Mechanisms of PKA-Dependent Potentiation of Kv7.5 Channel Activity in Human Airway Smooth Muscle Cells


Abstract
:1. Introduction
2. Results
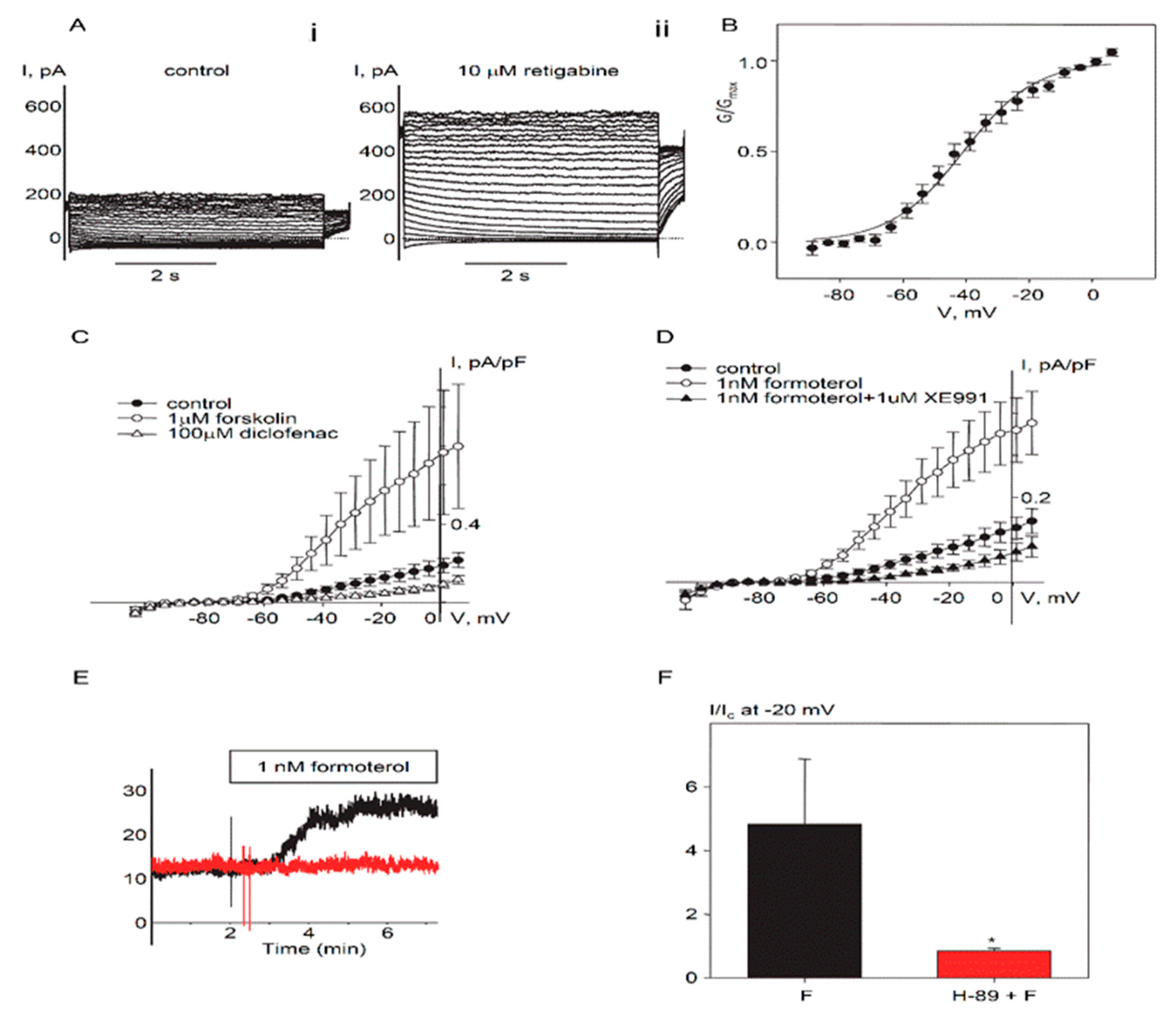
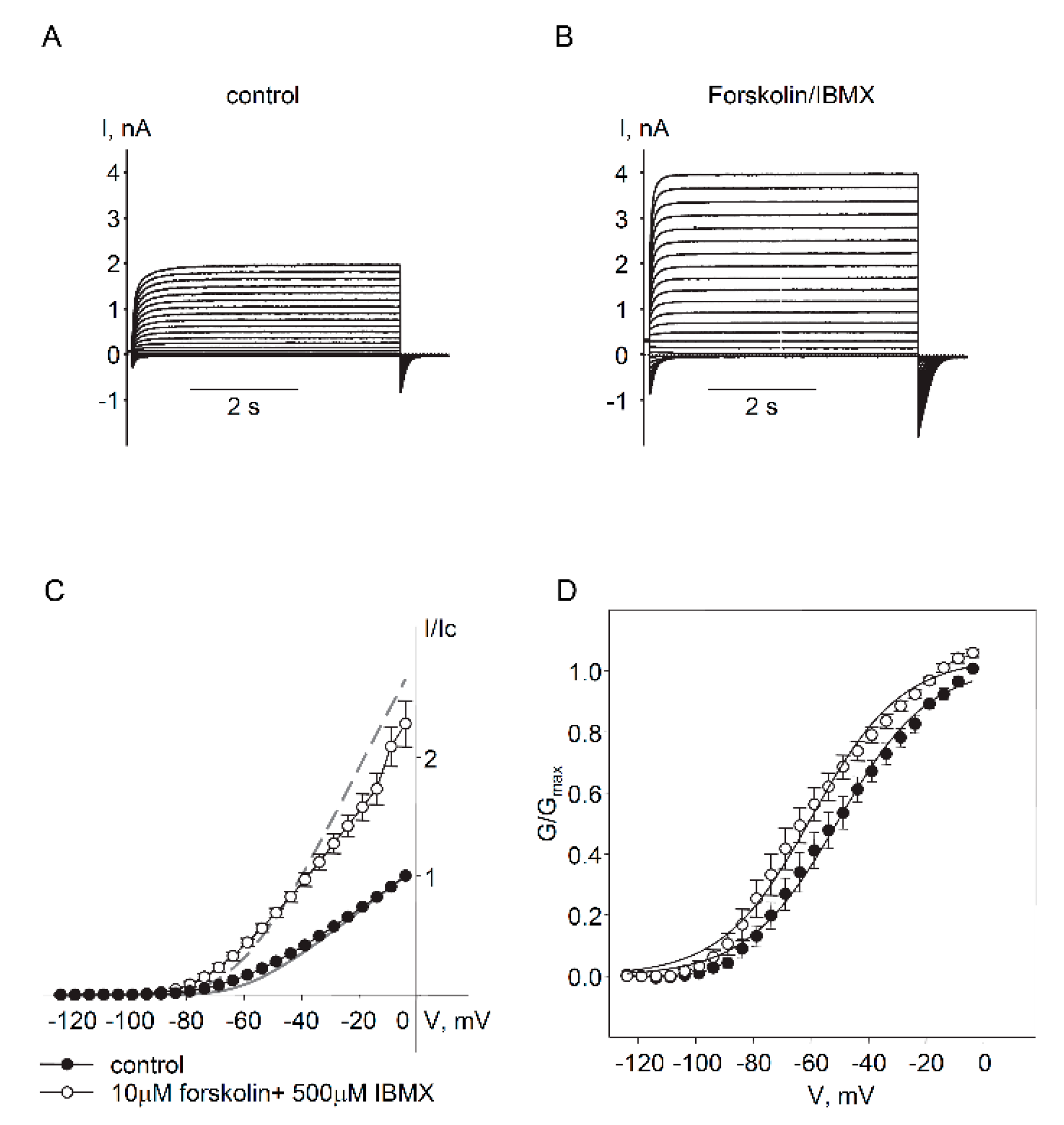
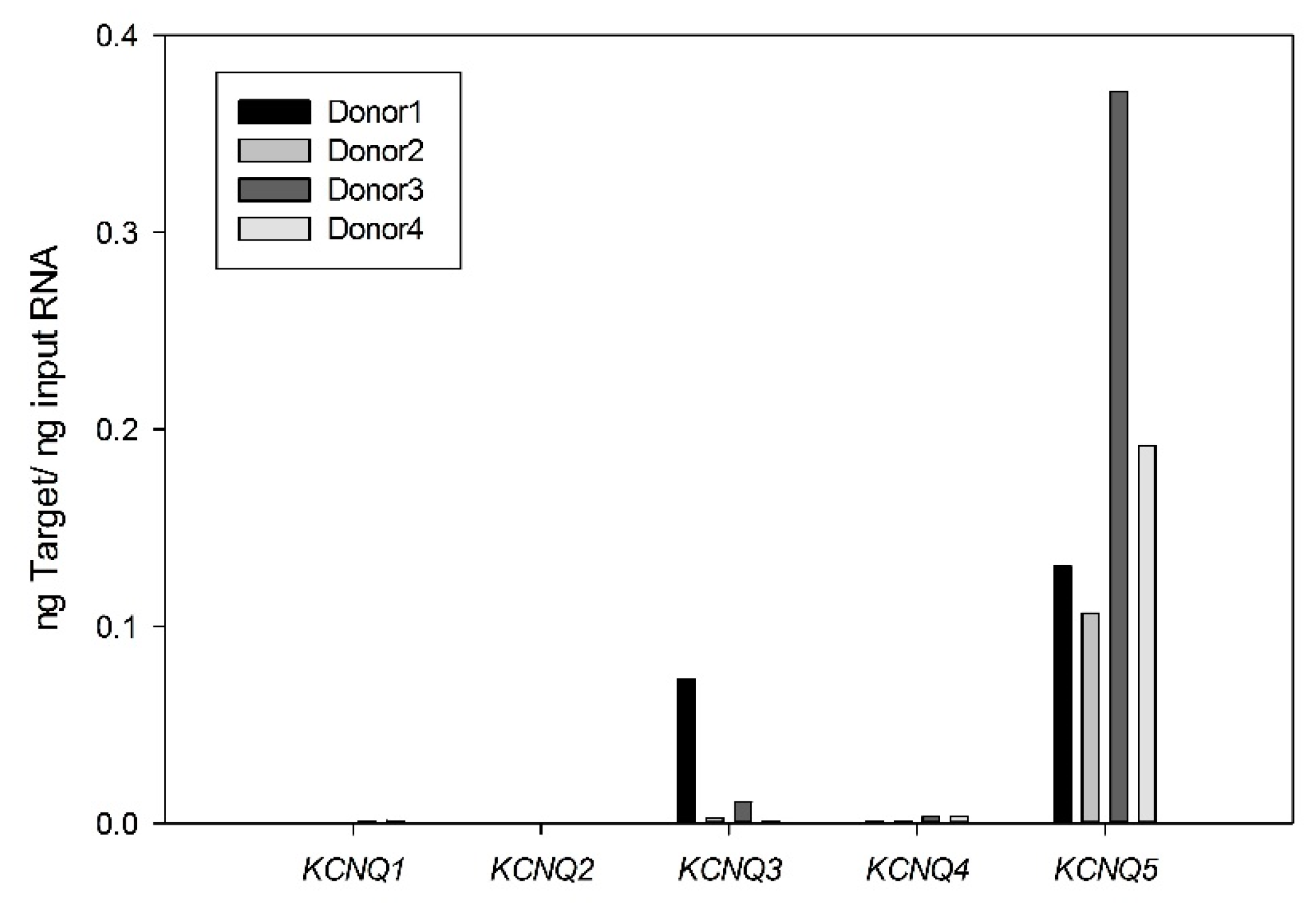
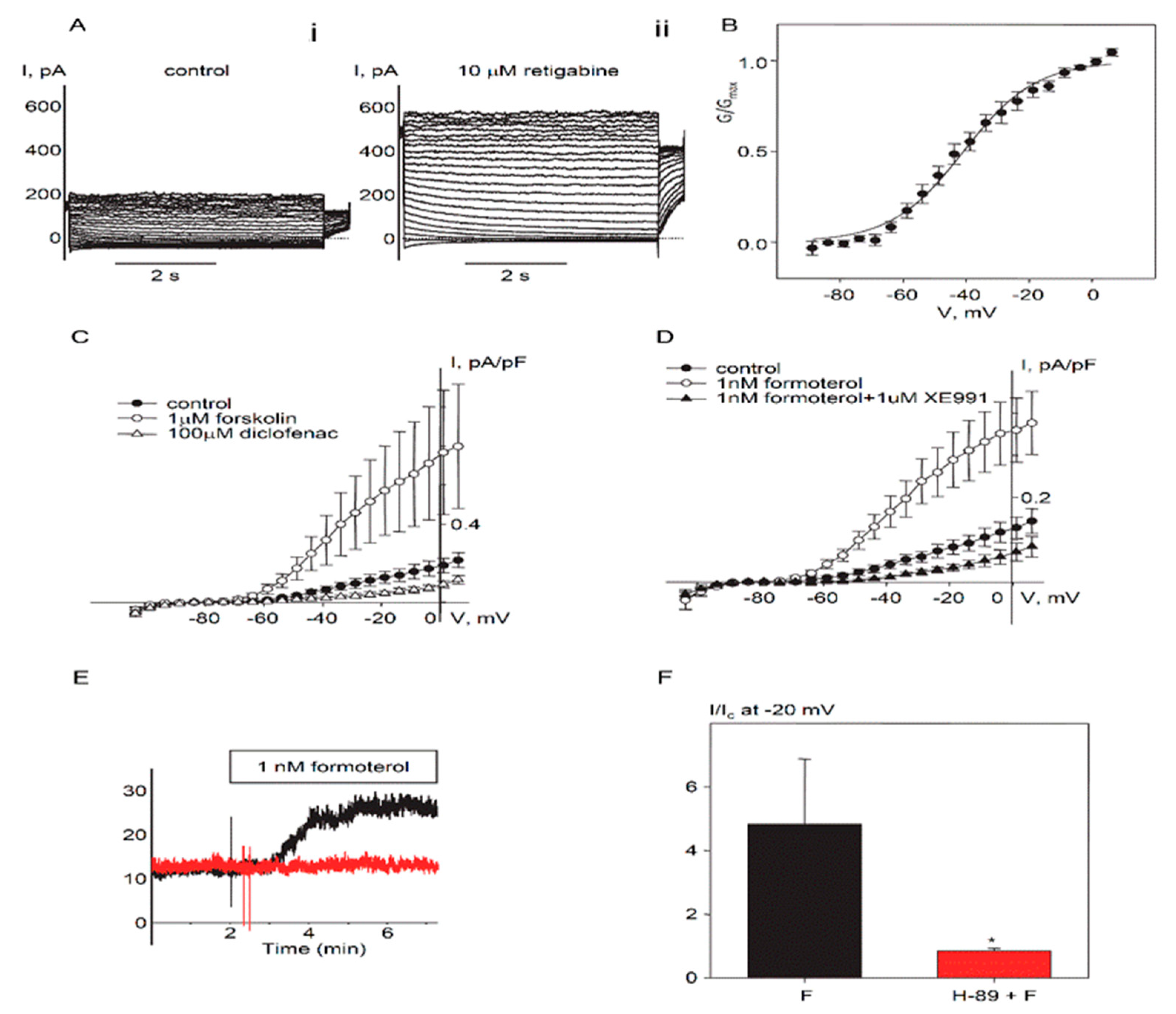
2.1. Expression and Functional Characteristics of Kv7 Channels in Cultured HASMCs
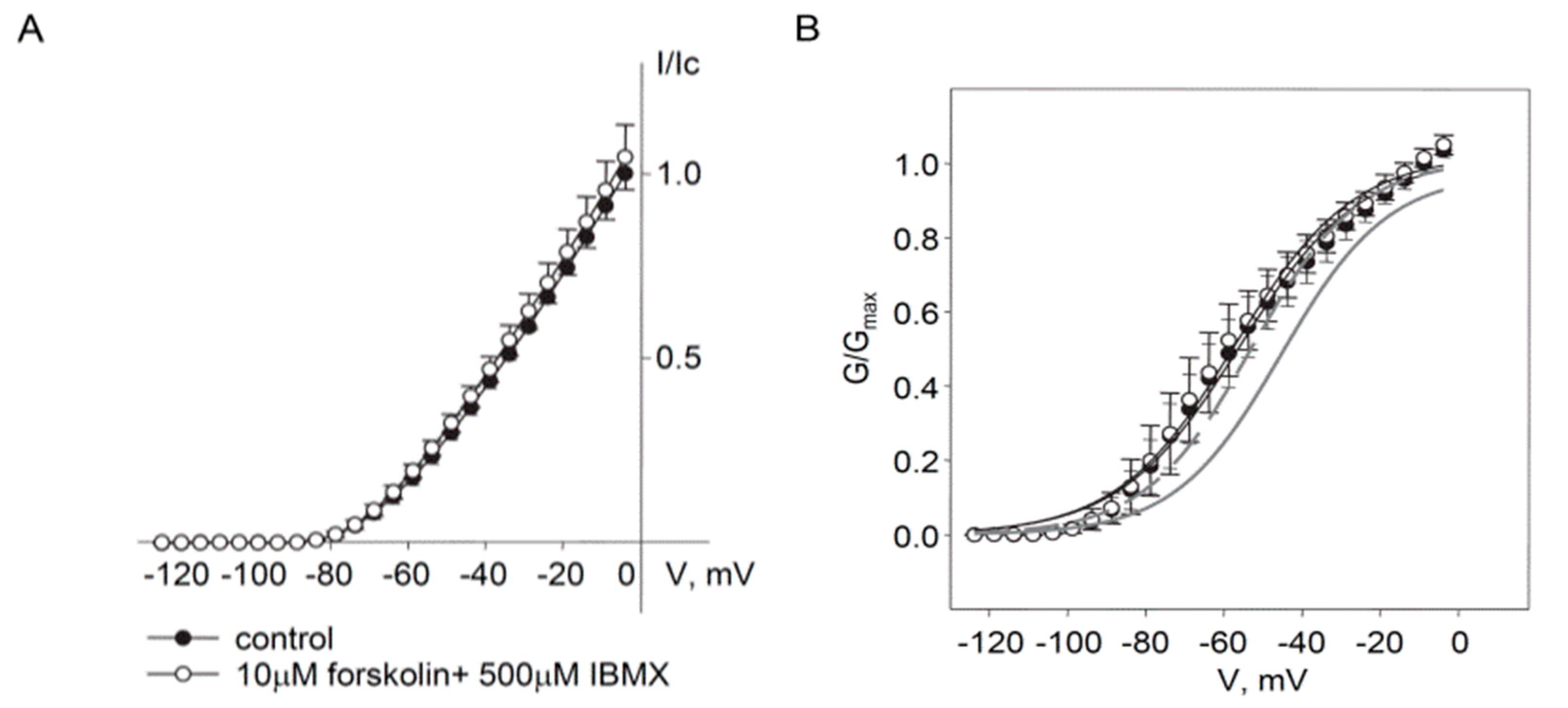
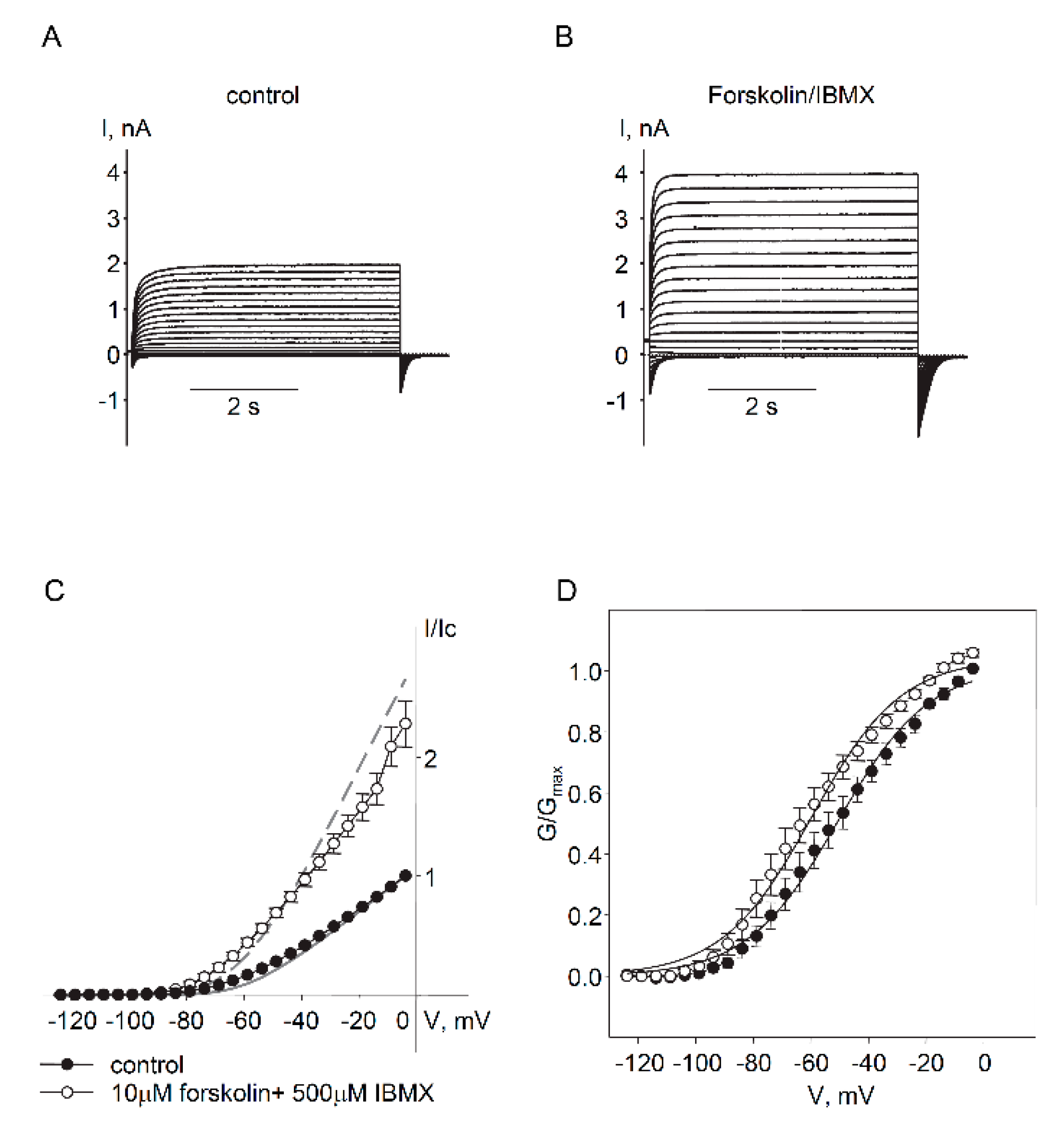
2.2. Regulation of Endogenous Kv7.5 Currents in Cultured HASMCs by β-Adrenergic/Gs/cAMP/PKA Pathway
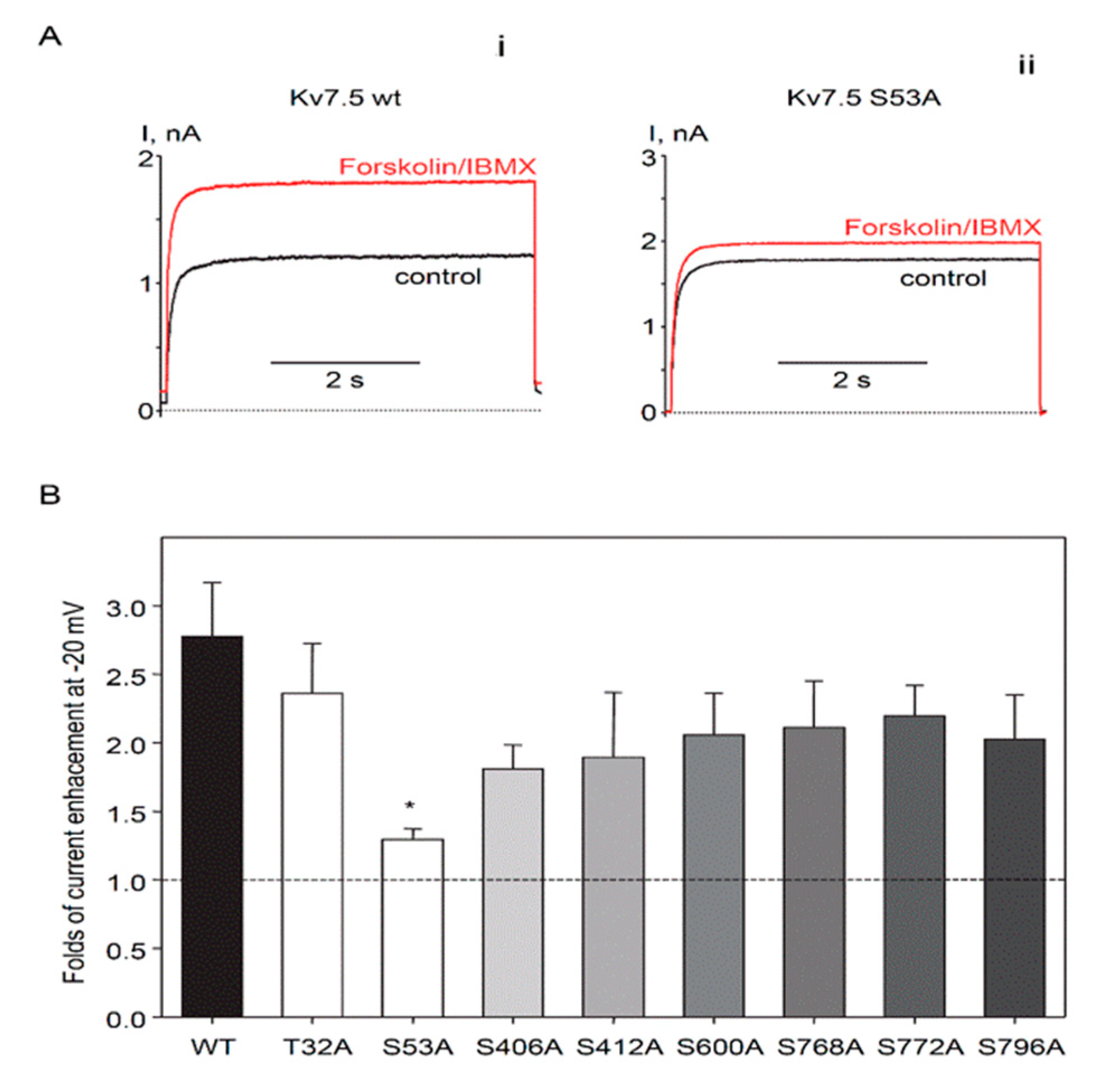
2.3. Identification of PKA Phosphorylation Sites on Human Kv7.5 Channels
3. Discussion
4. Materials and Methods
4.1. Expression Constructs
4.2. Cell Culture
4.3. Quantitative Real Time Reverse-Transcriptase Polymerase Chain Reaction (qRT-PCR)
4.4. Patch-Clamp
4.5. Statistics
4.6. Material
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ANOVA | analysis of variance |
| ASMC | Airway smooth muscle cell |
| cAMP | Cyclic adenosine monophosphate |
| HASMCs | Human airway smooth muscle cells |
| IBMX | 3-Isobutyl-1-methylxanthine |
| PIP2 | Phosphatidylinositol 4,5-bisphosphate |
| PKA | Protein kinase A |
| PKC | Protein kinase C |
| qRT-PCR | Quantitative real time reverse-transcriptase polymerase chain reaction |
| S.E. | Standard error of the mean |
| VSMC | Vascular smooth muscle cell |
| βAR | β-adrenergic receptor |
References
- Brown, D.A. Kv7 (KCNQ) potassium channels that are mutated in human diseases. J. Physiol. 2008, 586, 1781–1783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, D.A.; Passmore, G.M. Neural KCNQ (Kv7) channels. Br. J. Pharmacol. 2009, 156, 1185–1195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haick, J.M.; Byron, K.L. Novel treatment strategies for smooth muscle disorders: Targeting Kv7 potassium channels. Pharmacol. Ther. 2016, 165, 14–25. [Google Scholar] [CrossRef] [PubMed]
- Byron, K.L.; Brueggemann, L.I. Kv7 potassium channels as signal transduction intermediates in the control of microvascular tone. Microcirculation 2018, 25, e12419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwake, M.; Jentsch, T.J.; Friedrich, T. A carboxy-terminal domain determines the subunit specificity of KCNQ K. + channel assembly. EMBO. Rep. 2003, 4, 76–81. [Google Scholar] [CrossRef] [PubMed]
- Delmas, P.; Brown, D.A. Pathways modulating neural KCNQ/M (Kv7) potassium channels. Nat. Rev. Neurosci. 2005, 6, 850–862. [Google Scholar] [CrossRef] [PubMed]
- Stott, J.B.; Jepps, T.A.; Greenwood, I.A. K(V)7 potassium channels: A new therapeutic target in smooth muscle disorders. Drug. Discov. Today 2014, 19, 413–424. [Google Scholar] [CrossRef] [PubMed]
- Mani, B.K.; Robakowski, C.; Brueggemann, L.I.; Cribbs, L.L.; Tripathi, A.; Majetschak, M.; Byron, K.L. Kv7.5 Potassium Channel Subunits Are the Primary Targets for PKA-Dependent Enhancement of Vascular Smooth Muscle Kv7 Currents. Mol. Pharmacol. 2016, 89, 323–334. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, B.C.; Hechenberger, M.; Weinreich, F.; Kubisch, C.; Jentsch, T.J. KCNQ5, a Novel Potassium Channel Broadly Expressed in Brain, Mediates, M.-type Currents. J. Biol. Chem. 2000, 275, 24089–24095. [Google Scholar] [CrossRef] [PubMed]
- Obenauer, J.C.; Cantley, L.C.; Yaffe, M.B. Scansite 2.0: Proteome-wide prediction of cell signaling interactions using short sequence motifs. Nucleic Acids Res. 2003, 31, 3635–3641. [Google Scholar] [CrossRef] [PubMed]
- Haick, J.M.; Brueggemann, L.I.; Cribbs, L.L.; Denning, M.F.; Schwartz, J.; Byron, K.L. PKC-dependent regulation of Kv7.5 channels by the bronchoconstrictor histamine in human airway smooth muscle cells. Am. J. Physiol. Lung. Cell. Mol. Physiol. 2017, 312, L822–Ll834. [Google Scholar] [CrossRef] [PubMed]
- Brueggemann, L.I.; Moran, C.J.; Barakat, J.A.; Yeh, J.Z.; Cribbs, L.L.; Byron, K.L. Vasopressin stimulates action potential firing by protein kinase C-dependent inhibition of KCNQ5 in A7r5 rat aortic smooth muscle cells. Am. J. Physiol. Heart. Circ. Physiol. 2007, 292, H1352–H1363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brueggemann, L.I.; Mackie, A.R.; Martin, J.L.; Cribbs, L.L.; Byron, K.L. Diclofenac distinguishes among homomeric and heteromeric potassium channels composed of KCNQ4 and KCNQ5 subunits. Mol. Pharmacol. 2011, 79, 10–23. [Google Scholar] [CrossRef] [PubMed]
- Peretz, A.; Degani, N.; Nachman, R.; Uziyel, Y.; Gibor, G.; Shabat, D.; Attali, B. Meclofenamic Acid and Diclofenac, Novel Templates of KCNQ2/Q3 Potassium Channel Openers, Depress Cortical Neuron Activity and Exhibit Anticonvulsant Properties. Mol. Pharmacol. 2005, 67, 1053–1066. [Google Scholar] [CrossRef] [PubMed]
- Brueggemann, L.I.; Mackie, A.R.; Cribbs, L.L.; Freda, J.; Tripathi, A.; Majetschak, M.; Byron, K.L. Differential Protein Kinase C-Dependent Modulation of Kv7.4 and Kv7.5 Subunits of Vascular Kv7 Channels. J. Biol. Chem. 2013, 289, 2099–2111. [Google Scholar] [PubMed]
- Chadha, P.S.; Jepps, T.A.; Carr, G.; Stott, J.B.; Zhu, H.-L.; Cole, W.C.; Greenwood, I.A. Contribution of Kv7.4/Kv7.5 heteromers to intrinsic and calcitonin gene-related peptide–induced cerebral reactivity. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 887–893. [Google Scholar] [CrossRef] [PubMed]
- Oliveras, A.; Roura-Ferrer, M.; Sole, L.; de la Cruz, A.; Prieto, A.; Etxebarria, A.; Manils, J.; Morales-Cano, D.; Condom, E.; Soler, C.; Cogolludo, A.; Valenzuela, C.; Villarroel, A.; Comes, N.; Felipe, A. Functional assembly of Kv7.1/Kv7.5 channels with emerging properties on vascular muscle physiology. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 1522–1530. [Google Scholar] [CrossRef] [PubMed]
- Volkers, L.; Rook, M.B.; Das, J.H.; Verbeek, N.E.; Groenewegen, W.A.; van Kempen, M.J.; Lindhout, D.; Koeleman, B.P. Functional analysis of novel KCNQ2 mutations found in patients with Benign Familial Neonatal Convulsions. Neurosci. Lett. 2009, 462, 24–29. [Google Scholar] [CrossRef] [PubMed]
- Brueggemann, L.I.; Kakad, P.P.; Love, R.B.; Solway, J.; Dowell, M.L.; Cribbs, L.L.; Byron, K.L. Kv7 potassium channels in airway smooth muscle cells: Signal transduction intermediates and pharmacological targets for bronchodilator therapy. Am. J. Physiol. Lung. Cell. Mol. Physiol. 2012, 302, L120–L132. [Google Scholar] [CrossRef] [PubMed]
- Brueggemann, L.I.; Haick, J.M.; Neuburg, S.; Tate, S.; Randhawa, D.; Cribbs, L.L.; Byron, K.L. KCNQ (Kv7) potassium channel activators as bronchodilators: Combination with a β2-adrenergic agonist enhances relaxation of rat airways. Am. J. Physiol. Lung. Cell. Mol. Physiol. 2014, 306, L476–L486. [Google Scholar] [CrossRef] [PubMed]
- Evseev, A.I.; Semenov, I.; Archer, C.R.; Medina, J.L.; Dube, P.H.; Shapiro, M.S.; Brenner, R. Functional effects of KCNQ K + channels in airway smooth muscle. Front. Physiol. 2013, 4. [Google Scholar] [CrossRef] [PubMed]
- Penn, R.B.; Benovic, J.L. Regulation of Heterotrimeric G Protein Signaling in Airway Smooth Muscle. Proc. Am. Thorac. Soc. 2008, 5, 47–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Byron, K.L.; Brueggemann, L.I.; Kakad, P.P.; Haick, J.M. Kv7 (KCNQ) potassium channels and L-type calcium channels in the regulation of airway diameter. In Calcium Signaling In Airway Smooth Muscle Cells; Wang, Y.-X., Ed.; Springer: New York, NY, USA, 2014; pp. 21–33. [Google Scholar]
- Johnson, M. The beta-adrenoceptor. Am. J. Respir. Crit. Care. Med. 1998, 158 Pt 3, S146–S153. [Google Scholar] [CrossRef] [PubMed]
- Anderson, G.P. Current issues with β2-adrenoceptor agonists: Pharmacology and molecular and cellular mechanisms. Clin. Rev. Allergy. Immunol. 2006, 31, 119–130. [Google Scholar] [CrossRef]
- Voolstra, O.; Rhodes-Mordov, E.; Katz, B.; Bartels, J.P.; Oberegelsbacher, C.; Schotthofer, S.K.; Yasin, B.; Tzadok, H.; Huber, A.; Minke, B. The Phosphorylation State of the Drosophila TRP Channel Modulates the Frequency Response to Oscillating Light. In Vivo J. Neurosci. 2017, 37, 4213–4224. [Google Scholar] [CrossRef] [PubMed]
- Heijman, J.; Spatjens, R.L.; Seyen, S.R.; Lentink, V.; Kuijpers, H.J.; Boulet, I.R.; de Windt, L.J.; David, M.; Volders, P.G. Dominant-negative control of cAMP-dependent IKs upregulation in human long-QT syndrome type 1. Circ. Res. 2012, 110, 211–219. [Google Scholar] [CrossRef] [PubMed]
- Suh, B.C.; Hille, B. Regulation of KCNQ channels by manipulation of phosphoinositides. J. Physiol. 2007, 582 Pt 3, 911–916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Gamper, N.; Hilgemann, D.W.; Shapiro, M.S. Regulation of Kv7 (KCNQ) K + channel open probability by phosphatidylinositol 4,5-bisphosphate. J. Neurosci. 2005, 25, 9825–9835. [Google Scholar] [CrossRef] [PubMed]
- Lopes, C.M.; Remon, J.I.; Matavel, A.; Sui, J.L.; Keselman, I.; Medei, E.; Shen, Y.; Rosenhouse-Dantsker, A.; Rohacs, T.; Logothetis, D.E. Protein kinase A modulates PLC-dependent regulation and PIP2-sensitivity of K.+ channels. Channels 2007, 1, 124–134. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, B.C.; Kubisch, C.; Stein, V.; Jentsch, T.J. Moderate loss of function of cyclic-AMP-modulated KCNQ2/KCNQ3 K+ channels causes epilepsy. Nature 1998, 396, 687–690. [Google Scholar] [CrossRef] [PubMed]
- Salzer, I.; Erdem, F.A.; Chen, W.Q.; Heo, S.; Koenig, X.; Schicker, K.W.; Kubista, H.; Lubec, G.; Boehm, S.; Yang, J.W. Phosphorylation regulates the sensitivity of voltage-gated Kv7.2 channels towards phosphatidylinositol-4,5-bisphosphate. J. Physiol. 2017, 595, 759–776. [Google Scholar] [CrossRef] [PubMed]
- Stott, J.B.; Povstyan, O.V.; Carr, G.; Barrese, V.; Greenwood, I.A. G-protein βγ subunits are positive regulators of Kv7.4 and native vascular Kv7 channel activity. Proc. Natl. Acad. Sci. USA 2015, 112, 6497–6502. [Google Scholar] [CrossRef] [PubMed]
- Povstyan, O.V.; Barrese, V.; Stott, J.B.; Greenwood, I.A. Synergistic interplay of Gβγ and phosphatidylinositol 4,5-bisphosphate dictates Kv7.4 channel activity. Pflugers. Arch. 2017, 469, 213–223. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Primer Sequence | Product Size (bp) |
|---|---|---|
| KCNQ1 | F: 5′-AAC CTC ATG GTG CGC ATC AAG-3′ R: CCG CGA TCC TTG CTC TTT TCT G -3′ | 101 |
| KCNQ2 | F: 5′-CGG AAA CCG TTC TGT GTG ATT GAC-3′ R:5′-ATC C GCA GAA TCT GCA GGA AG C-3′ | 131 |
| KCNQ3 | F: 5′-CCA CGC CAA AAC ACA AGA AGT CT-3′ R: 5′-TGA TGT GGA TGG TCT GGC TAC A-3′ | 101 |
| KCNQ4 | F: 5′-TGCG ACC GTA CGA CGT GAA G-3′ R: 5′-CAA TTT GGT CCA CCC GAG TTT GC-3′ | 102 |
| KCNQ5 | F: 5′-CCATCCCTGAGCACACAAAATTGGC -3′ R: CACCCTGACACATAAACCCTG-3′ | 109 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brueggemann, L.I.; Cribbs, L.L.; Schwartz, J.; Wang, M.; Kouta, A.; Byron, K.L. Mechanisms of PKA-Dependent Potentiation of Kv7.5 Channel Activity in Human Airway Smooth Muscle Cells. Int. J. Mol. Sci. 2018, 19, 2223. https://doi.org/10.3390/ijms19082223
Brueggemann LI, Cribbs LL, Schwartz J, Wang M, Kouta A, Byron KL. Mechanisms of PKA-Dependent Potentiation of Kv7.5 Channel Activity in Human Airway Smooth Muscle Cells. International Journal of Molecular Sciences. 2018; 19(8):2223. https://doi.org/10.3390/ijms19082223
Chicago/Turabian StyleBrueggemann, Lyubov I., Leanne L. Cribbs, Jeffrey Schwartz, Minhua Wang, Ahmed Kouta, and Kenneth L. Byron. 2018. "Mechanisms of PKA-Dependent Potentiation of Kv7.5 Channel Activity in Human Airway Smooth Muscle Cells" International Journal of Molecular Sciences 19, no. 8: 2223. https://doi.org/10.3390/ijms19082223
APA StyleBrueggemann, L. I., Cribbs, L. L., Schwartz, J., Wang, M., Kouta, A., & Byron, K. L. (2018). Mechanisms of PKA-Dependent Potentiation of Kv7.5 Channel Activity in Human Airway Smooth Muscle Cells. International Journal of Molecular Sciences, 19(8), 2223. https://doi.org/10.3390/ijms19082223