Restraining Akt1 Phosphorylation Attenuates the Repair of Radiation-Induced DNA Double-Strand Breaks and Reduces the Survival of Irradiated Cancer Cells


 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. DNA-PKcs Participates in Radiation-Induced Akt S473 Phosphorylation in Glioblastoma Cells
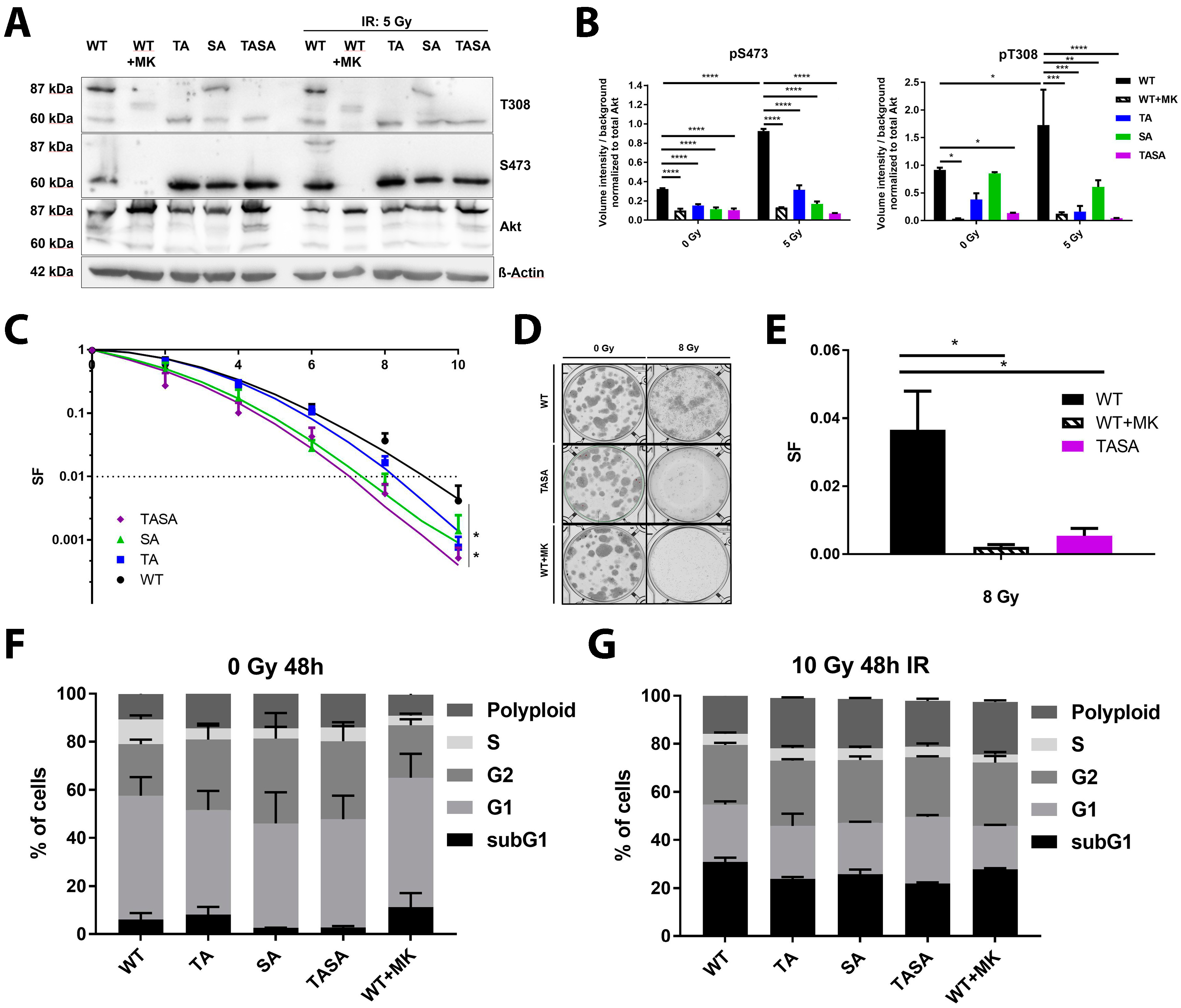
2.2. Phosphorylation-Deficient Mutants Akt1-SA and -TASA Enhance the Radiosensitivity of TrC1 Prostate Cancer Cells
2.3. Phosphorylation Status of Akt Is Not Crucial for Nuclear Localization and Its Translocation Upon IR
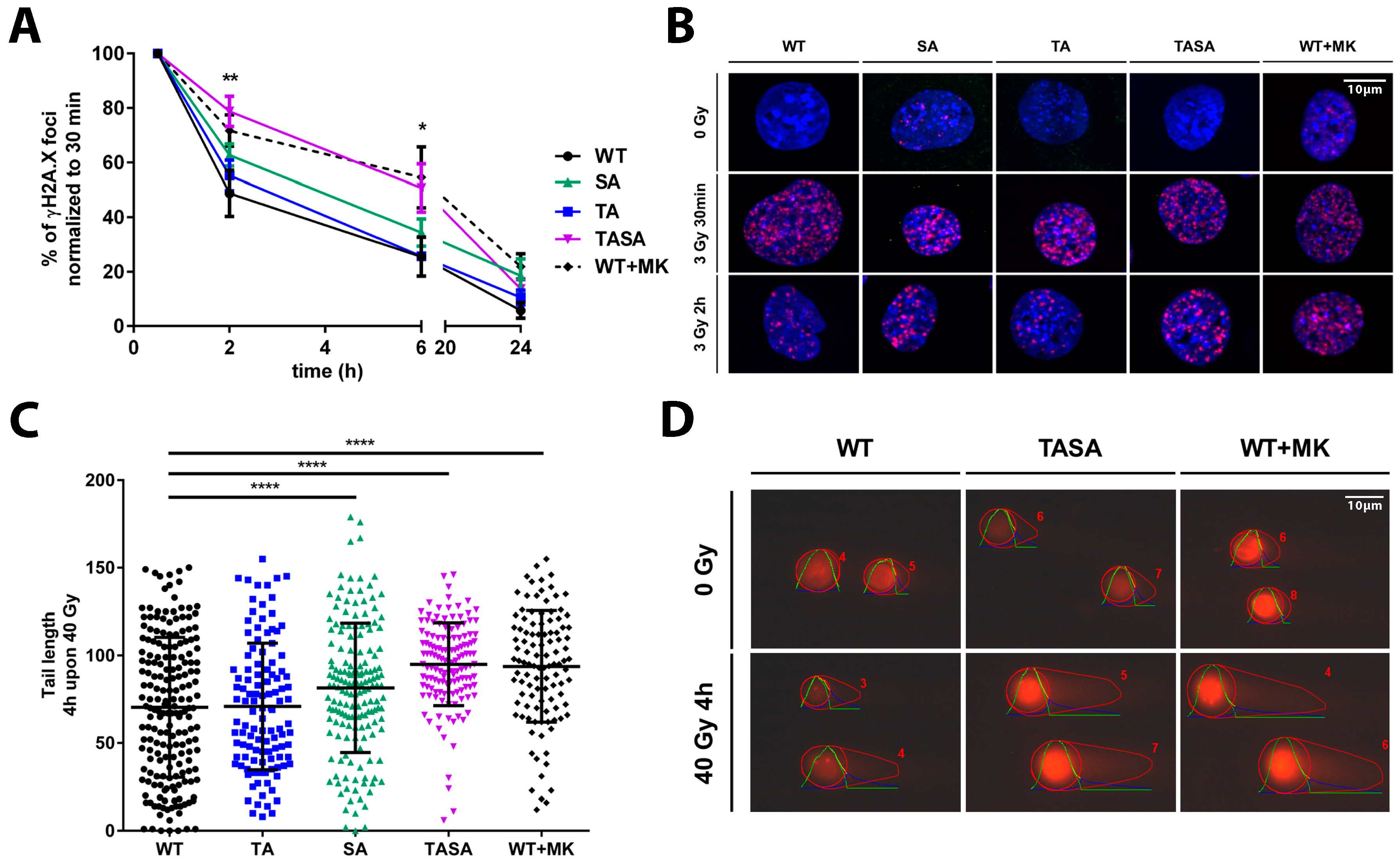
2.4. Overexpression of the Phosphorylation-Deficient Akt1-TASA Mutant Delays the Kinetics of DNA Repair Upon Irradiation, Potentially via Decreased Phosphorylation of Effector Proteins with an Impact on DSB Repair
3. Discussion
4. Materials and Methods
4.1. Chemicals, Antibodies, and Drugs
4.2. Cell Culture, Drug Treatment, and Irradiation
4.3. Generating Stably Akt1 Mutant Expressing Cells
4.4. Immunofluorescence Staining
4.5. Colony Formation Assay
4.6. Comet Assay
4.7. Flow Cytometry
4.8. Statistical Analysis
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Fruman, D.A.; Rommel, C. PI3K and Cancer: Lessons, Challenges and Opportunities. Nat. Rev. Drug Discov. 2014, 13, 140–156. [Google Scholar] [CrossRef] [PubMed]
- Brognard, J.; Clark, A.S.; Ni, Y.; Brognard, J.; Clark, A.S.; Ni, Y.; Dennis, P.A. Akt/Protein Kinase B Is Constitutively Active in Non-Small Cell Lung Cancer Cells and Promotes Cellular Survival and Resistance to Chemotherapy and Radiation Akt/Protein Kinase B Is Constitutively Active in Non-Small Cell Lung Cancer Cells and Promote. Cancer Res. 2001, 61, 3986–3997. [Google Scholar] [PubMed]
- Danielsen, S.A.; Eide, P.W.; Nesbakken, A.; Guren, T.; Leithe, E.; Lothe, R.A. Portrait of the PI3K/AKT pathway in colorectal cancer. Biochim. Biophys. Acta 2015, 1855, 104–121. [Google Scholar] [CrossRef] [PubMed]
- Manning, B.D.; Toker, A. AKT/PKB Signaling: Navigating the Network. Cell 2017, 169, 381–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szymonowicz, K.; Oeck, S.; Malewicz, N.M.; Jendrossek, V. New insights into protein kinase B/Akt signaling: Role of localized Akt activation and compartment-specific target proteins for the cellular radiation response. Cancers 2018, 10, 78. [Google Scholar] [CrossRef] [PubMed]
- Bozulic, L.; Surucu, B.; Hynx, D.; Hemmings, B.A. PKBα/Akt1 Acts Downstream of DNA-PK in the DNA Double-Strand Break Response and Promotes Survival. Mol. Cell 2008, 30, 203–213. [Google Scholar] [CrossRef] [PubMed]
- Oeck, S.; Al-Refae, K.; Riffkin, H.; Wiel, G.; Handrick, R.; Klein, D.; Iliakis, G.; Jendrossek, V. Activating Akt1 mutations alter DNA double strand break repair and radiosensitivity. Sci. Rep. 2017, 7, 42700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toulany, M.; Schickfluss, T.-A.; Fattah, K.R.; Lee, K.-J.; Chen, B.P.C.; Fehrenbacher, B.; Schaller, M.; Chen, D.J.; Rodemann, H.P. Function of erbB receptors and DNA-PKcs on phosphorylation of cytoplasmic and nuclear Akt at S473 induced by erbB1 ligand and ionizing radiation. Radiother. Oncol. 2011, 101, 140–146. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Li, X.; Liu, Q.; Xu, J.; Ge, H.; Wang, Z.; Wang, H.; Wang, Z.; Shi, C.; Xu, X.; et al. UBE2S, a novel substrate of Akt1, associates with Ku70 and regulates DNA repair and glioblastoma multiforme resistance to chemotherapy. Oncogene 2017, 36, 1145–1156. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Gan, W.; Guo, C.; Xie, A.; Gao, D.; Guo, J.; Zhang, J.; Willis, N.; Su, A.; Asara, J.M.; et al. Akt-mediated phosphorylation of XLF impairs non-homologous end-joining DNA repair. Mol. Cell 2015, 57, 648–661. [Google Scholar] [CrossRef] [PubMed]
- Ezell, S.A.; Polytarchou, C.; Hatziapostolou, M.; Guo, A.; Sanidas, I.; Bihani, T.; Comb, M.J.; Sourvinos, G.; Tsichlis, P.N. The protein kinase Akt1 regulates the interferon response through phosphorylation of the transcriptional repressor EMSY. Proc. Natl. Acad. Sci. USA 2012, 109, E613–E621. [Google Scholar] [CrossRef] [PubMed]
- Mueck, K.; Rebholz, S.; Harati, M.; Rodemann, H.; Toulany, M. Akt1 Stimulates Homologous Recombination Repair of DNA Double-Strand Breaks in a Rad51-Dependent Manner. Int. J. Mol. Sci. 2017, 18, 2473. [Google Scholar] [CrossRef] [PubMed]
- Holler, M.; Grottke, A.; Mueck, K.; Manes, J.; Jücker, M.; Rodemann, H.P.; Toulany, M. Dual Targeting of Akt and mTORC1 Impairs Repair of DNA Double-Strand Breaks and Increases Radiation Sensitivity of Human Tumor Cells. PLoS ONE 2016, 11, e0154745. [Google Scholar] [CrossRef] [PubMed]
- Shao, G.; Patterson-Fortin, J.; Messick, T.E.; Feng, D.; Shanbhag, N.; Wang, Y.; Greenberg, R.A. MERIT40 controls BRCA1-Rap80 complex integrity and recruitment to DNA double-strand breaks. Genes Dev. 2009, 23, 740–754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, K.K.; Montaser-Kouhsari, L.; Beck, A.H.; Toker, A. MERIT40 Is an Akt Substrate that Promotes Resolution of DNA Damage Induced by Chemotherapy. Cell Rep. 2015, 11, 1358–1366. [Google Scholar] [CrossRef] [PubMed]
- DiBiase, S.J.; Zeng, Z.C.; Chen, R.; Hyslop, T.; Curran, W.J.; Iliakis, G. DNA-dependent protein kinase stimulates an independently active, nonhomologous, end-joining apparatus. Cancer Res. 2000, 60, 1245–1253. [Google Scholar] [PubMed]
- Dibiase, S.J.; Zeng, Z.-C.; Chen, R.; Hyslop, T.; Curran, W.J.; Iliakis, G. Active, Nonhomologous, End-Joining Apparatus DNA-dependent Protein Kinase Stimulates an Independently DNA-dependent Protein Kinase Stimulates an Independently Active, Nonhomologous, End-Joining Apparatus. Cancer Res. 2000, 60, 1245–1253. [Google Scholar] [PubMed]
- Kloet, D.E.A.; Burgering, B.M.T. The PKB/FOXO switch in aging and cancer. Biochim. Biophys. Acta 2011, 1813, 1926–1937. [Google Scholar] [CrossRef] [PubMed]
- Lamprecht, M.R.; Sabatini, D.M.; Carpenter, A.E. CellProfiler TM: Free, versatile software for automated biological image analysis. Biotechniques 2007, 42, 71–75. [Google Scholar] [CrossRef] [PubMed]
- Oeck, S.; Malewicz, N.M.; Hurst, S.; Al-Refae, K.; Krysztofiak, A.; Jendrossek, V. The Focinator v2-0—Graphical Interface, Four Channels, Colocalization Analysis and Cell Phase Identification. Radiat. Res. 2017, 188, 114–120. [Google Scholar] [CrossRef] [PubMed]
- Vikrant; Sawant, U.U.; Varma, A.K. Role of MERIT40 in stabilization of BRCA1 complex: A protein-protein interaction study. Biochem. Biophys. Res. Commun. 2014, 446, 1139–1144. [Google Scholar] [CrossRef] [PubMed]
- Gyorffy, B.; Lánczky, A.; Szállási, Z. Implementing an online tool for genomewide validation of survival-associated biomarkers in ovarian-cancer using microarray data from 1287 patients. Endocr. Relat. Cancer 2012, 19, 197–208. [Google Scholar] [CrossRef] [PubMed]
- Szász, A.M.; Lánczky, A.; Nagy, Á.; Förster, S.; Hark, K.; Green, J.E.; Boussioutas, A.; Busuttil, R.; Szabó, A.; Győrffy, B. Cross-validation of survival associated biomarkers in gastric cancer using transcriptomic data of 1065 patients. Oncotarget 2016, 7, 49322–49333. [Google Scholar] [CrossRef] [PubMed]
- Surucu, B.; Bozulic, L.; Hynx, D.; Parcellier, A.; Hemmings, B.A. In vivo analysis of protein kinase B (PKB)/Akt regulation in DNA-PKcs-null mice reveals a role for PKB/Akt in DNA damage response and tumorigenesis. J. Biol. Chem. 2008, 283, 30025–30033. [Google Scholar] [CrossRef] [PubMed]
- Stronach, E.A.; Chen, M.; Maginn, E.N.; Agarwal, R.; Mills, G.B.; Wasan, H.; Gabra, H. DNA-PK Mediates AKT Activation and Apoptosis Inhibition in Clinically Acquired Platinum Resistance. Neoplasia 2011, 13, 1069–1080. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhang, L.; Liu, Y.; Sun, C.; Zhang, H.; Miao, G.; Di, C.X.; Zhou, X.; Zhou, R.; Wang, Z. DNA-PKcs deficiency inhibits glioblastoma cell-derived angiogenesis after ionizing radiation. J. Cell. Physiol. 2015, 230, 1094–1103. [Google Scholar] [CrossRef] [PubMed]
- Toulany, M.; Kehlbach, R.; Florczak, U.; Sak, A.; Wang, S.; Chen, J.; Lobrich, M.; Rodemann, H.P. Targeting of AKT1 enhances radiation toxicity of human tumor cells by inhibiting DNA-PKcs-dependent DNA double-strand break repair. Mol. Cancer Ther. 2008, 7, 1772–1781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, T.P.; Suman, S.; Alatassi, H.; Ankem, M.K.; Damodaran, C. Inhibition of AKT promotes FOXO3a-dependent apoptosis in prostate cancer. Cell Death Dis. 2016, 7, e2111. [Google Scholar] [CrossRef] [PubMed]
- Hou, T.; Li, Z.; Zhao, Y.; Zhu, W.-G. Mechanisms controlling the anti-neoplastic functions of FoxO proteins. Semin. Cancer Biol. 2018, 50, 101–114. [Google Scholar] [CrossRef] [PubMed]
- Boronenkov, I.V.; Loijens, J.C.; Umeda, M.; Anderson, R.A. Phosphoinositide signaling pathways in nuclei are associated with nuclear speckles containing pre-mRNA processing factors. Mol. Biol. Cell 1998, 9, 3547–3560. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Redondo-Muñoz, J.; Perez-García, V.; Cortes, I.; Chagoyen, M.; Carrera, A.C. Nuclear but not cytosolic phosphoinositide 3-kinase beta has an essential function in cell survival. Mol. Cell. Biol. 2011, 31, 2122–2133. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Fernandez-Capetillo, O.; Fernadez-Capetillo, O.; Carrera, A.C. Nuclear phosphoinositide 3-kinase beta controls double-strand break DNA repair. Proc. Natl. Acad. Sci. USA 2010, 107, 7491–7496. [Google Scholar] [CrossRef] [PubMed]
- Alessi, D.R.; Andjelkovic, M.; Caudwell, B.; Cron, P.; Morrice, N.; Cohen, P.; Hemmings, B.A. Mechanism of activation of protein kinase B by insulin and IGF-1. EMBO J. 1996, 15, 6541–6551. [Google Scholar] [PubMed]
- Saji, M.; Vasko, V.; Kada, F.; Allbritton, E.H.; Burman, K.D.; Ringel, M.D. Akt1 contains a functional leucine-rich nuclear export sequence. Biochem. Biophys. Res. Commun. 2005, 332, 167–173. [Google Scholar] [CrossRef] [PubMed]
- Adini, I.; Rabinovitz, I.; Sun, J.F.; Prendergast, G.C.; Benjamin, L.E. RhoB controls Akt trafficking and stage-specific survival of endothelial cells during vascular development. Genes Dev. 2003, 17, 2721–2732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xuan Nguyen, T.L.; Choi, J.W.; Lee, S.B.; Ye, K.; Woo, S.-D.; Lee, K.-H.; Ahn, J.-Y. Akt phosphorylation is essential for nuclear translocation and retention in NGF-stimulated PC12 cells. Biochem. Biophys. Res. Commun. 2006, 349, 789–798. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Hu, C.; Li, J.; Xue, P.; He, X.; Ge, C.; Qin, W.; Yao, G.; Gu, J. Real-time imaging nuclear translocation of Akt1 in HCC cells. Biochem. Biophys. Res. Commun. 2007, 356, 1038–1043. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Q.; Paramasivam, M.; Aressy, B.; Wu, J.; Bellani, M.; Tong, W.; Seidman, M.M.; Greenberg, R.A. MERIT40 cooperates with BRCA2 to resolve DNA interstrand cross-links. Genes Dev. 2015, 29, 1955–1968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, L.; Huang, T.; Chen, J. MERIT40 facilitates BRCA1 localization and DNA damage repair. Genes Dev. 2009, 23, 719–728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olive, P.L.; Banáth, J.P. The comet assay: A method to measure DNA damage in individual cells. Nat. Protoc. 2006, 1, 23–29. [Google Scholar] [CrossRef] [PubMed]





© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Szymonowicz, K.; Oeck, S.; Krysztofiak, A.; Van der Linden, J.; Iliakis, G.; Jendrossek, V. Restraining Akt1 Phosphorylation Attenuates the Repair of Radiation-Induced DNA Double-Strand Breaks and Reduces the Survival of Irradiated Cancer Cells. Int. J. Mol. Sci. 2018, 19, 2233. https://doi.org/10.3390/ijms19082233
Szymonowicz K, Oeck S, Krysztofiak A, Van der Linden J, Iliakis G, Jendrossek V. Restraining Akt1 Phosphorylation Attenuates the Repair of Radiation-Induced DNA Double-Strand Breaks and Reduces the Survival of Irradiated Cancer Cells. International Journal of Molecular Sciences. 2018; 19(8):2233. https://doi.org/10.3390/ijms19082233
Chicago/Turabian StyleSzymonowicz, Klaudia, Sebastian Oeck, Adam Krysztofiak, Jansje Van der Linden, George Iliakis, and Verena Jendrossek. 2018. "Restraining Akt1 Phosphorylation Attenuates the Repair of Radiation-Induced DNA Double-Strand Breaks and Reduces the Survival of Irradiated Cancer Cells" International Journal of Molecular Sciences 19, no. 8: 2233. https://doi.org/10.3390/ijms19082233
APA StyleSzymonowicz, K., Oeck, S., Krysztofiak, A., Van der Linden, J., Iliakis, G., & Jendrossek, V. (2018). Restraining Akt1 Phosphorylation Attenuates the Repair of Radiation-Induced DNA Double-Strand Breaks and Reduces the Survival of Irradiated Cancer Cells. International Journal of Molecular Sciences, 19(8), 2233. https://doi.org/10.3390/ijms19082233