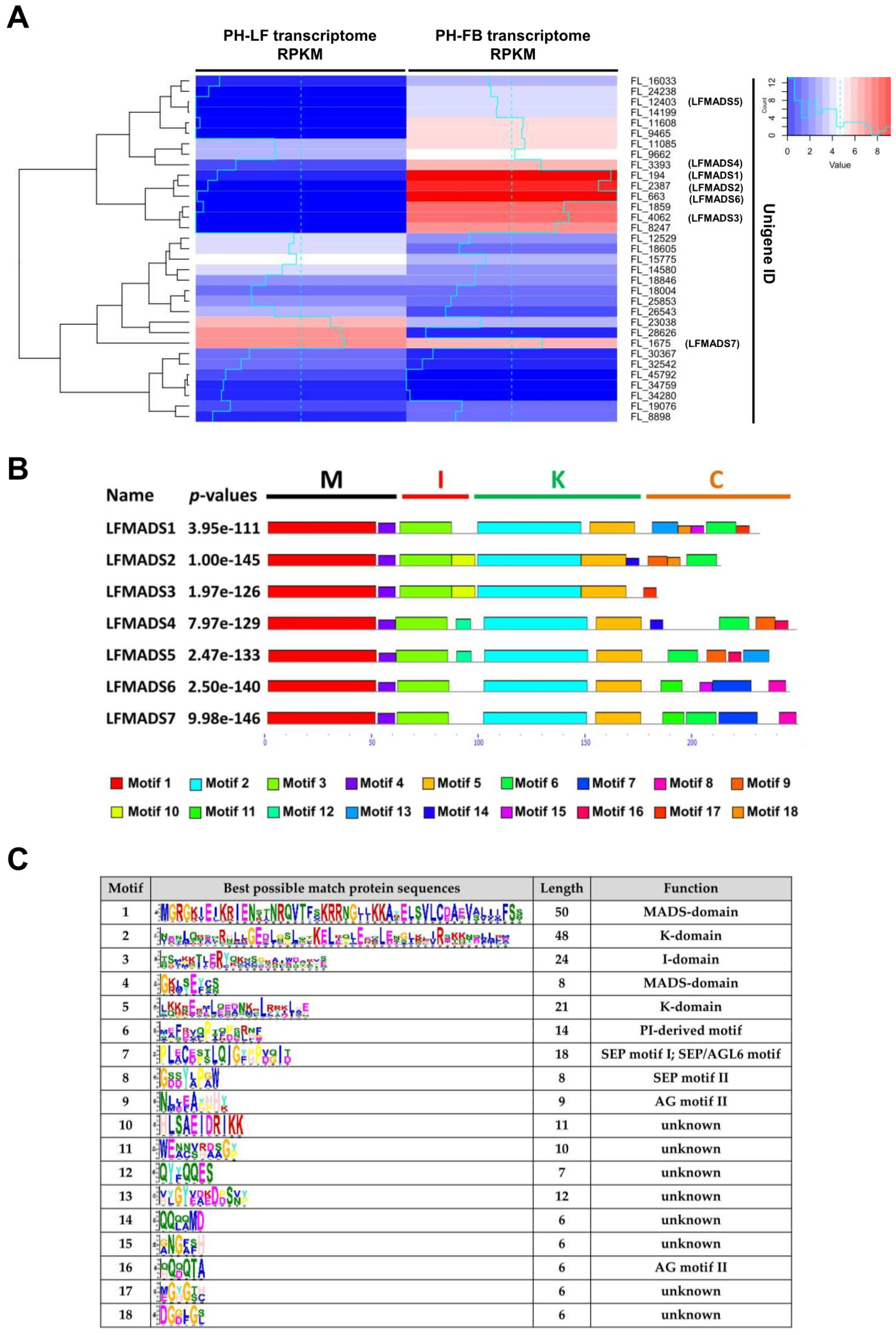
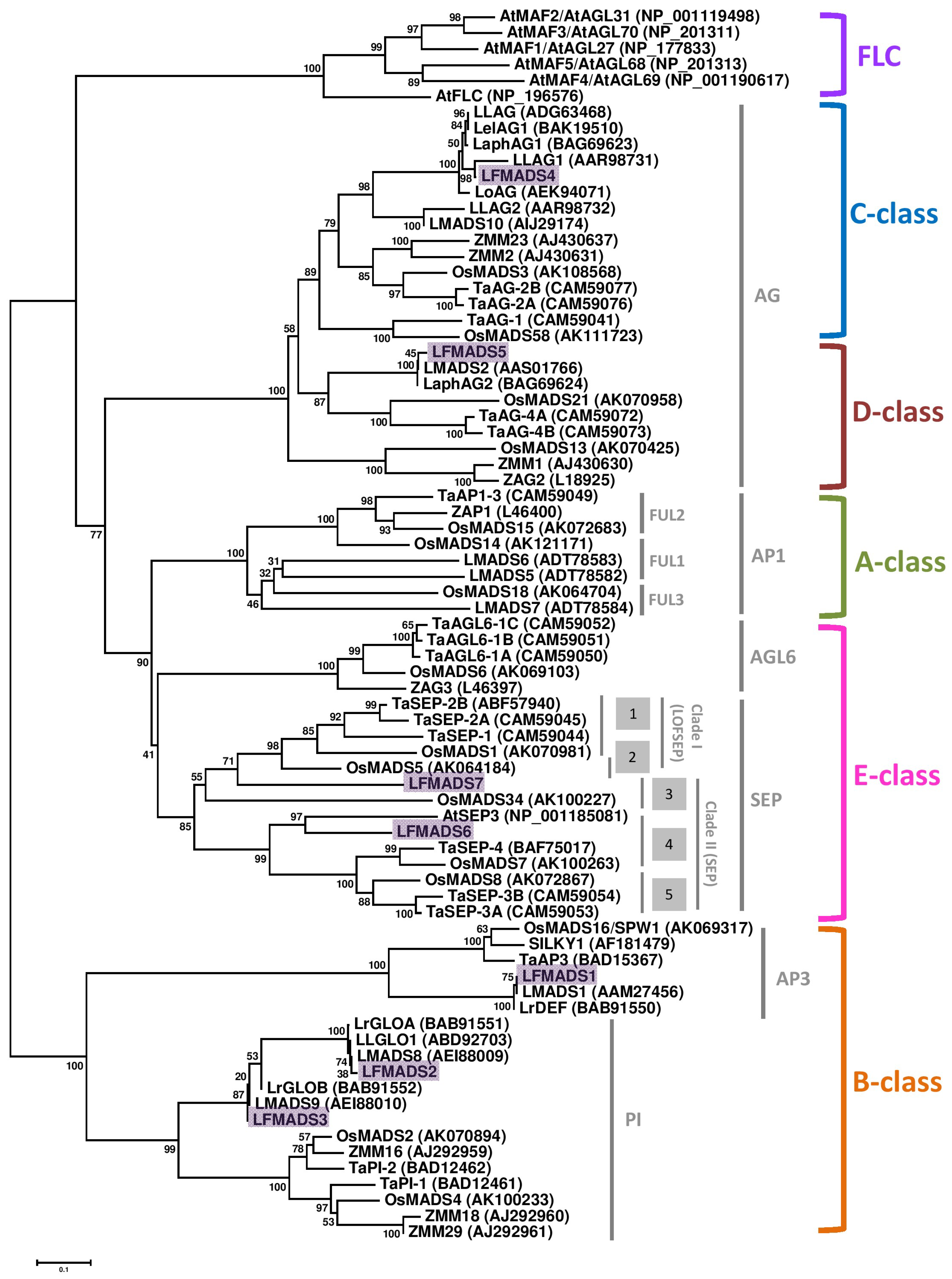
2.3. Identifying LFMADS Genes in L. formosanum Transcriptome
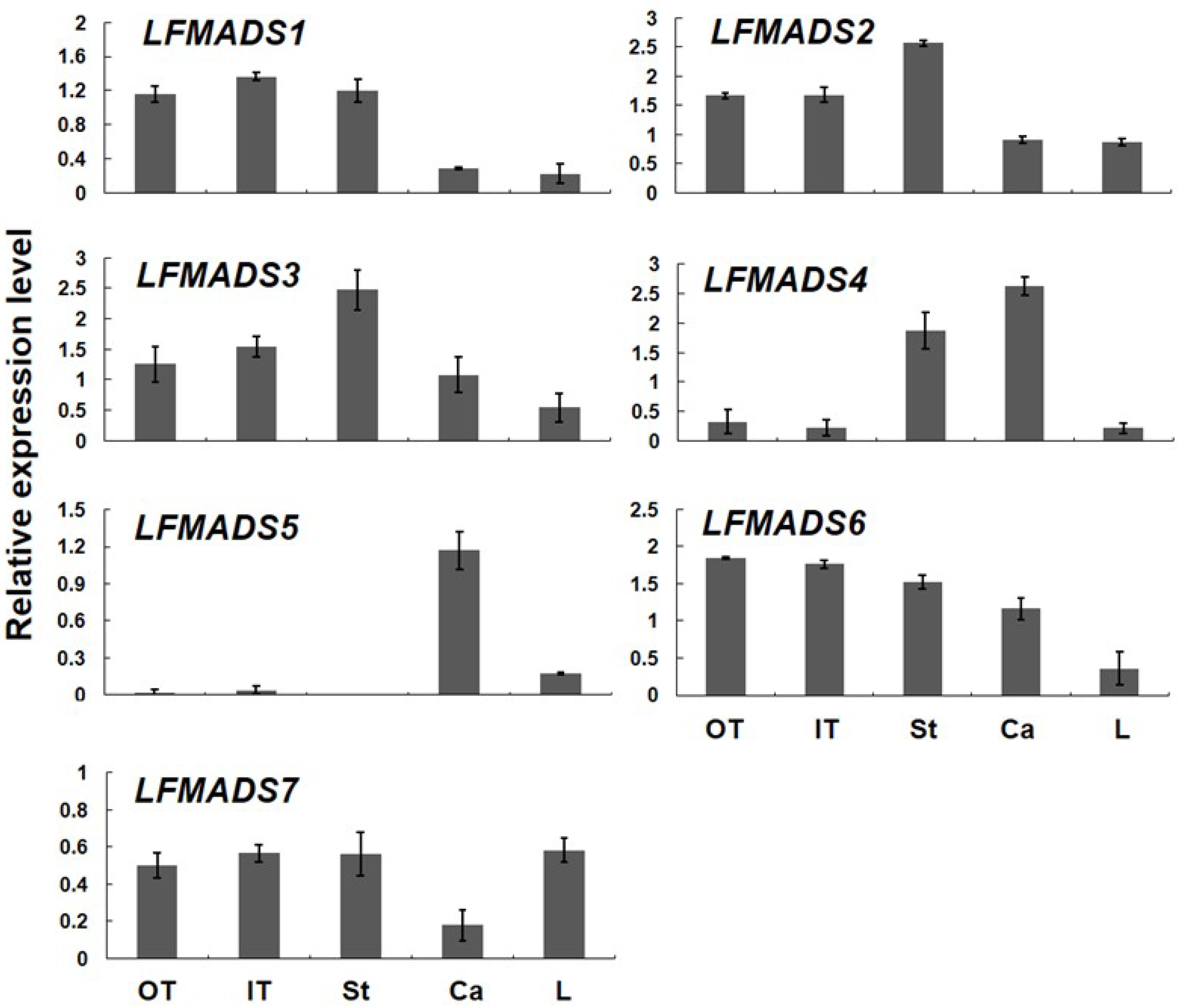
2.4. Expression Patterns of Seven MADS-Box Genes of L. formosanum
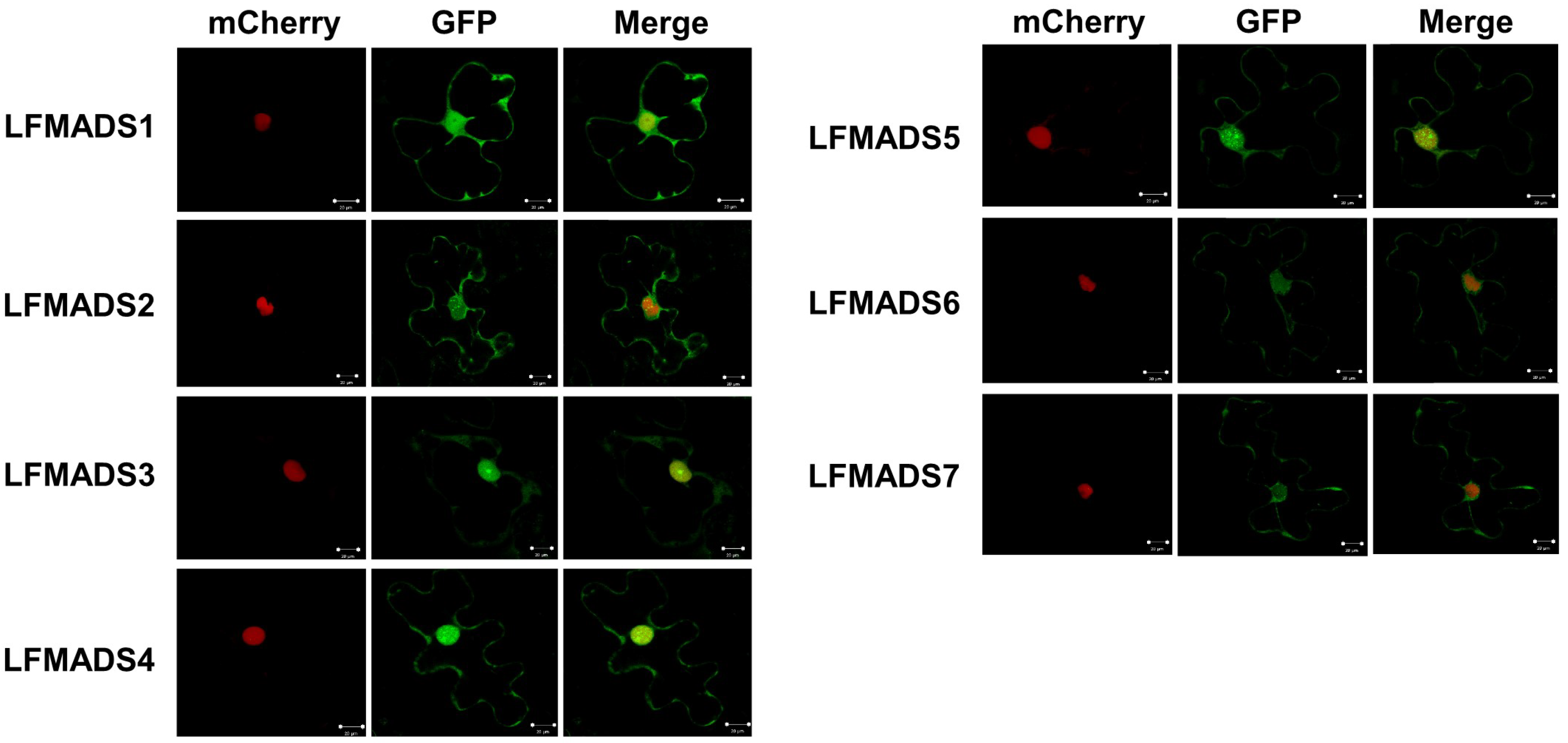
2.5. Nuclear Localization of Seven lily MADS-Box Proteins
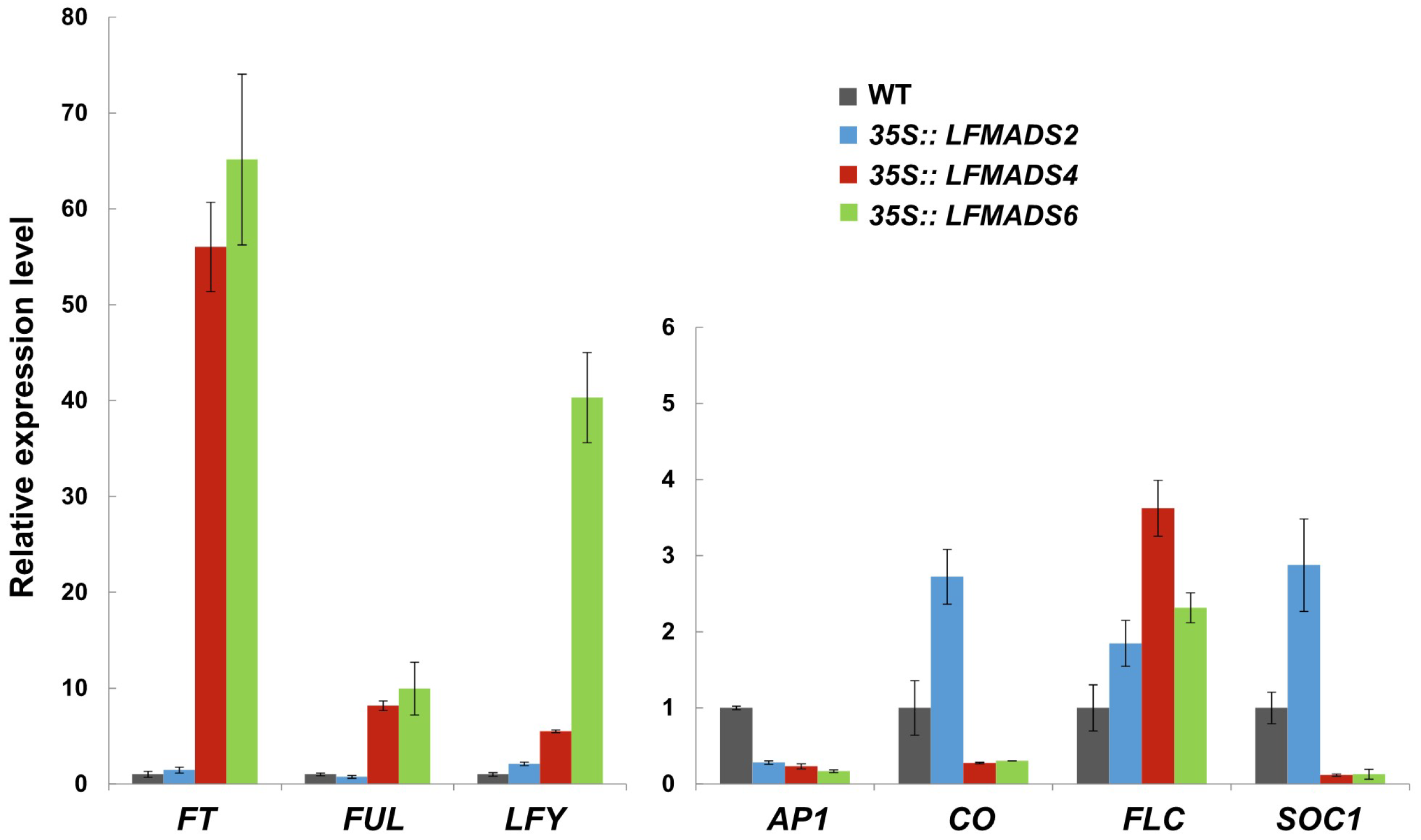
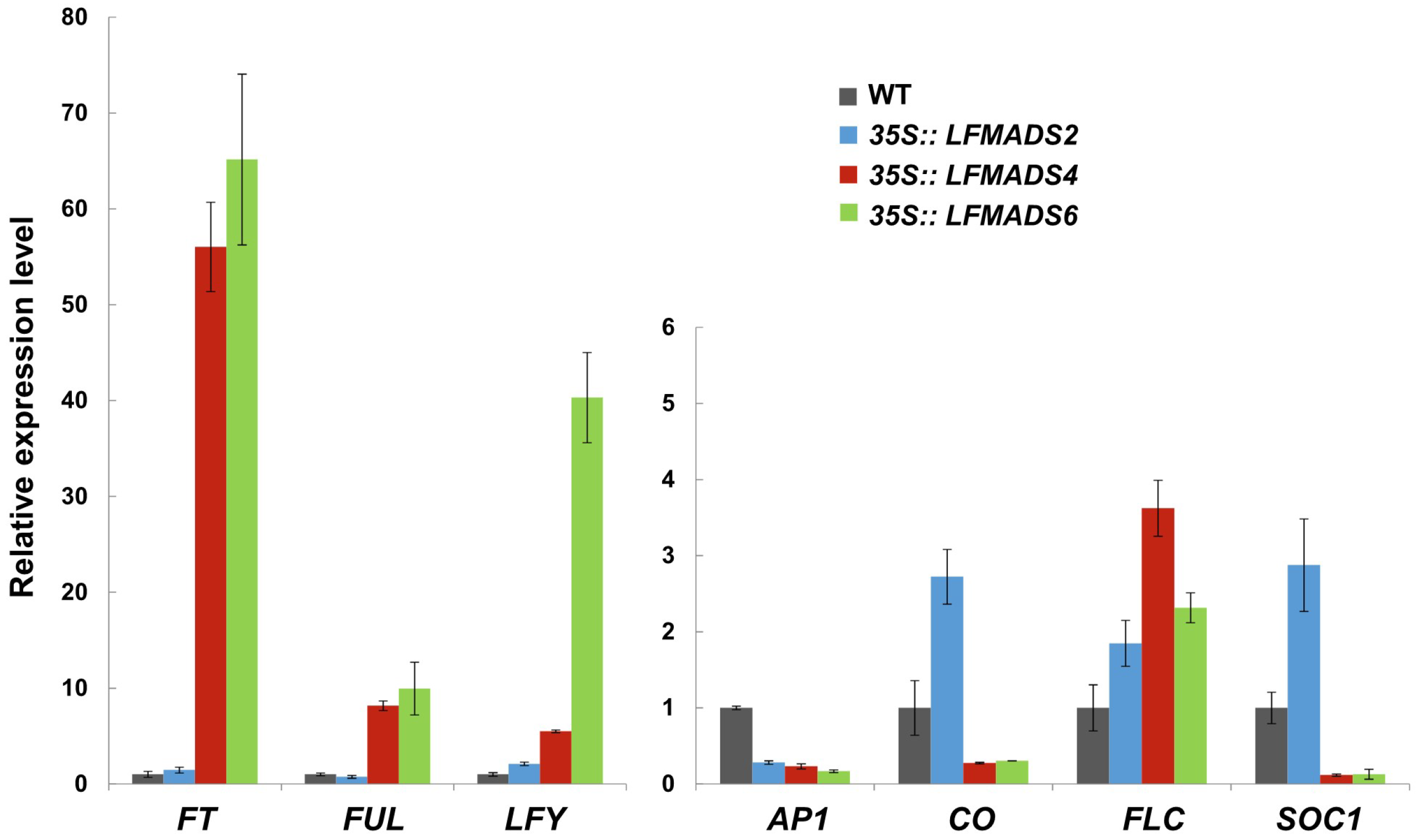
2.6. Ectopic Expression of LFMADS2, LFMADS4, and LFMADS6 Cause Floral Defects in Transgenic Arabidopsis, with LFMADS4 and LFMADS6, Further Causing Early Flowering
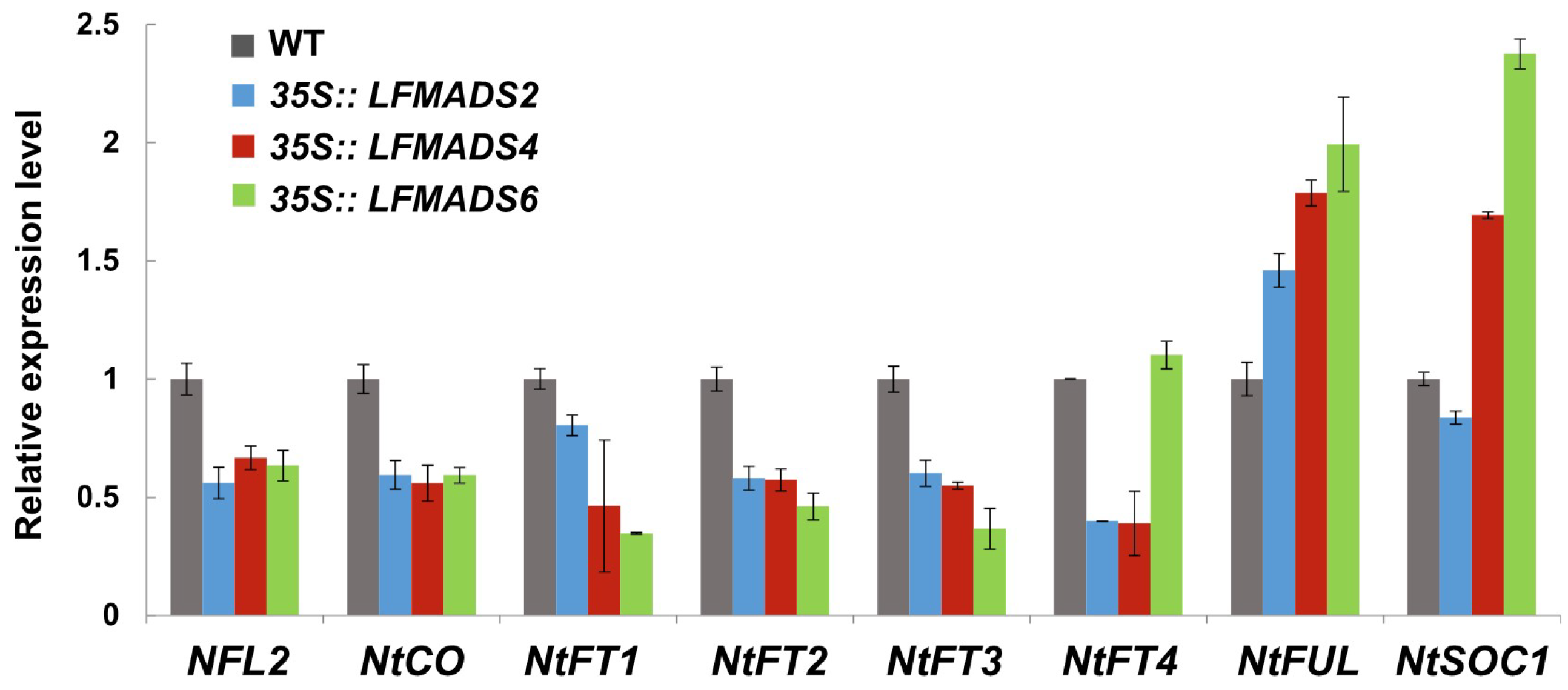
2.7. Overexpression of the LFMADS2, LFMADS4, and LFMADS6 Genes Induced Early Flowering in Transgenic Tobacco
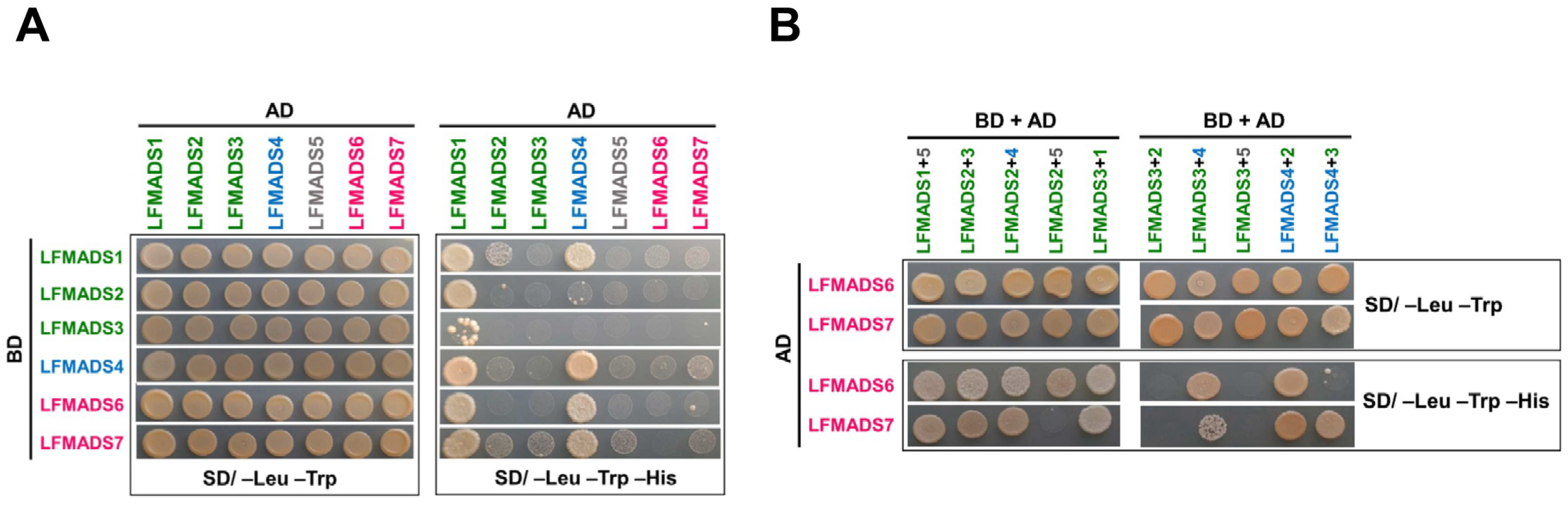
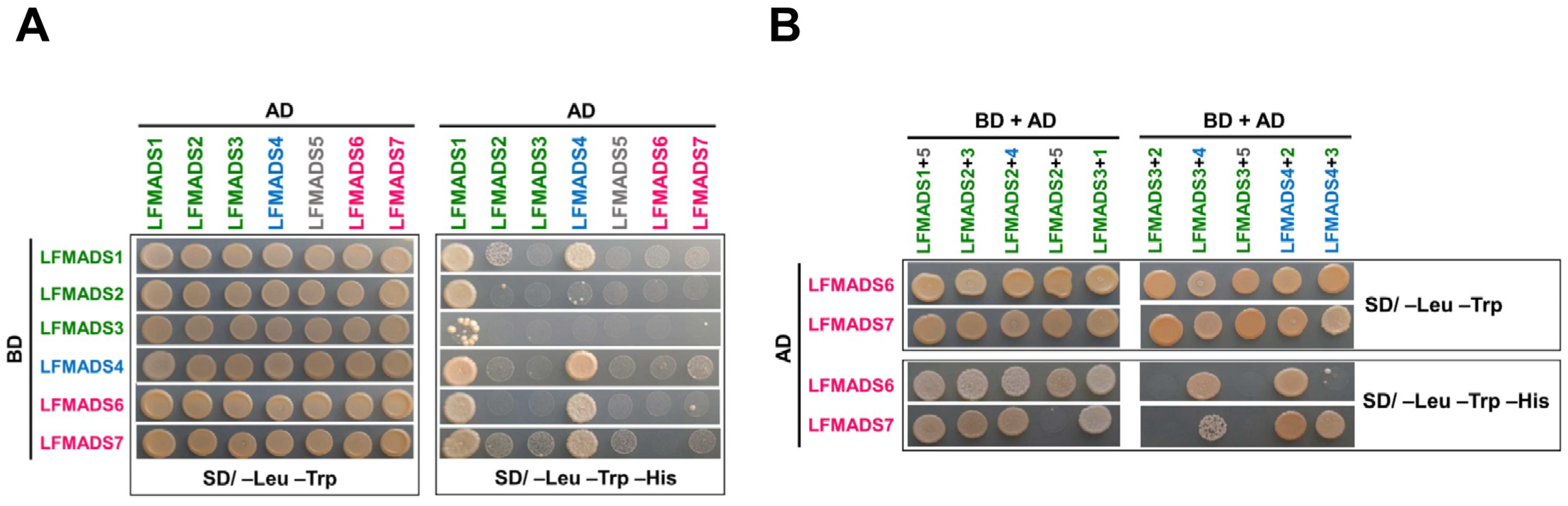
2.8. LFMADSs form Protein Complex with Various MADS-Box Proteins
3. Materials and Methods
3.1. Plant Materials and Growth Conditions
3.2. RNA Isolation, Illumina Sequencing, Sequence Annotation, and Differential Gene Expression (DGE) Analysis
3.3. Cloning of the cDNA for LFMADS Genes from the Formosa Lily
3.4. Sequence Alignment and Phylogenetic Analyses
3.5. RT-PCR and Real-Time Quantitative RT-PCR (qRT-PCR)
3.6. Genomic DNA-PCR and Genotype Analysis
3.7. Subcellular Localization of Green Fluorescent LFMADSs Fusion Proteins
3.8. Binary Vector Construction, Plant Transformation, and Analysis of Transgenic Plants
3.9. Scanning Electron Microscopy
3.10. Yeast-Two Hybrid Analysis and Spotting Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| DGE | differential gene expression |
| TF | transcription factor |
| NCBI | National Center for Biotechnology Information |
| SRA | Sequence Read Archive |
| FDR | False Discovery Rate |
| DEGs | differential expressed genes |
| SEM | scanning electron microscope |
References
- Weigel, D.; Nilsson, O. A developmental switch sufficient for flower initiation in diverse plants. Nature 1995, 377, 495–500. [Google Scholar] [CrossRef] [PubMed]
- Fitter, A.H.; Fitter, R.S. Rapid changes in flowering time in British plants. Science 2002, 296, 1689–1691. [Google Scholar] [CrossRef] [PubMed]
- Roldán, M.; Gómez-Mena, C.; Ruiz-García, L.; Salinas, J.; Martínez-Zapater, J.M. Sucrose availability on the aerial part of the plant promotes morphogenesis and flowering of Arabidopsis in the dark. Plant J. 1999, 20, 581–590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mouradov, A.; Cremer, F.; Coupleland, G. Control of flowering time: Interacting pathways as a basis of diversity. Plant Cell 2002, 14, S111–S130. [Google Scholar] [CrossRef] [PubMed]
- Blazquez, M.A.; Ahn, J.H.; Weigel, D. A thermosensory pathway controlling flowering time in Arabidopsis thaliana. Nat. Genet. 2003, 33, 168–171. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.V.; Lucyshyn, D.; Jaeger, K.E.; Alos, E.; Alvey, E.; Harberd, N.P.; Wigge, P.A. Transcription factor PIF4 controls the thermosensory activation of flowering. Nature 2012, 484, 242–245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kardailsky, I.; Shukla, V.K.; Ahn, J.H.; Dagenais, N.; Christensen, S.K.; Nguyen, J.T.; Chory, J.; Harrison, M.J.; Weigel, D. Activation tagging of the floral inducer FT. Science 1999, 286, 1962–1965. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, Y.; Kaya, H.; Goto, K.; Iwabuchi, M.; Araki, T. A pair of related genes with antagonistic roles in mediating flowering signals. Science 1999, 286, 1960–1962. [Google Scholar] [CrossRef] [PubMed]
- Moon, J.; Suh, S.S.; Lee, H.; Choi, K.R.; Hong, C.B.; Paek, N.C.; Kim, S.G.; Lee, I. The SOC1 MADS-box gene integrates vernalization and gibberellin signals for flowering in Arabidopsis. Plant J. 2003, 35, 613–623. [Google Scholar] [CrossRef] [PubMed]
- Corbesier, L.; Vincent, C.; Jang, S.; Fornara, F.; Fan, Q.; Searle, I.; Giakountis, A.; Farrona, S.; Gissot, L.; Turnbull, C.; Coupland, G. FT protein movement contributes to long-distance signaling in floral induction of Arabidopsis. Science 2007, 316, 1030–1033. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Oh, M.; Park, H.; Lee, I. SOC1 translocated to the nucleus by interaction with AGL24 directly regulates LEAFY. Plant J. 2008, 55, 832–843. [Google Scholar] [CrossRef] [PubMed]
- Messenguy, F.; Dubois, E. Role of MADS box proteins and their cofactors in combinatorial control of gene expression and cell development. Gene 2003, 316, 1–21. [Google Scholar] [CrossRef]
- Alvarez-Buylla, E.R.; Pelaz, S.; Liljegren, S.J.; Gold, S.E.; Burgeff, C.; Ditta, G.S.; Ribas de Pouplana, L.; Martinez-Castilla, L.; Yanofsky, M.F. An ancestral MADS-box gene duplication occurred before the divergence of plants and animals. Proc. Natl. Acad. Sci. USA 2000, 97, 5328–5333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krizek, B.A.; Fletcher, J.C. Molecular mechanisms of flower development: An armchair guide. Nat. Rev. Genet. 2005, 6, 688–698. [Google Scholar] [CrossRef] [PubMed]
- Theissen, G.; Melzer, R. Molecular mechanisms underlying origin and diversification of the angiosperm flower. Ann. Bot. 2007, 100, 603–619. [Google Scholar] [CrossRef] [PubMed]
- Egea-Cortines, M.; Saedler, H.; Sommer, H. Ternary complex formation between the MADS-box proteins SQUAMOSA, DEFICIENS and GLOBOSA is involved in the control of floral architecture in Antirrhinum majus. EMBO J. 1999, 18, 5370–5379. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, K.; Melzer, R.; Theissen, G. MIKC-type MADS-domain proteins: Structural modularity, protein interactions and network evolution in land plants. Gene 2005, 347, 183–198. [Google Scholar] [CrossRef] [PubMed]
- Parenicova, L.; de Folter, S.; Kieffer, M.; Horner, D.S.; Favalli, C.; Busscher, J.; Cook, H.E.; Ingram, R.M.; Kater, M.M.; Davies, B.; Angenent, G.C.; Colombo, L. Molecular and phylogenetic analyses of the complete MADS-box transcription factor family in Arabidopsis: New openings to the MADS world. Plant Cell 2003, 15, 1538–1551. [Google Scholar] [CrossRef] [PubMed]
- Arora, R.; Agarwal, P.; Ray, S.; Singh, A.K.; Singh, V.P.; Tyagi, A.K.; Kapoor, S. MADS-box gene family in rice: Genome-wide identification, organization and expression profiling during reproductive development and stress. BMC Genomics. 2007, 8, 242. [Google Scholar] [CrossRef] [PubMed]
- Pelaz, S.; Ditta, G.S.; Baumann, E.; Wisman, E.; Yanofsky, M.F. B and C floral organ identity functions require SEPALLATA MADS-box genes. Nature 2000, 405, 200–203. [Google Scholar] [CrossRef] [PubMed]
- Pelaz, S.; Tapia-López, R.; Alvarez-Buylla, E.R.; Yanofsky, M.F. Conversion of leaves into petals in Arabidopsis. Curr. Biol. 2001, 11, 182–184. [Google Scholar] [CrossRef] [Green Version]
- Theissen, G. Development of floral organ identity: Stories from the MADS house. Curr. Opin. Plant. Biol. 2001, 4, 75–85. [Google Scholar] [CrossRef]
- Ditta, G.; Pinyopich, A.; Robles, P.; Pelaz, S.; Yanofsky, M.F. The SEP4 gene of Arabidopsis thaliana functions in floral organ and meristem identity. Curr. Biol. 2004, 14, 1935–1940. [Google Scholar] [CrossRef] [PubMed]
- Immink, R.G.H.; Kaufmann, K.; Angement, G.C. The “ABC” of MADS domain protein behaviour and interactions. Semin. Cell Dev. Biol. 2010, 21, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Malcomber, S.T.; Kellogg, E.A. SEPALLATA gene diversification: Brave new whorls. Trends Plant Sci. 2005, 10, 427–435. [Google Scholar] [CrossRef] [PubMed]
- Honma, T.; Goto, K. Complexes of MADS-box proteins are sufficient to convert leaves into floral organs. Nature 2001, 409, 525–529. [Google Scholar] [CrossRef] [PubMed]
- Kanno, A.; Nakada, M.; Akita, Y.; Hirai, M. Class B gene expression and the modified ABC model in nongrass monocots. Sci. World J. 2007, 7, 268–279. [Google Scholar] [CrossRef] [PubMed]
- Tang, Q.; Ma, X.J.; Mo, C.M.; Wilson, I.W.; Song, C. An efficient approach to finding Siraitia grosvenorii triterpene biosynthetic genes by RNA-seq and digital gene expression analysis. BMC Genomics 2011, 12, 343. [Google Scholar] [CrossRef] [PubMed]
- Surget-Groba, Y.; Montoya-Burgos, J.I. Optimization of de novo transcriptome assembly from next-generation sequencing data. Genome Res. 2011, 20, 1432–1440. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Tzeng, T.Y.; Yang, C.H. A MADS box gene from lily (Lilium Longiflorum) is sufficient to generate dominant negative mutation by interacting with PISTILLATA (PI) in Arabidopsis thaliana. Plant Cell Physiol. 2001, 42, 1156–1168. [Google Scholar] [CrossRef] [PubMed]
- Tzeng, T.Y.; Chen, H.Y.; Yang, C.H. Ectopic expression of carpel-specific MADS box genes from lily and lisianthus causes similar homeotic conversion of sepal and petal in Arabidopsis. Plant Physiol. 2002, 130, 1827–1836. [Google Scholar] [CrossRef] [PubMed]
- Tzeng, T.Y.; Hsiao, C.C.; Chi, P.J.; Yang, C.H. Two lily SEPALLATA-like genes cause different effects on floral formation and floral transition in Arabidopsis. Plant Physiol. 2003, 133, 1091–1101. [Google Scholar] [CrossRef] [PubMed]
- Tzeng, T.Y.; Liu, H.C.; Yang, C.H. The C-terminal sequence of LMADS1 is essential for the formation of homodimers for B function proteins. J Biol Chem. 2004, 279, 10747–10755. [Google Scholar] [CrossRef] [PubMed]
- Benedito, V.A.; Visser, P.B.; van Tuyl, J.M.; Angenent, G.C.; de Vries, S.C.; Krens, F.A. Ectopic expression of LLAG1, an AGAMOUS homologue from lily (Lilium longiflorum Thunb.) causes floral homeotic modifications in Arabidopsis. J. Exp. Bot. 2004, 55, 1391–1399. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.K.; Lin, I.C.; Yang, C.H. Functional analysis of three lily (Lilium longiflorum) APETALA1-like MADS box genes in regulating floral transition and formation. Plant Cell Physiol. 2008, 49, 704–717. [Google Scholar] [CrossRef] [PubMed]
- Thiruvengadam, M.; Yang, C.H. Ectopic expression of two MADS box genes from orchid (Oncidium Gower Ramsey) and lily (Lilium longiflorum) alters flower transition and formation in Eustoma grandiflorum. Plant Cell Rep. 2009, 28, 1463–1473. [Google Scholar] [CrossRef] [PubMed]
- Hsu, H.F.; Hsieh, W.P.; Chen, M.K.; Chang, Y.Y.; Yang, C.H. C/D class MADS box genes from two monocots, orchid (Oncidium Gower Ramsey) and lily (Lilium longiflorum), exhibit different effects on floral transition and formation in Arabidopsis thaliana. Plant Cell Physiol. 2010, 51, 1029–1045. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.K.; Hsieh, W.P.; Yang, C.H. Functional analysis reveals the possible role of the C-terminal sequences and PI motif in the function of lily (Lilium longiflorum) PISTILLATA (PI) orthologues. J. Exp. Bot. 2012, 63, 941–961. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.K.; Liu, C.C.; Chiang, T.Y.; Chen, M.T.; Chou, C.H.; Yeh, C.H. Characterization of expressed sequence tags from flower buds of alpine Lilium formosanum using a subtractive cDNA library. Plant Mol. Bio. Rep. 2011, 29, 88–97. [Google Scholar] [CrossRef]
- Huang, J.; Liu, X.; Wang, J.; Lü, Y. Transcriptomic analysis of Asiatic lily in the process of vernalization via RNA-seq. Mol. Biol. Rep. 2014, 41, 3839–3852. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wang, Q.; Gu, J.; Lü, Y. Vernalization of Oriental hybrid lily “Sorbonne”: Changes in physiology metabolic activity and molecular mechanism. Mol. Biol. Rep. 2014, 41, 6619–6634. [Google Scholar] [CrossRef] [PubMed]
- Hamo, M.L.-B.; Martin, C.V.; Zaccai, M. Characterization of expressed sequence tags from Lilium longiflorum in vernalized and non-vernalized bulbs. J. Plant Physiol. 2015, 173, 72–81. [Google Scholar] [CrossRef] [PubMed]
- Villacorta-Martin, C.; de Cáceres González, F.F.N.; de Haan, J.; Huijben, K.; Passarinho, P.; Hamo, M.L.B.; Zaccai, M. Whole transcriptome profiling of the vernalization process in Lilium longiflorum (cultivar White Heaven) bulbs. BMC Genomics 2015, 16, 1. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, H. Acceleration of flowering by night break and heating treatment for harvesting in April and May in Lilium x foromolongi cv. Hayachine. Hortic. Res. 2005, 4, 191–195. [Google Scholar] [CrossRef]
- Lazare, S.; Zaccai, M. Flowering pathway is regulated by bulb size in Lilium longiflorum (Easter lily). Plant Biol. J. 2016, 18, 577–584. [Google Scholar] [CrossRef] [PubMed]
- Leeggangers, H.A.C.F.; Rosilio-Brami, T.; Bigas-Nadal, J.; Rubin, N.; van Dijk, A.D.J.; Nunez de Caceres Gonzalez, F.F.; Saadon-Shitrit, S.; Nijveen, H.; Hilhorst, H.W.M.; Immink, R.G.H.; et al. Tulipa gesneriana and Lilium longiflorum PEBP genes and their putative roles in flowering time control. Plant Cell Physiol. 2018, 59, 90–106. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.F.; Zhao, Y.Q.; Zhang, M.; Jia, G.X.; Zaccai, M. Functional and evolutionary characterization of the CONSTANS-like family in Lilium x formolongi. Plant Cell Physiol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.F.; Zhang, M.F.; Zhang, M.; Jia, G.X. Analysis of global gene expression profiles during the flowering initiation process of Lilium × formolongi. Plant Mol. Biol. 2017, 94, 361–379. [Google Scholar] [CrossRef] [PubMed]
- Norman, C.; Runswick, M.; Pollock, R.; Treisman, R. Isolation and properties of cDNA clones encoding SRF, a transcription factor that binds to the c-fos serum response element. Cell 1988, 55, 989–1003. [Google Scholar] [CrossRef]
- De Bodt, S.; Maere, S.; Van de Peer, Y. Genome duplication and the origin of angiosperms. Trends Ecol. Evol. 2005, 20, 591–597. [Google Scholar] [CrossRef] [PubMed]
- Smaczniak, C.; Immink, R.G.; Angenent, G.C.; Kaufmann, K. Developmental and evolutionary diversity of plant MADS-domain factors: Insights from recent studies. Development 2012, 139, 3081–3098. [Google Scholar] [CrossRef] [PubMed]
- van Tunen, A.J.; Eikeboom, W.; Angenent, G.C. Floral organogenesis in Tulipa. Flow. Newsl. 1993, 16, 33–38. [Google Scholar]
- Kanno, A.; Saeki, H.; Kameya, T.; Saedler, H.; Theissen, G. Heterotopic expression of class B floral homeotic genes supports a modified ABC model for tulip (Tulipa gesneriana). Plant Mol. Biol. 2003, 52, 831–841. [Google Scholar] [CrossRef] [PubMed]
- Theissen, G.; Becker, A.; Di Rosa, A.; Kanno, A.; Kim, J.T.; Münster, T.; Winter, K.-U.; Saedler, H. A short history of MADS-box genes in plants. Plant Mol. Biol. 2000, 42, 115–149. [Google Scholar] [CrossRef] [PubMed]
- Winter, K.U.; Weiser, C.; Kaufmann, K.; Bohne, A.; Kirchner, C.; Kanno, A.; Saedler, H.; Theissen, G. Evolution of class B floral homeotic proteins: Obligate heterodimerization originated from homodimerization. Mol. Biol. Evol. 2002, 19, 587–596. [Google Scholar] [CrossRef] [PubMed]
- Pan, I.L.; McQuinn, R.; Giovannoni, J.J.; Irish, V.F. Functional diversification of AGAMOUS lineage genes in regulating tomato flower and fruit development. J. Exp. Bot. 2010, 61, 1795–1806. [Google Scholar] [CrossRef] [PubMed]
- Wei, B.; Liu, D.; Guo, J.; Leseberg, C.H.; Zhang, X.; Mao, L. Functional divergence of two duplicated D-lineage MADS-box genes BdMADS2 and BdMADS4 from Brachypodium distachyon. J. Plant Physiol. 2013, 170, 424–431. [Google Scholar] [CrossRef] [PubMed]
- Tsaftaris, A.; Pasentsis, K.; Makris, A.; Darzentas, N.; Polidoros, A.; Kalivas, A.; Argiriou, A. The study of the E-class SEPALLATA3-like MADS-box genes in wild-type and mutant flowers of cultivated saffron crocus (Crocus sativus L.) and its putative progenitors. J. Plant Physiol. 2011, 168, 1675–1684. [Google Scholar] [CrossRef] [PubMed]
- Paolacci, A.R.; Tanzarella, O.A.; Porceddu, E.; Varotto, S.; Ciaffi, M. Molecular and phylogenetic analysis of MADS-box genes of MIKC type and chromosome location of SEP-like genes in wheat (Triticum aestivum L.). Mol. Genet. Genomics 2007, 278, 689–708. [Google Scholar] [CrossRef] [PubMed]
- Immink, R.G.; Gadella, T.W., Jr.; Ferrario, S.; Busscher, M.; Angenent, G.C. Analysis of MADS box protein-protein interactions in living plant cells. Proc. Natl. Acad. Sci. USA. 2002, 99, 2416–2421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gramzow, L.; Theißen, G. A hitchhiker’s guide to the MADS world of plants. Genome Biol. 2010, 11, 214. [Google Scholar] [CrossRef] [PubMed]
- Shih, M.C.; Chou, M.L.; Yue, J.J.; Hsu, C.T.; Chang, W.J.; Ko, S.S.; Liao, D.C.; Huang, Y.T.; Chen, J.J.; Yuan, J.L.; Gu, X.P.; Lin, C.S. BeMADS1 is a key to delivery MADSs into nucleus in reproductive tissues- De novo characterization of Bambusa edulis transcriptome and study of MADS genes in bamboo floral development. BMC Plant Biol. 2014, 14, 179. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.S.; Hsu, C.T.; Liao, D.C.; Chang, W.J.; Chou, M.L.; Huang, Y.T.; Chen, J.J.W.; Ko, S.S.; Chan, M.T.; Shih, M.C. Transcriptome-wide analysis of the MADS-box gene family in the orchid Erycina pusilla. Plant Biotech. J. 2016, 14, 284–298. [Google Scholar] [CrossRef] [PubMed]
- Benfey, P.N.; Chua, N.H. The cauliflower mosaic virus 35S promoter: Combinatorial regulation of transcription in plants. Science 1990, 250, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, N.; Wada, M.; Komori, S.; Bessho, H.; Suzuki, A. Functional analysis of MdPI, the PISTILLATA gene homologue of apple, in Arabidopsis. J. Jpn. Soc. Hort. Sci. 2007, 76, 125–132. [Google Scholar] [CrossRef]
- Nakamura, T.; Fukuda, T.; Nakano, M.; Hasebe, M.; Kameya, T.; Kanno, A. The modified ABC model explains the development of the petaloid perianth of Agapanthus praecox ssp. orientalis (Agapanthaceae) flowers. Plant Mol. Biol. 2005, 58, 435–445. [Google Scholar] [CrossRef] [PubMed]
- Jing, D.; Xia, Y.; Chen, F.; Wang, Z.; Zhang, S.; Wang, J. Ectopic expression of a Catalpa bungei (Bignoniaceae) PISTILLATA homologue rescues the petal and stamen identities in Arabidopsis pi-1 mutant. Plant Sci. 2015, 231, 40–51. [Google Scholar] [CrossRef] [PubMed]
- Mandel, M.A.; Bowman, J.L.; Kempin, S.A.; Ma, H.; Meyerowitz, E.M.; Yanofsky, M.F. Manipulation of flower structure in transgenic tobacco. Cell 1992, 71, 133–143. [Google Scholar] [CrossRef]
- Mizukami, Y.; Ma, H. Ectopic expression of the floral homeotic gene AGAMOUS in transgenic Arabidopsis plants alters floral organ identity. Cell 1992, 71, 119–131. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, X.; Liu, Z.; Zhang, D.; Wang, J.; Liu, D.; Li, F.; Lu, H. Isolation and characterization of an AGAMOUS-like gene from Hosta plantaginea. Mol. Biol. Rep. 2012, 39, 2875–2881. [Google Scholar] [CrossRef] [PubMed]
- Shitsukawa, N.; Tahira, C.; Kassai, K.; Hirabayashi, C.; Shimizu, T.; Takumi, S.; Mochida, K.; Kawaura, K.; Ogihara, Y.; Murai, K. Genetic and epigenetic alteration among three homoeologous genes of a class E MADS box gene in hexaploid wheat. Plant Cell 2007, 19, 1723–1737. [Google Scholar] [CrossRef] [PubMed]
- Qu, G.-Z.; Zheng, T.; Liu, G.; Wang, W.; Zang, L.; Liu, H.; Yang, C. Overexpression of a MADS-Box gene from Birch (Betula platyphylla) promotes flowering and enhances chloroplast development in transgenic tobacco. PLoS ONE 2013, 8, e63398. [Google Scholar]
- Qin, C.; Chen, W.; Shen, J.; Cheng, L.; Akande, F.; Zhang, K.; Yuan, C.; Li, C.; Zhang, P.; Shi, N.; et al. A virus-induced assay for functional dissection and analysis of monocot and dicot flowering time genes. Plant Physiol. 2017, 174, 875–885. [Google Scholar] [CrossRef] [PubMed]
- Harig, L.; Beinecke, F.A.; Oltmanns, J.; Muth, J.; Müller, O.; Rüping, B.; Twyman, R.M.; Fischer, R.; Prüfer, D.; Noll, G.A. Proteins from the FLOWERING LOCUS T-like subclade of the PEBP family act antagonistically to regulate floral initiation in tobacco. Plant J. 2012, 72, 908–921. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.; Xu, G.; Chi, Y.; Liu, H.; Xue, Q.; Zhao, T.; Gai, J.; Yu, D. A soybean MADS-box protein modulates floral organ numbers, petal identity and sterility. BMC Plant Biol. 2014, 14, 89. [Google Scholar] [CrossRef] [PubMed]
- Guo, B.; Hexige, S.; Zhang, T.; Pittman, J.K.; Chen, D.; Ming, F. Cloning and characterization of a PI-like MADS-box gene in Phalaenopsis orchid. J. Biochem. Mol. Biol. 2007, 40, 845–852. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Wang, Y.; Wang, Y.; Wang, L.; Chen, S.; Li, H. Identification of a sugar beet BvM14-MADS box gene through differential gene expression analysis of monosomic addition line M14. J. Plant Physiol. 2011, 168, 1980–1986. [Google Scholar] [CrossRef] [PubMed]
- Theißen, G.; Saedler, H. Floral quartets. Nature 2001, 409, 469–471. [Google Scholar] [CrossRef] [PubMed]
- Immink, R.G.; Tonaco, I.A.; de Folter, S.; Shchennikova, A.; van Dijk, A.D.; Busscher-Lange, J.; Borst, J.W.; Angenent, G.C. SEPALLATA3: The “glue” for MADS box transcription factor complex formation. Genome Biol. 2009, 10, 24. [Google Scholar] [CrossRef] [PubMed]
- Pan, Z.J.; Chen, Y.Y.; Du, J.S.; Chen, Y.Y.; Chung, M.C.; Tsai, W.C.; Wang, C.N.; Chen, H.H. Flower development of Phalaenopsis orchid involves functionally divergent SEPALLATA-like genes. New Phytol. 2014, 202, 1024–1042. [Google Scholar] [CrossRef] [PubMed]
- Murashige, T.; Skoog, F. A revised medium for rapid growth and bio-assays with tobacco tissue cultures. Physiol. Plant. 1962, 15, 473–497. [Google Scholar] [CrossRef]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Nagalakshmi, U.; Wang, Z.; Waern, K.; Shou, C.; Raha, D.; Gerstein, M. The transcriptional landscape of the yeast genome defined by RNA sequencing. Science 2008, 320, 1344–1349. [Google Scholar] [CrossRef] [PubMed]
- American Association for the Advancement of Science. A simple and general method for transferring genes into plants. Science 1985, 227, 1229–1231. [Google Scholar]
- Thompson, J.D.; Higgins, D.G.; Gibson, T.J. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994, 22, 4673–4680. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, T.; Bressan, D.A.; Shinohara, M.; Haber, J.E.; Shinohara, A. In vivo assembly and disassembly of Rad51 and Rad52 complexes during double-strand break repair. EMBO J. 2004, 23, 939–949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimura, M. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol. 1980, 16, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.T.; Liao, D.C.; Wu, F.H.; Liu, N.T.; Shen, S.C.; Chou, S.J.; Tung, S.Y.; Yang, C.H.; Chan, M.T.; Lin, C.S. Integration of molecular biology tools for identifying promoters and genes abundantly expressed in flowers of Oncidium Gower Ramsey. BMC Plant Biol. 2011, 11, 60. [Google Scholar] [CrossRef] [PubMed]
- Clough, S.J.; Bent, A.F. Floral dip: A simplified method for Agrobacterium-mediated transformation of Arabidopsis thaliana. Plant J. 1998, 16, 735–743. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | ALL Transcriptome Unigene ID | PH-FB Total Counts | PH-LF Total Counts | Gene Length (bp) | Number of Amino Acids | MW (kDa) 1 | Isoelectric Point 1 | Accession Number |
|---|---|---|---|---|---|---|---|---|
| LFMADS1 | FL_194 | 8071 | 13 | 687 | 228 | 26.18 | 8.68 | AHY82568 |
| LFMADS2 | FL_2387 | 6698 | 7 | 633 | 210 | 24.53 | 8.99 | AHY82569 |
| LFMADS3 | FL_4062 | 2144 | 0 | 546 | 181 | 20.98 | 9.14 | AHY82570 |
| LFMADS4 | FL_3393 | 1067 | 44 | 738 | 245 | 28.62 | 9.21 | AHY82571 |
| LFMADS5 | FL_12403 | 291 | 0 | 699 | 232 | 26.72 | 9.03 | AHY82572 |
| LFMADS6 | FL_663 | 15291 | 1 | 729 | 242 | 27.58 | 9.16 | AHY82573 |
| LFMADS7 | FL_1675 | 1219 | 1897 | 741 | 246 | 28.17 | 7.05 | AHY82574 |
| Plant Genotype 1 | Number of Plants (n) | Days to Flowering 2 |
|---|---|---|
| 35S::LFMADS2 | 45 | 28.8 ± 0.8 |
| 35S::LFMADS4 | 45 | 19.8 ± 0.8 |
| 35S::LFMADS6 | 45 | 24.0 ± 0.7 |
| Columbia wild-type | 45 | 29.2 ± 1.3 |
| Transgenic Tobacco Line | Number of Plants (n) | Flowering Time (days) |
|---|---|---|
| WT | 30 | 81.8 ± 0.8 |
| 35S::LFMADS2 | 30 | 45.2 ± 0.7 |
| 35S::LFMADS4 | 30 | 39.6 ± 0.5 |
| 35S::LFMADS6 | 30 | 41.6 ± 0.5 |
| B-Class | C-Class | D-Class | E-Class | ||||
|---|---|---|---|---|---|---|---|
| LFMADS1 1 | LFMADS2 | LFMADS3 | LFMADS4 | LFMADS5 | LFMADS6 | LFMADS7 | |
| LFMADS1 2 | +++++ 3 | ++ | – | ++++ | – | + | + |
| LFMADS2 | +++++ | – | – | – | – | – | – |
| LFMADS3 | +++ | – | – | – | – | – | – |
| LFMADS4 | +++++ | – | – | +++++ | – | – | + |
| LFMADS5 | n.d. 4 | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. |
| LFMADS6 | +++++ | – | – | ++++ | – | – | – |
| LFMADS7 | +++++ | + | + | ++++ | + | – | + |
| LFMADS 1 1+5 2 | LFMADS 2+3 | LFMADS 2+4 | LFMADS 2+5 | LFMADS 3+1 | |
|---|---|---|---|---|---|
| LFMADS6 3 | +++++ 4 | ++++ | ++++ | +++++ | +++++ |
| LFMADS7 | +++++ | +++++ | +++++ | – | +++++ |
| LFMADS 3+2 | LFMADS 3+4 | LFMADS 3+5 | LFMADS 4+2 | LFMADS 4+3 | |
| LFMADS6 | – | +++++ | – | +++++ | – |
| LFMADS7 | – | ++ | – | +++++ | +++++ |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liao, W.-Y.; Lin, L.-F.; Lin, M.-D.; Hsieh, S.-C.; Li, A.Y.-S.; Tsay, Y.-S.; Chou, M.-L. Overexpression of Lilium formosanum MADS-box (LFMADS) Causing Floral Defects While Promoting Flowering in Arabidopsis thaliana, Whereas Only Affecting Floral Transition Time in Nicotiana tabacum. Int. J. Mol. Sci. 2018, 19, 2217. https://doi.org/10.3390/ijms19082217
Liao W-Y, Lin L-F, Lin M-D, Hsieh S-C, Li AY-S, Tsay Y-S, Chou M-L. Overexpression of Lilium formosanum MADS-box (LFMADS) Causing Floral Defects While Promoting Flowering in Arabidopsis thaliana, Whereas Only Affecting Floral Transition Time in Nicotiana tabacum. International Journal of Molecular Sciences. 2018; 19(8):2217. https://doi.org/10.3390/ijms19082217
Chicago/Turabian StyleLiao, Wan-Yu, Lee-Fong Lin, Ming-Der Lin, Sheng-Che Hsieh, Althea Yi-Shan Li, Yueh-Shiah Tsay, and Ming-Lun Chou. 2018. "Overexpression of Lilium formosanum MADS-box (LFMADS) Causing Floral Defects While Promoting Flowering in Arabidopsis thaliana, Whereas Only Affecting Floral Transition Time in Nicotiana tabacum" International Journal of Molecular Sciences 19, no. 8: 2217. https://doi.org/10.3390/ijms19082217
APA StyleLiao, W.-Y., Lin, L.-F., Lin, M.-D., Hsieh, S.-C., Li, A. Y.-S., Tsay, Y.-S., & Chou, M.-L. (2018). Overexpression of Lilium formosanum MADS-box (LFMADS) Causing Floral Defects While Promoting Flowering in Arabidopsis thaliana, Whereas Only Affecting Floral Transition Time in Nicotiana tabacum. International Journal of Molecular Sciences, 19(8), 2217. https://doi.org/10.3390/ijms19082217